 Runzhi Zhang
Runzhi Zhang Susmita Datta
Susmita Datta- Department of Biostatistics, University of Florida, Gainesville, FL, United States
Introduction: With the advancement of high-throughput studies, an increasing wealth of high-dimensional multi-omics data is being collected from the same patient cohort. However, leveraging this multi-omics data to predict survival outcomes poses a significant challenge due to its complex structure.
Methods: In this article, we present a novel approach, the Adaptive Sparse Multi-Block Partial Least Squares (asmbPLS) Regression model, which introduces a dynamic assignment of penalty factors to distinct blocks within various PLS components, facilitating effective feature selection and prediction.
Results: We compared the proposed method with several state-of-the-art algorithms encompassing prediction performance, feature selection and computation efficiency. We conducted comprehensive evaluations using both simulated data with various scenarios and a real dataset from the melanoma patients to validate the effectiveness and efficiency of the asmbPLS method. Additionally, we applied the lung squamous cell carcinoma (LUSC) dataset from The Cancer Genome Atlas (TCGA) to further assess the feature selection capability of asmbPLS.
Discussion: The inherent nature of asmbPLS imparts it with higher sensitivity in feature selection compared to other methods. Furthermore, an R package called asmbPLS implementing this method is made publicly available.
1 Introduction
The high-throughput technology has experienced a tremendous improvement, yielding the rapid and cost-effective generation of extensive omics data. This extensive data spans multiple platforms, such as genomics, transcriptomics, epigenomics, proteomics, microbiomics, and metabolomics (Hasin et al., 2017). This collectively enriches our understanding of the molecular mechanism behind different diseases. For instance, molecular phenotyping utilizing genomics and epigenomics data is poised to facilitate timely and accurate disease diagnosis and prediction, thereby enhancing the accuracy of prognostic assessments and the discernment of disease progression (Bell, 2004). Numerous studies have consistently demonstrated that genomic risks play a significant role in the development and progression and finally the patient survival of various types of diseases. Genomic factors can contribute substantially to a range of health conditions including but not limited to Alzheimer’s disease (Kamboh, 2022), congenital heart disease (Morton et al., 2022), and even certain types of cancers (Duijf et al., 2019; Wadowska et al., 2020; Haffner et al., 2021). In addition, microbiome-derived metabolites have been identified as biomarkers contributing to a wide range of diseases such as inflammatory bowel disease (Wlodarska et al., 2017), colorectal cancer (Louis et al., 2014), type II diabetes (Patterson et al., 2016), asthma (Lee-Sarwar et al., 2020; Chen and Blaser, 2007), as well as obesity (Ejtahed et al., 2020).
In the past decade, many bioinformatics tools have been developed to enable the analysis of individual omics data (Blekherman et al., 2011; Roumpeka et al., 2017; Calderón-González et al., 2016). As high-throughput studies advance, acquiring multiple types of omics data for the same patient becomes achievable. Hence, researchers have increasingly shifted their focus from single-omics analysis to multi-omics analysis. In multi-omics analysis, datasets are organized into blocks, with each block representing variables from a particular type of omics data observed across a cohort of individuals. Usually, multi-omics data has several important characteristics: 1) high dimensionality: the number of features typically vastly outnumbers the sample size; 2) sparsity: some features are detectable only in a minority of samples; 3) Collinearity: the features across different blocks are not independent. In addition, variables from different blocks can have different numbers of features and their structures. Therefore, integrating diverse omics datasets from various platforms and technologies while ensuring data quality is a complex yet an essential task. Integrating multi-omics data is particularly significant, as each type of omics data can offer distinct insights, allowing for a comprehensive and systematic understanding of the complex relationship between the omics data and the host. Moreover, leveraging multi-omics data provides an opportunity to develop prediction models with high accuracy, empowering us to make informed predictions about various phenotypic outcomes, such as patients’ survival time (Yang et al., 2022; Ribeiro et al., 2022; Lin et al., 2022). Predicting patient survival using multi-omics data can greatly aid healthcare professionals in crafting precise treatment strategies, thus becoming a cornerstone of personalized medicine. By pinpointing patients at higher risk of unfavorable outcomes, clinicians can customize interventions and therapies to enhance patient survival rates. This tailored approach maximizes the efficacy of treatments, ultimately improving patient care and prognosis. Furthermore, when dealing with a substantial number of features in each block, deciphering their collective contribution to the process can prove challenging. Thus, performing feature selection and dimension reduction becomes essential to pinpoint the most pertinent and predictive features within each block. This approach streamlines analysis, enhancing the interpretability and effectiveness of multi-omics data in predicting outcomes accurately.
