 Lei Liu1†
Lei Liu1† Tingying Lei
Tingying Lei- 1Department of Obstetrics, Guangzhou Women and Children’s Medical Center, Guangzhou Medical University, Guangzhou, China
- 2Prenatal Diagnostic Center, Guangzhou Women and Children’s Medical Center, Guangzhou Medical University, Guangzhou, China
- 3Prenatal Diagnostic Center, Guangzhou Women and Children’s Medical Center, Southern Medical University, Guangzhou, China
Objective: The recurrent 1q21.1 microdeletion syndrome is an autosomal dominant disorder and is characterized by dysmorphic facial features, microcephaly, developmental delay, and congenital defects. However, most studies on the distal deletions in the 1q21.1 region were diagnosed postnatally. This study aimed to provide a better understanding of the ultrasound and molecular findings of fetuses with recurrent 1q21.1 microdeletions in prenatal diagnosis.
Methods: In this retrospective study, we reported 21 cases with the recurrent 1q21.1 microdeletion syndrome diagnosed at our prenatal diagnostic center from January 2016 to January 2023. The clinical data were reviewed for these cases, including the maternal demographics, indications for invasive testing, ultrasound findings, CMA results, and pregnancy outcomes.
Results: In the study, a total of 21 cases with recurrent 1q21.1 microdeletions were diagnosed prenatally by CMA. Fifteen cases were described with ultrasound indications, and the most common findings are as follows: increased nuchal translucency (NT) (26.7%), intrauterine growth retardation (IUGR) (26.7%), congenital heart defects (CHD) (20%), and congenital anomalies of the kidney and urinary tract (CAKUT) (13.3%). All the cases with the distal 1q21.1 deletions contain the common minimal region (located between BP3 and BP4) and eight OMIM genes. Parental studies to determine the inheritance of the deletion were performed for eight cases, and half of the cases were inherited from one of the parents. Pregnancy outcomes were available for nine cases; eight (88.9%) pregnancies were determined to be terminated and one (11.1%) was full-term delivery.
Conclusion: To our knowledge, this is the largest study to find that fetuses with recurrent 1q21.1 microdeletions were closely associated with increased NT, CHD, IUGR, and CAKUT. In addition, ours is the first study to report that cerebral ventriculomegaly might be associated with recurrent 1q21.1 microdeletions. More comprehensive studies are needed for a better understanding of the prenatal phenotype–genotype relationship of the recurrent 1q21.1 microdeletion syndrome in future.
Introduction
The recurrent 1q21.1 microdeletion syndrome (OMIM # 612474) is an autosomal dominant contiguous gene deletion syndrome, which occurs at the 1q21.1 distal region, extending from BP3 to BP4 (GRCh37/hg19: chr1:146533376–147883376), with a size range of 800 kb to 2 Mb, and includes at least eight genes: PRKAB2, FMO5, CHD1L, BCL9, ACP6, GJA5, GJA8, and GPR89B (Mefford et al., 2008). Individuals with the recurrent 1q21.1 microdeletion have a wide range of clinical manifestations, ranging from unaffected to severely affected. The most common findings include mildly dysmorphic but nonspecific facial features (>75%), mild intellectual disability or learning disabilities (25%), microcephaly (43%), and eye abnormalities (26%) (Edwards et al., 2021; Bourgois et al., 2024). Other findings include congenital heart defects (CHD), congenital anomalies of the kidney and urinary tract (CAKUT), skeletal malformations, joint laxity, seizures (∼23%), and psychiatric conditions such as attention deficit hyperactivity disorder (ADHD), autism spectrum disorder (ASD), and behavioral anomalies (Bernier et al., 2016; Brunetti-Pierri et al., 2008; Digilio et al., 2013; Buse et al., 2017; Upadhyai et al., 2020). However, the cause of the phenotypic variability associated with the deletions remains largely unexplained. Thus, it is challenging to provide genetic counseling for these patients. In addition, most studies on the distal deletions in the 1q21.1 region were diagnosed postnatally, and prenatal reports involving the recurrent 1q21.1 microdeletions were limited (Edwards et al., 2021; Bourgois et al., 2024; Bernier et al., 2016). To better understand these prenatally detected chromosomal microscopic imbalances, we present the findings of ultrasound and molecular analysis of 21 cases with recurrent 1q21.1 microdeletions, especially in pregnant women undergoing prenatal invasive testing, and provide a systematic summary of prenatal phenotypes for such genomic disorders.
Materials and methods
This study was a retrospective study approved by the Institutional Review Board of the Ethics Committee of Guangzhou Women and Children’s Medical Center, and it met with the ethical standards of experiments on human subjects. Twenty-one prenatal cases of recurrent 1q21.1 microdeletions were recruited in the Prenatal Diagnostic Center, Guangzhou Women and Children’s Medical Center from January 2016 to January 2023. Only the cases with pure distal 1q21.1 deletions were included, and those involving other pathogenic chromosomal microscopic imbalances were excluded. The clinical data were reviewed for these cases, including the maternal demographics, indications for invasive testing, ultrasound findings, chromosomal microarray analysis (CMA) results, and pregnancy outcomes. The data are given as the median (range) or n (%).