To date, there exists a wealth of literature for dimension reduction and feature selection methods and some of them were successfully applied to this context. For example, suitable feature selection methods include Sparse Group Lasso (SGL) (Simon et al., 2013), which incorporates a convex combination of the standard Lasso penalty and the group-Lasso penalty (Yuan and Lin, 2006). Integrative Lasso with Penalty Factors (IPF-Lasso), an extension of standard Lasso, accounts for group structure by assigning different penalty factors to different blocks for feature selection and prediction (Boulesteix et al., 2017). Priority-Lasso (Klau et al., 2018) is another Lasso-based method that incorporates different groups of variables by defining a priority order for them. Additionally, Random Forest (Breiman, 2001), a powerful prediction algorithm, is known for capturing complex dependency patterns between predictors and outcome. An extension of Random Forest, called Block Forest (Hornung and Wright, 2019), has been developed for the consideration of the group structure in the data.
On the other hand, Partial least squares (PLS) regression (Geladi and Kowalski, 1986) has been used for dimension reduction and prediction using high-dimensional data. Unlike traditional linear regression, PLS is a statistical technique crafted to address scenarios featuring high-dimensional and correlated predictors within regression models. It accomplishes this by constructing latent variables, which are linear combinations of the original predictors. These latent variables aim to capture the maximum covariances between the predictors and the response variable, thus facilitating more effective modeling despite high dimensionality and correlation among predictors. Multi-block Partial Least Squares (mbPLS) (Wangen and Kowalski, 1989; Wold, 1987), initially developed for chemical systems, can be adapted for outcome prediction using multi-omics data due to the shared group structure between the multi-omics data. Furthermore, sparse mbPLS (smbPLS) (Li et al., 2012) has been developed to apply in the bioinformatics field due to the sparse nature of the data. However, smbPLS algorithm utilizes fixed penalty factors for different blocks regardless of the PLS components. This may not guarantee the optimal prediction performance. In addition, an inappropriate penalty factor may lead to the exclusion of important features from some specific blocks. This limitation hampers both biomarker identification and outcome prediction. Driven by these considerations, we introduce a novel multi-omics prediction model called adaptive sparse multi-block partial least squares (asmbPLS). This innovative framework involves the assignment of distinct penalty factors to the blocks across different PLS components by utilizing the specific quantile of feature weights as the penalty factor. This approach guarantees consistent access to insights regarding the relative significance of features within each respective block. The objective of the proposed method is to identify the subset of features most strongly associated with the outcome and subsequently predict the outcome using these selected features. Moreover, employing the quantile of feature weights as the penalty factor will enhance the interpretability of the results, making them more straightforward to understand and apply in practice. Several methodologies have emerged for predicting phenotypic features like patient survival and the onset time of severe illness. Many of these methods leverage the Cox model (Team RC, 2013; Chung and Kang, 2019; Spencer et al., 2021), whose efficacy hinges on the proportional hazard assumption. When the proportional hazard assumption is breached, an alternative to the Cox model is the accelerated failure time (AFT) model. In the AFT model, if