The main indications for prenatal diagnosis included ultrasound anomalies, serum screening results for aneuploidy, and advanced maternal age. Fetal samples were collected using amniocentesis (<25 weeks) or cord blood sampling (≥25 weeks or if oligohydramnios was present). Parental blood samples were obtained at the same time. During the study period, with very few exceptions, the CMA was used as a first-tier technology for prenatal diagnosis in fetuses. The CMA platform used was the CytoScan 750 K Array (Affymetrix Inc., Santa Clara, CA, United States), containing 750,436 25-85-mer oligonucleotide probes, including 550,000 nonpolymorphic (NP) probes and 200,436 single-nucleotide polymorphic (SNP) probes (100 Kb resolution). Genomic coordinates are given in GRCh37/hg19. Genomic coordinates in GRCh38/hg38 were remapped to GRCh37/hg19 using the UCSC LiftOver tool (https://genome.ucsc.edu/cgi-bin/hgLiftOver). All patients were offered counseling by a maternal–fetal medicine team, including genetic counselors, before testing and after the diagnosis of 1q21.1 microdeletion.
Results
In the study, a total of 21 cases with recurrent 1q21.1 microdeletions were diagnosed prenatally by CMA (Figure 1). The major fetal and parental clinical indications are summarized in Table 1. The median maternal age was 30 (21–42) years. The median gestational age at prenatal diagnosis was 19 (17–31) weeks. Fifteen cases manifested mild-to-moderate prenatal ultrasound indications at different gestational weeks, including increased nuchal translucency (NT) (cases 8–11, 26.7%), intrauterine growth retardation (IUGR) (cases 12–15, 26.7%), CHD (cases 16–18, 20%), CAKUT (cases 16 and 20, 13.3%), bilateral ventriculomegaly (case 19, 6.7%), and polyhydramnios (case 21, 6.7%). For the other seven cases (cases 1–7), prenatal diagnosis was carried out not based on ultrasound indications but on the advanced maternal age (cases 1–5) and the high risk of results of the serological screening (trisomy 21) (cases 6 and 7). Interestingly, there was no obvious abnormality on ultrasound in any of the seven cases according to the subsequent examinations.
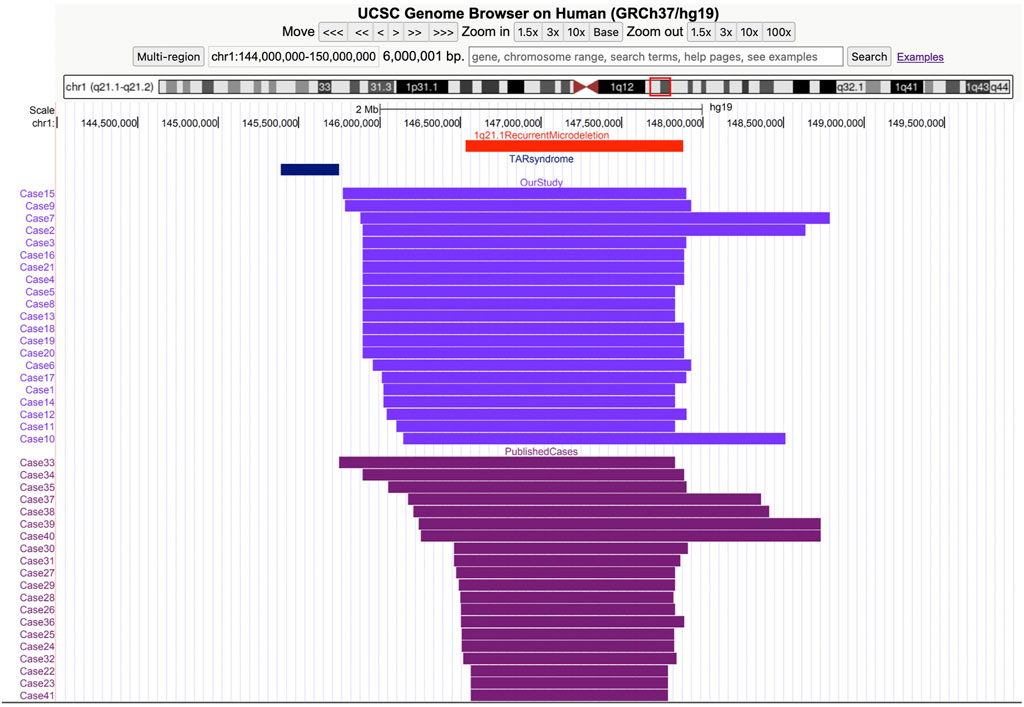
Figure 1. Screenshot from the UCSC Genome Browser (GRCh37/hg19 assembly) showing the coordinates of the 1q21.1 recurrent microdeletion syndrome. Thrombocytopenia absent radius (TAR) syndrome and the 1q21.1 deletions of cases in our study and the previous study.
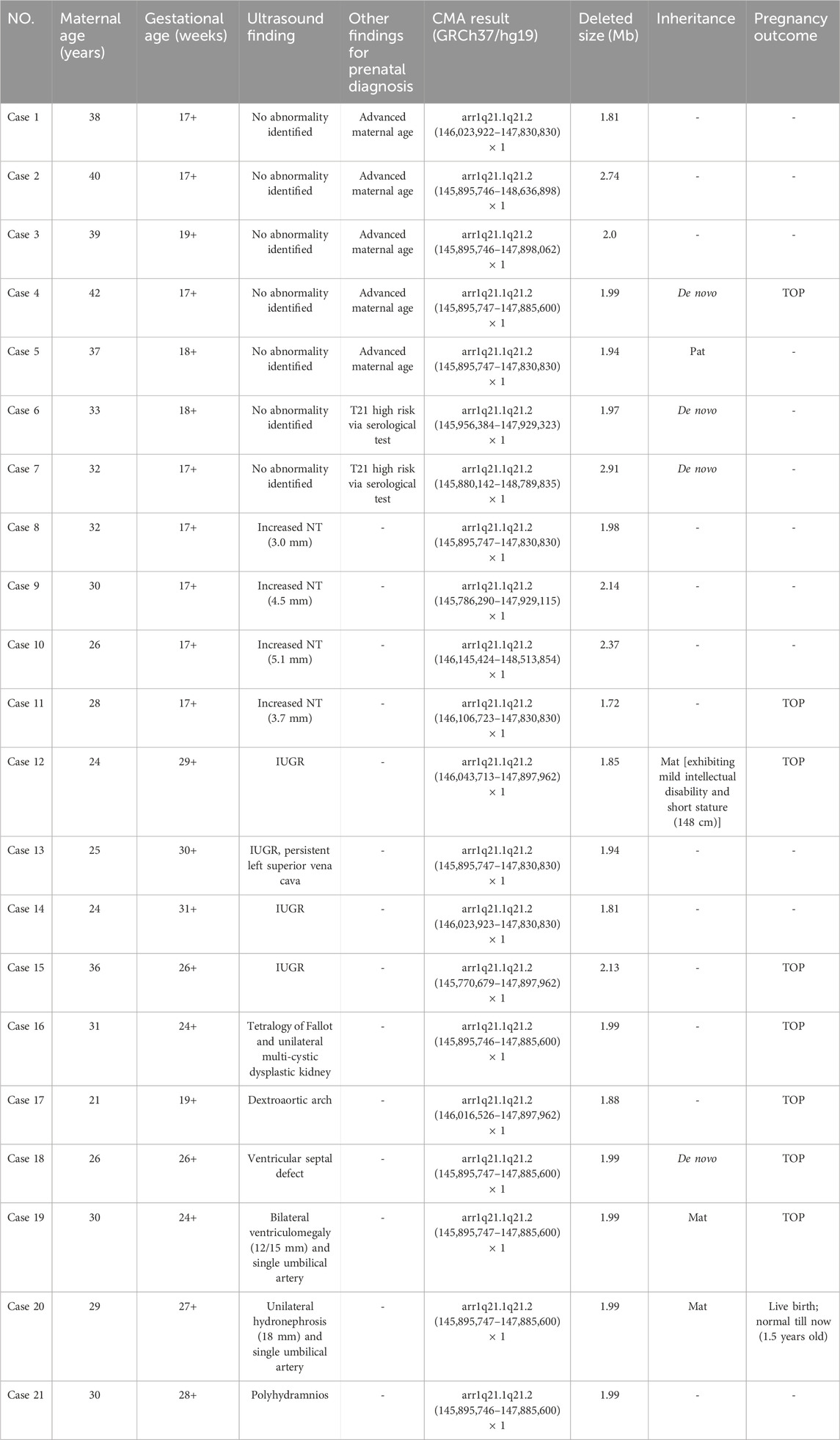
Table 1. Clinical data of fetuses with recurrent 1q21.1 microdeletions detected by CMA in our study.
In the 21 cases with distal 1q21.1 deletions, the minimum fragment length is 1.72 Mb (Case 11) and the maximum is 2.91 Mb (Case 7). All the segmental deletions contain the common minimal region (1.35 Mb, located between BP3 and BP4) and eight OMIM genes that are unique to the region: GJA5, GJA8, ACP6, BCL9, CHD1L, FMO5, GPR89B, and PRKAB2 (Figure 2). Parental studies to determine the inheritance of the deletion were performed for eight cases. Of these, four cases (50%, 4/8) were inherited from one of the parents (cases 5, 12, 19, and 20), and four (50%, 4/8) were confirmed to be de novo (cases 4, 6, 7, and 18). The mother of Case 12 exhibited mild intellectual disability and short stature (148 cm), and the parents of seven other cases were asymptomatic. Parental specimens were not available for the remaining 13 cases. Pregnancy outcomes were available for nine cases. Of these, eight (88.9%, 8/9) pregnancies were determined to be terminated; and one (11.1%, 1/9) was full-term delivery, which was a baby girl diagnosed with hydronephrosis and ureteropelvic stenosis. The renal function was normal after surgery, and she is 1.5 years old now and has normal growth.