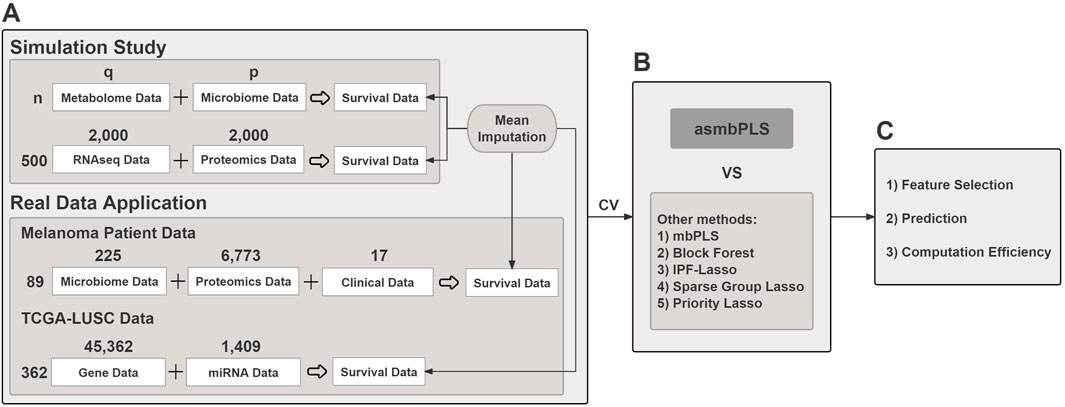
Figure 1. The overview of the study. (A) Both simulated and real data are used; (B) Cross validation is used for parameter tuning for different models. asmbPLS models and other competitive models with the optimal parameters settings are fitted; (C) Results comparison in terms of feature selection, prediction, and computation efficiency.
2 Materials and methods
2.1 Right censored data imputation
In survival analysis, censoring occurs when the time-to-event information for a subject is incomplete. Among various types of censoring, right-censoring is the most prevalent. For instance, right-censoring occurs when a patient is lost to follow-up before experiencing the event of interest. Let
Under this scheme, we keep observed
2.2 Adaptive sparse multi-block PLS algorithm
In mbPLS regression, the input matrix including
max cov(
Once all the parameters are estimated for the first PLS component, X and Y are deflated and then the deflated X and Y are used for the calculation of the second PLS component and so on.
Furthermore, smbPLS (Li et al., 2012), a sparse version of the mbPLS, can be achieved by adding an
Unlike smbPLS, asmbPLS allows different penalty factors

Table 1. Pseudocode for the asmbPLS algorithm.
2.3 Parameter tuning and model selection
We let the block size

Table 2. CV procedure for the asmbPLS algorithm.
The selection of folds for the CV,
With the information from the CV, we can determine the number of PLS components used for prediction also. Usually, the optimal number of PLS components is the one that corresponds to the lowest mean squared error (MSE) in CV. However, due to the over-fitting issue, we allow the selection of fewer components if the decrease of MSE is minimal when one more component is included. The strategy for selecting the number of PLS components is summarized: 1) Let the initial number of components be
2.4 Technicalities and implementation
All the implementations were conducted using R 4.1.0 (Team RC, 2013) in the “HiperGator 3.0” high-performance computing cluster, which includes 70,320 cores with 8 GB of RAM on average for each core, at the University of Florida. We compared the proposed method, i.e. asmbPLS, with mbPLS, Block Forest, IPF-Lasso, Sparse Group Lasso, and Priority-Lasso using both the simulated and the real data. The choice of parameters for each method followed the suggestion from the corresponding R package tutorial. All these methods make use of group structure information and enable us to do the prediction.
2.5 Data Source
The R codes for the simulation study have been made publicly accessible on https://github.com/RunzhiZ/Runzhi_Susmita_asmbPLS_2024. The simulated ovarian cancer dataset was generated with OmicsSIMLA (Chung and Kang, 2019) (https://omicssimla.sourceforge.io/simuomicsTCGA.html), which includes RNA-seq data and proteomics data. The melanoma dataset was obtained from (Spencer et al., 2021) (https://github.com/mda-primetr/Spencer_et_al_2021), which includes progression-free survival interval/status, clinical covariates, and various types of omics data such as microbiome data and proteomics data for melanoma patients. The lung squamous cell carcinoma (LUSC) from The Cancer Genome Atlas (TCGA) was obtained via the Genomic Data Commons (GDC) data portal (https://portal.gdc.cancer.gov/). Gene and miRNA expression data, along with survival data, were downloaded and pre-processed using R package TCGAbiolinks (Colaprico et al., 2016).