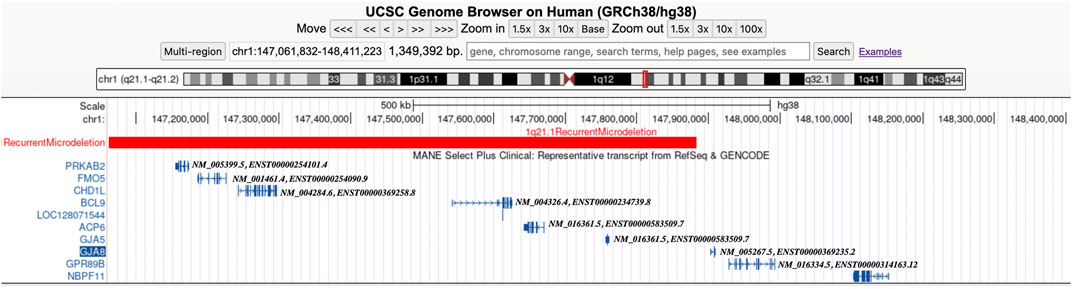
Figure 2. Screenshot from the UCSC Genome Browser (GRCh38/hg38 assembly) showing the location of the 1q21.1 recurrent microdeletion syndrome and the OMIM morbid genes (including MANE Select Plus Clinical transcripts).
Discussion
The recurrent 1q21.1 microdeletion syndrome (OMIM # 612474) is an autosomal dominant disorder usually caused by a recurrent 1.35-Mb deletion in the distal BP3–BP4 region and displays a diversity of clinical phenotypes. In 2008, Brunetti-Pierri et al. (2008) and Mefford et al. (2008) first reported findings of 1q21.1 recurrent deletions in 52 clinical cases, who were associated with certain dysmorphic facial features (such as frontal bossing, deep-set eyes, and bulbous nose), mild-to-moderate intellectual disability, microcephaly, cardiac abnormalities, and cataracts. Since that time, a total of 102 probands with recurrent 1q21.1 microdeletions have been identified by CMA postnatally (Edwards et al., 2021; Bourgois et al., 2024; Bernier et al., 2016). It is estimated that the frequency of the recurrent 1q21.1 microdeletion syndrome is approximately 0.2% of individuals with developmental delays, intellectual disabilities, and/or congenital anomalies (Rosenfeld et al., 2013), following the most commonly reported recurrent deletions: 22q11.2 deletions, proximal 16p11.2 deletions, Williams–Beuren syndrome deletions (7q11.23), and 15q13.3 BP4–BP5 deletions (Dittwald et al., 2013). However, most research studies involving the recurrent 1q21.1 microdeletions focused on postnatal cases, and the prenatal genotype–phenotype association was still unclear due to inadequate reports in the clinic (Yue et al., 2023; Wen et al., 2022). Hence, to provide a better understanding of the deletions in the prenatal setting, we present the ultrasound and molecular findings of 21 cases with recurrent 1q21.1 microdeletions in pregnant women undergoing prenatal invasive testing in the study. To our knowledge, this is the largest study to explore the genotype–phenotype association of the recurrent 1q21.1 microdeletion syndrome in prenatal diagnosis.
The clinical phenotype of the recurrent 1q21.1 microdeletion syndrome is highly variable. It is characterized by dysmorphic facial features, microcephaly, and developmental delay. Several congenital defects, including cardiac, genitourinary, ocular, skeletal anomalies, congenital hypothyroidism, and psychiatric or behavioral abnormalities, have also been described (Buse et al., 2017; Upadhyai et al., 2020). Compared with postnatal phenotypes, prenatal phenotypes involving recurrent 1q21.1 microdeletions were limited in the clinic. To our knowledge, until now, only 20 prenatal cases have been reported (Figure 1) (Bourgois et al., 2024; Yue et al., 2023; Wen et al., 2022; Wang et al., 2017; Chen et al., 2018; Egloff et al., 2018; Su et al., 2022). Prenatal ultrasound findings according to the literature are summarized in Table 2; the common ultrasound features are as follows: increased NT (23.1%, 3/13), CHD (23.1%, 3/13), central nervous system (CNS) abnormalities (23.1%, 3/13), CAKUT (15.4%, 2/13), IUGR (15.4%, 2/13), skeletal anomalies (7.7%, 1/13), and oligohydramnios (7.7%, 1/13). In addition, in the 21 cases, the common ultrasound findings are as follows: increased NT (26.7%, 4/15), IUGR (26.7%, 4/15), CHD (20%, 3/15), CAKUT (13.3%, 2/15), bilateral ventriculomegaly (6.7%, 1/15), and polyhydramnios (6.7%, 1/15). Our findings are mostly consistent with those of prior literature, with the exception that IUGR was more prevalent among our study cohort. Based on the findings mentioned above, we assumed that recurrent 1q21.1 microdeletions were closely associated with fetuses with increased NT (25%, 7/28), CHD (21.4%, 6/28), IUGR (21.4%, 6/28), CAKUT (14.3%, 4/28), and CNS (14.3%, 4/28) in prenatal diagnosis. Additionally, imaging results demonstrate cerebral ventriculomegaly for some 1q21.1 duplication cases, but not deletion cases, in the literature before (Bernier et al., 2016; Yue et al., 2023). In our study, Case 19 was described as having bilateral ventriculomegaly, which will be the first case to be reported with the recurrent 1q21.1 microdeletion, but more evidence should be collected. In summary, our study findings will not only expand the prenatal phenotypes of the deletions but also bring forward the diagnoses of these syndromes and allow couples to make informed decisions about the pregnancy. However, due to the small sample size of the current studies, more efforts should be taken to confirm the prenatal genotype–phenotype association of recurrent 1q21.1 microdeletions in the future.