3 Results
3.1 Simulation study 1
3.1.1 Simulation strategies
We simulated
3.1.1.1 Microbiome data
We simulated the microbiome data using the Dirichlet-multinomial (DM) distribution (Chen and Li, 2013) to accurately capture the over-dispersed taxon counts. We denote
where
The DM distribution assumes that proportions
where
In summary, to generate microbiome data, we first generated
3.1.1.2 Metabolome data
Once microbiome data was generated, it can be used to simulate the metabolome data due to the associations between microbiome data and metabolome data. Notice that
where
3.1.1.3 Censored survival time
Following the generation of the metabolome data, a similar scaling process was applied. Subsequently, the scaled metabolome data was integrated with the scaled microbiome data to facilitate the generation of survival time. Under the AFT framework, we assumed that both microbiome data and metabolome data collectively impact the actual survival time:
where
For

Table 3. Different settings for
3.1.2 Simulation results
A variety of simulation settings were considered to account for different scenarios. In low dimensional setting,
Since the microbial data were presented in the form of relative abundance, we imputed the zero value with the small pseudo value 0.5 and then implemented centered log-ratio transformation (Aitchison, 1982) to the relative data. We conducted the same procedure for the real data.
The number of PLS components used for asmbPLS and mbPLS were both taken to be 3 in the simulation study. For asmbPLS, the quantile combinations used for cross validation (CV) in low dimension setting were
3.1.2.1 Prediction performance
The performances of prediction of all the methods were measured in terms of the scaled MS
Figure 2 displays the results for low dimension setting with lognormal distributed survival time and
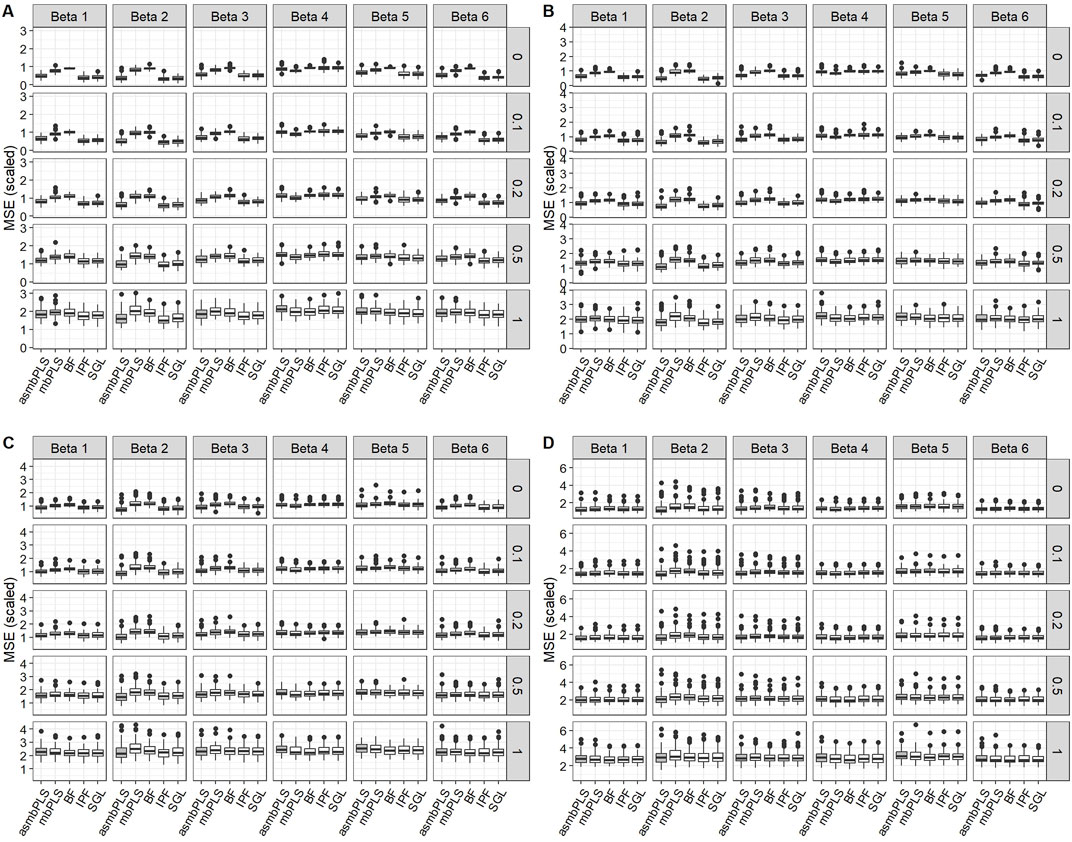
Figure 2. Prediction results for low dimension setting with lognormal distributed survival time and
We observed comparable patterns in low dimension setting with lognormal survival time and
3.1.2.2 Feature selection
In this section, only results for
For different