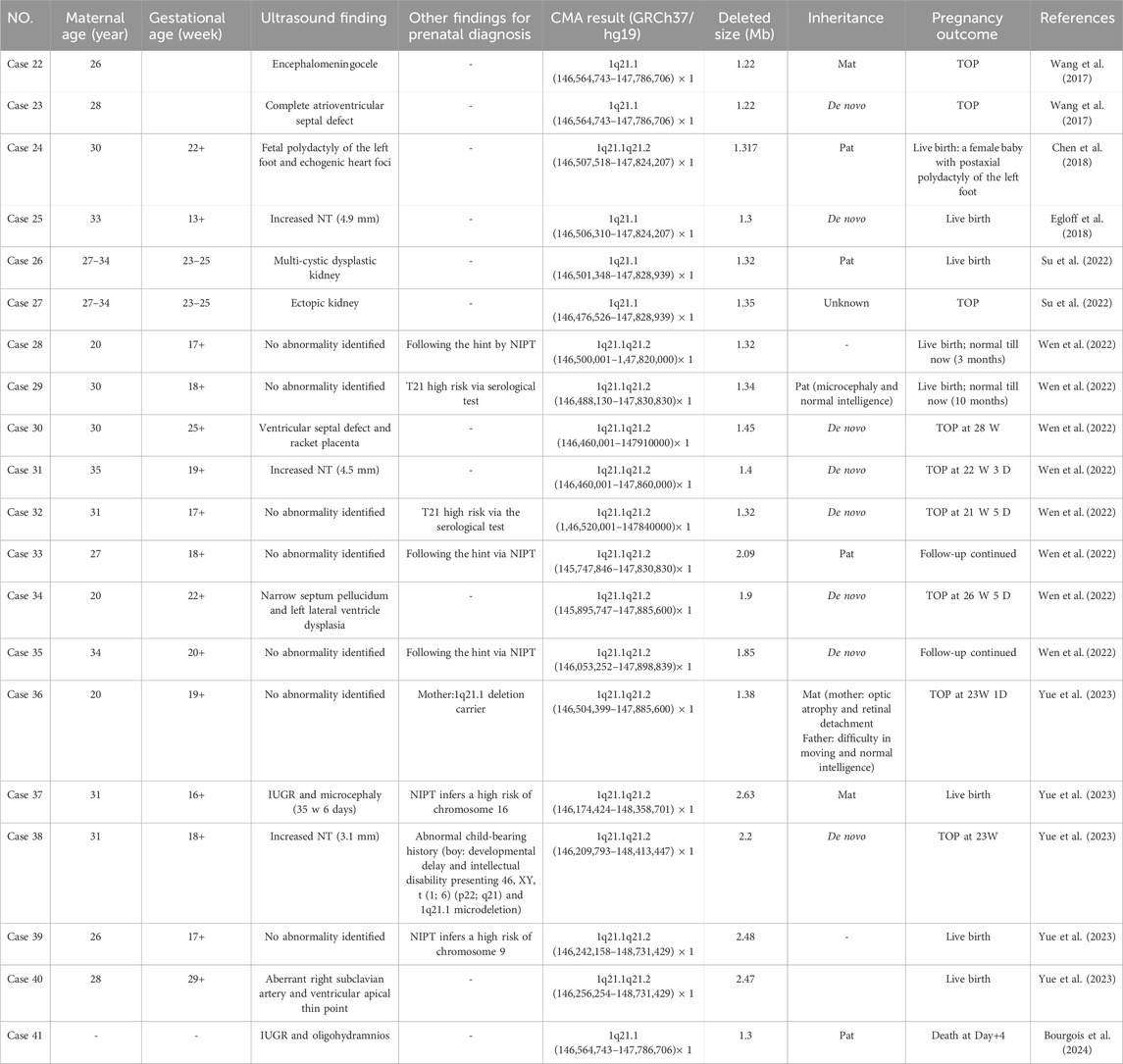
Table 2. Clinical data of fetuses with recurrent 1q21.1 microdeletions detected by CMA in the published literature.