Figure 3 presents the sensitivity and specificity of the feature selection for low dimension setting with lognormal distributed survival time. According to Figure 3, the performances of different methods on feature selection vary in different types of omics data and all methods show a similar trend in different
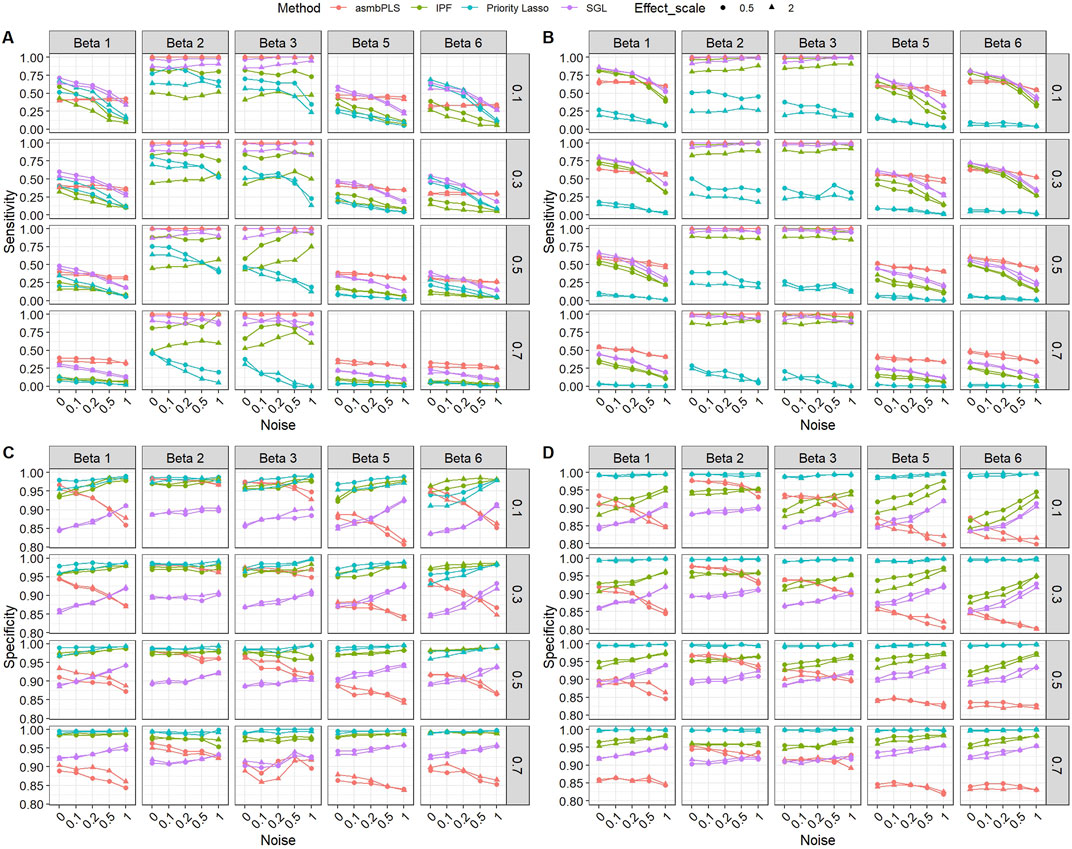
Figure 3. Sensitivity and specificity of the feature selection for low dimension setting with lognormal distributed survival time. (A) Sensitivity for microbiome block; (B) Sensitivity for metabolome block; (C) Specificity for microbiome block; (D) Specificity for metabolome block. The x-axis indicates different noise levels, and the y-axis indicates sensitivity or specificity. The vertical facet titles indicate different
3.1.2.3 Computational efficiency
Table 4 displays the mean computational time for each method in different dimension settings. The computational time is measured as the time needed for model fitting including the CV. Among all the methods, mbPLS, which does not require the CV step, is the fastest procedure, followed by Priority-Lasso, asmbPLS and IPF-Lasso. As the special case of asmbPLS, the computational time of mbPLS is equal to asmbPLS given selected quantile combinations for all the PLS components. For asmbPLS, the CV step takes the longest time, which largely depends on the number of quantile combinations and the number of folds for the CV. Block Forest and SGL run much slower compared to other methods, especially as the size of data increases. It is noteworthy that IPF-Lasso and Priority-Lasso shows a decreased computational time in the mixed dimension and high dimension settings compared to low dimension setting. The computational time of all the other methods increase in different degrees with the increases of the dimension. SGL and Block Forest are two of the slowest methods among all those evaluated.
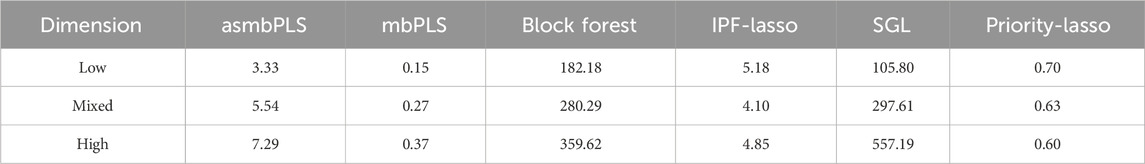
Table 4. Average computation time (in seconds) for different methods in different dimension settings.
3.2 Simulation study 2
3.2.1 Simulation strategies