In the study, we detected 21 cases with distal 1q21.1 deletions, and there are eight OMIM genes of interest within the following regions: GJA5, GJA8, ACP6, BCL9, CHD1L, FMO5, GPR89B, and PRKAB2. Haploinsufficiency of one or more of the deleted genes likely contributes to the phenotypes associated with pathogenic variants in these genes. Among them, GJA5 (OMIM * 121013) encodes gap junction protein, a5, and heterozygous pathogenic variants in GJA5 is associated with familial atrial fibrillation 11 (OMIM # 614049) and atrial standstill (OMIM # 108770) (Gollob et al., 2006). Several studies also found that heterozygous mutations and deletions in GJA5 have been identified in patients with structural cardiac defects (especially aortic arch anomalies and tetralogy of Fallot) (Guida et al., 2013; Soemedi et al., 2012; Christiansen et al., 2004) and essential hypertension (Wang et al., 2023). Therefore, we speculated that the fetuses (cases 16–18) with CHD may be associated with heterozygous deletions of GJA5 in the 1q21.1 region. BCL9 (OMIM * 602597) encodes B-cell CLL/lymphoma 9 and plays a causal role in the development of B-cell malignancies (Kramps et al., 2002). Li et al. (2011) demonstrated that common pathogenic variants in BCL9 may be associated with schizophrenia, bipolar disorder, and major depressive disorder in the Chinese Han population. A heterozygous pathogenic variant has also been identified in a patient with a left ventricular outflow tract abnormality (Zaidi et al., 2013). Case 17 presented with dextro-aortic arch, which might also be attributed to the loss of the BCL9 gene in addition to the GJA5 gene. CHD1L (OMIM * 613039) encodes chromodomain helicase DNA-binding protein 1-like, which is highly expressed in the brain, heart, lung, kidney, and stomach (Ahel et al., 2009). Dou et al. (2017) speculated that CHD1L can promote neuronal differentiation in hESCs and play an important role in nervous system development. Case 19 was associated with bilateral ventriculomegaly, which might be correlated with CHD1L to some degree, but the reliable correlation between CHD1L and CNS abnormalities still needs further investigation. Brockschmidt et al. (2012) also found that CHD1L plays a role in kidney development and suggested that CHD1L may be a candidate gene for CAKUT. Hwang et al. (2014) identified pathogenic variants in the CHD1L gene in patients associated with CAKUT. In our study, cases 16 and 20 were associated with CAKUT, which will further confirm that heterozygous CHD1L deletion might be associated with fetal CAKUT. Nevertheless, no genes within the distal 1q21.1 region were found to be associated with IUGR in fetuses.
The recurrent 1q21.1 microdeletions have mostly been described as having incomplete penetrance and variable expressivity (Bernier et al., 2016). They can either occur de novo or be inherited from a parent. It was reported that 18%–50% of patients with recurrent 1q21.1 microdeletions were de novo and 50%–82% were inherited from their parents (Wang et al., 2017). Like several other recurrent microdeletions (e.g., 16p11.2 and 15q13.3), the 1q21.1 recurrent microdeletions can be inherited from asymptomatic or mildly affected parents (Mefford et al., 2008; Bernier et al., 2016). In our study, parental studies were performed to determine the inheritance of the deletion in eight cases, and four (50%) cases were inherited from one of the parents. Of them, one case (Case 5) was with no abnormality identified prenatally and inherited from the asymptomatic father. Unfortunately, Case 5 was lost to follow-up. The other three cases were associated with abnormal ultrasound findings. Case 12 was associated with IUGR inherited from the mother, who was with mild intellectual disability and of short stature. Case 19 was associated with bilateral ventriculomegaly, which was inherited from the asymptomatic mother. Both pregnancies were chosen to be terminated after genetic counseling. Case 20 was associated with unilateral hydronephrosis, which was inherited from the asymptomatic mother. A baby girl was delivered at term and diagnosed with hydronephrosis and ureteropelvic stenosis. The renal function is normal after surgery, and she is 1.5 years old now and has normal growth. The findings in our study again reconfirmed the ambiguity of disease penetrance, diversity of expressivity, and lack of clear genotype–phenotype correlation, especially in prenatal diagnosis. It will be difficult to offer precise genetic counseling and prognostic assessment for prenatal cases. Hence, multi-center collaboration should be adopted to enlarge the sample size to establish a clearer relationship between recurrent 1q21.1 microdeletions and prenatal–postnatal phenotypes in the future. Furthermore, long-term follow-up should also be guaranteed after birth, including autism, intellectual disability, hearing impairments, attention-deficit hyperactivity disorder, seizures, cardiac disease, and motor difficulties.
Conclusion
In summary, we described the findings of ultrasound and molecular analysis in 21 fetuses, aiming to investigate the relationship between the recurrent 1q21.1 microdeletion syndrome and prenatal phenotypes. This is the largest study to found that fetuses with recurrent 1q21.1 microdeletions were closely associated with increased NT, CHD, IUGR, and CAKUT. In addition, we are the first to report that cerebral ventriculomegaly might be associated with recurrent 1q21.1 microdeletions. More comprehensive studies are needed for a better understanding of the prenatal phenotype–genotype relationship of the recurrent 1q21.1 microdeletion syndrome in the future.
Data availability statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics statement
The studies involving humans were conducted in accordance with the Declaration of Helsinki and approved by the Institutional Review Board of the Ethics Committee of Guangzhou Women and Children’s Medical Center (Approval code [2019]11600). The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author contributions
LL: methodology and writing–original draft, TL: funding acquisition and writing–original draft. FG: data curation and writing–review and editing. CM: data curation and writing–review and editing. LZe: data curation, investigation, and writing–review and editing. LZa: data curation and writing–review and editing. DL: resources, writing–review and editing, and formal analysis.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This research was funded by the Project of Guangzhou Science and Technology (202201020604 and 20231A011030).
Acknowledgments
The authors would like to thank the couples for participating in the present study.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Ahel, D., Horejsi, Z., Wiechens, N., Polo, S. E., Garcia-Wilson, E., Ahel, I., et al. (2009). Poly(ADP-ribose)-dependent regulation of DNA repair by the chromatin remodeling enzyme ALC1. Science 325 (5945), 1240–1243. doi:10.1126/science.1177321
Bernier, R., Steinman, K. J., Reilly, B., Wallace, A. S., Sherr, E. H., Pojman, N., et al. (2016). Clinical phenotype of the recurrent 1q21.1 copy-number variant. Genet. Med. 18 (4), 341–349. doi:10.1038/gim.2015.78
Bourgois, A., Bizaoui, V., Colson, C., Vincent-Devulder, A., Molin, A., Gerard, M., et al. (2024). Phenotypic and genotypic characterization of 1q21.1 copy number variants: a report of 34 new individuals and literature review. Am. J. Med. Genet. A 194 (3), e63457. doi:10.1002/ajmg.a.63457
Brockschmidt, A., Chung, B., Weber, S., Fischer, D. C., Kolatsi-Joannou, M., Christ, L., et al. (2012). CHD1L: a new candidate gene for congenital anomalies of the kidneys and urinary tract (CAKUT). Nephrol. Dial. Transpl. 27 (6), 2355–2364. doi:10.1093/ndt/gfr649
Brunetti-Pierri, N., Berg, J. S., Scaglia, F., Belmont, J., Bacino, C. A., Sahoo, T., et al. (2008). Recurrent reciprocal 1q21.1 deletions and duplications associated with microcephaly or macrocephaly and developmental and behavioral abnormalities. Nat. Genet. 40 (12), 1466–1471. doi:10.1038/ng.279
Buse, M., Cuttaia, H. C., Palazzo, D., Mazara, M. V., Lauricella, S. A., Malacarne, M., et al. (2017). Expanding the phenotype of reciprocal 1q21.1 deletions and duplications: a case series. Ital. J. Pediatr. 43 (1), 61. doi:10.1186/s13052-017-0380-x
Chen, C. P., Chang, S. Y., Chen, Y. N., Chern, S. R., Wu, P. S., Chen, S. W., et al. (2018). Prenatal diagnosis of a familial 1q21.1-q21.2 microdeletion in a fetus with polydactyly of left foot on prenatal ultrasound. Taiwan J. Obstet. Gynecol. 57 (5), 739–744. doi:10.1016/j.tjog.2018.08.024
Christiansen, J., Dyck, J. D., Elyas, B. G., Lilley, M., Bamforth, J. S., Hicks, M., et al. (2004). Chromosome 1q21.1 contiguous gene deletion is associated with congenital heart disease. Circ. Res. 94 (11), 1429–1435. doi:10.1161/01.RES.0000130528.72330.5c
Digilio, M. C., Bernardini, L., Consoli, F., Lepri, F. R., Giuffrida, M. G., Baban, A., et al. (2013). Congenital heart defects in recurrent reciprocal 1q21.1 deletion and duplication syndromes: rare association with pulmonary valve stenosis. Eur. J. Med. Genet. 56 (3), 144–149. doi:10.1016/j.ejmg.2012.12.004
Dittwald, P., Gambin, T., Szafranski, P., Li, J., Amato, S., Divon, M. Y., et al. (2013). NAHR-mediated copy-number variants in a clinical population: mechanistic insights into both genomic disorders and Mendelizing traits. Genome Res. 23 (9), 1395–1409. doi:10.1101/gr.152454.112
Dou, D., Zhao, H., Li, Z., Xu, L., Xiong, X., Wu, X., et al. (2017). CHD1L Promotes Neuronal Differentiation in Human Embryonic Stem Cells by Upregulating PAX6. Stem Cells Dev. 26 (22), 1626–1636. doi:10.1089/scd.2017.0110
Edwards, S. D., Schulze, K. V., Rosenfeld, J. A., Westerfield, L. E., Gerard, A., Yuan, B., et al. (2021). Clinical characterization of individuals with the distal 1q21.1 microdeletion. Am. J. Med. Genet. A 185 (5), 1388–1398. doi:10.1002/ajmg.a.62104
Egloff, M., Herve, B., Quibel, T., Jaillard, S., Le Bouar, G., Uguen, K., et al. (2018). Diagnostic yield of chromosomal microarray analysis in fetuses with isolated increased nuchal translucency: a French multicenter study. Ultrasound Obstet. Gynecol. 52 (6), 715–721. doi:10.1002/uog.18928
Gollob, M. H., Jones, D. L., Krahn, A. D., Danis, L., Gong, X. Q., Shao, Q., et al. (2006). Somatic mutations in the connexin 40 gene (GJA5) in atrial fibrillation. N. Engl. J. Med. 354 (25), 2677–2688. doi:10.1056/NEJMoa052800
Guida, V., Ferese, R., Rocchetti, M., Bonetti, M., Sarkozy, A., Cecchetti, S., et al. (2013). A variant in the carboxyl-terminus of connexin 40 alters GAP junctions and increases risk for tetralogy of Fallot. Eur. J. Hum. Genet. 21 (1), 69–75. doi:10.1038/ejhg.2012.109
Hwang, D. Y., Dworschak, G. C., Kohl, S., Saisawat, P., Vivante, A., Hilger, A. C., et al. (2014). Mutations in 12 known dominant disease-causing genes clarify many congenital anomalies of the kidney and urinary tract. Kidney Int. 85 (6), 1429–1433. doi:10.1038/ki.2013.508
Kramps, T., Peter, O., Brunner, E., Nellen, D., Froesch, B., Chatterjee, S., et al. (2002). Wnt/wingless signaling requires BCL9/legless-mediated recruitment of pygopus to the nuclear beta-catenin-TCF complex. Cell 109 (1), 47–60. doi:10.1016/s0092-8674(02)00679-7
Li, J., Zhou, G., Ji, W., Feng, G., Zhao, Q., Liu, J., et al. (2011). Common variants in the BCL9 gene conferring risk of schizophrenia. Arch. Gen. Psychiatry 68 (3), 232–240. doi:10.1001/archgenpsychiatry.2011.1
Mefford, H. C., Sharp, A. J., Baker, C., Itsara, A., Jiang, Z., Buysse, K., et al. (2008). Recurrent rearrangements of chromosome 1q21.1 and variable pediatric phenotypes. N. Engl. J. Med. 359 (16), 1685–1699. doi:10.1056/NEJMoa0805384
Rosenfeld, J. A., Coe, B. P., Eichler, E. E., Cuckle, H., and Shaffer, L. G. (2013). Estimates of penetrance for recurrent pathogenic copy-number variations. Genet. Med. 15 (6), 478–481. doi:10.1038/gim.2012.164
Soemedi, R., Topf, A., Wilson, I. J., Darlay, R., Rahman, T., Glen, E., et al. (2012). Phenotype-specific effect of chromosome 1q21.1 rearrangements and GJA5 duplications in 2436 congenital heart disease patients and 6760 controls. Hum. Mol. Genet. 21 (7), 1513–1520. doi:10.1093/hmg/ddr589
Su, J., Qin, Z., Fu, H., Luo, J., Huang, Y., Huang, P., et al. (2022). Association of prenatal renal ultrasound abnormalities with pathogenic copy number variants in a large Chinese cohort. Ultrasound Obstet. Gynecol. 59 (2), 226–233. doi:10.1002/uog.23702
Upadhyai, P., Amiri, E. F., Guleria, V. S., Bielas, S. L., Girisha, K. M., and Shukla, A. (2020). Recurrent 1q21.1 deletion syndrome: report on variable expression, nonpenetrance and review of literature. Clin. Dysmorphol. 29 (3), 127–131. doi:10.1097/MCD.0000000000000327
Wang, H. D., Liu, L., Wu, D., Li, T., Cui, C. Y., Zhang, L. Z., et al. (2017). Clinical and molecular cytogenetic analyses of four families with 1q21.1 microdeletion or microduplication. J. Gene Med. 19 (4), e2948. doi:10.1002/jgm.2948
Wang, J., Wang, X. C., Gu, Z. H., Ren, G. W., Zhao, X. H., Qu, X. K., et al. (2023). A novel GJA5 variant associated with increased risk of essential hypertension. Am. J. Transl. Res. 15 (2), 1259–1270.
Wen, X., Xing, H., Qi, K., Wang, H., Li, X., Zhu, J., et al. (2022). Analysis of 17 prenatal cases with the chromosomal 1q21.1 copy number variation. Dis. Markers 2022, 5487452. doi:10.1155/2022/5487452
Yue, F., Yang, X., Jiang, Y., Li, S., Liu, R., and Zhang, H. (2023). Prenatal phenotypes and pregnancy outcomes of fetuses with recurrent 1q21.1 microdeletions and microduplications. Front. Med. (Lausanne) 10, 1207891. doi:10.3389/fmed.2023.1207891
Keywords: distal 1q21.1 deletions, prenatal diagnosis, chromosomal microarray analysis, ultrasound findings, recurrent 1q21.1 microdeletion syndrome
Citation: Liu L, Lei T, Guo F, Ma C, Zhen L, Zhang L and Li D (2024) Prenatal diagnosis of the recurrent 1q21.1 microdeletions in fetuses with ultrasound anomalies and review of the literature. Front. Genet. 15:1448341. doi: 10.3389/fgene.2024.1448341
Received: 13 June 2024; Accepted: 13 August 2024;
Published: 29 August 2024.
Edited by:
Thomas Liehr, Friedrich Schiller University Jena, GermanyReviewed by:
Anja Weise, University Hospital Jena, GermanyRincic Martina, University of Zagreb, Croatia
Copyright © 2024 Liu, Lei, Guo, Ma, Zhen, Zhang and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dongzhi Li, bGlkb25nemhpMjAxM0BhbGl5dW4uY29t
†These authors share first authorship