We utilized a simulated multi-omics data generated with OmicsSIMLA (Chung and Kang, 2019), based on the ovarian cancer data from the TCGA project (https://omicssimla.sourceforge.io/simuomicsTCGA.html). OmicsSIMLA, a multi-omics data simulator, can simulate various types of omics data while modeling the relationships between omics data. 50 batches of samples were simulated, with each batch containing normalized RNA-seq data of 12,000 genes and normalized proteomics data of 12,000 proteins across 1,000 patients, comprising 500 cases and 500 controls. For simplicity, we selected 500 cases and the first 2,000 features from each omics data in each batch. In each batch, we utilized RNA-seq data and proteomics data to generate the censored survival time as described in section “Censored Survival Time”, with the first 400 patients treated as the training set and the remaining 100 patients as the test set. Among the 400 patients in the training set, 50% of the patients were designated to be censored. In each type of omics data, only the first 5 features were set as relevant, with the coefficients of all remaining features set to zero. The noise to signal ratio,
3.2.2 Simulation results
The numbers of PLS components used for asmbPLS and mbPLS were both taken to be 3 in the simulation study. For asmbPLS, the quantile combinations used for cross validation (CV) were
Table 5 outlines the comparative results for prediction and feature selection capabilities. Regarding prediction, IPF-Lasso is the top performer among the five evaluated methods, closely followed by asmbPLS (no significant difference between IPF-Lasso and asmbPLS). Conversely, mbPLS and Block Forest are noted for their inferior performance. For feature selection, asmbPLS and IPF-Lasso each achieve perfect sensitivity for RNA-seq data, with Priority-Lasso showing a sensitivity of 0.84, albeit with a higher specificity. For proteomics data, Priority-Lasso has the highest sensitivity, followed by asmbPLS, while IPF-Lasso’s sensitivity is markedly lower. Notably, Priority-Lasso attains the highest specificity for proteomics data also. Overall, asmbPLS demonstrates the highest average sensitivity across both omics datasets, with its prediction performance close to the best.
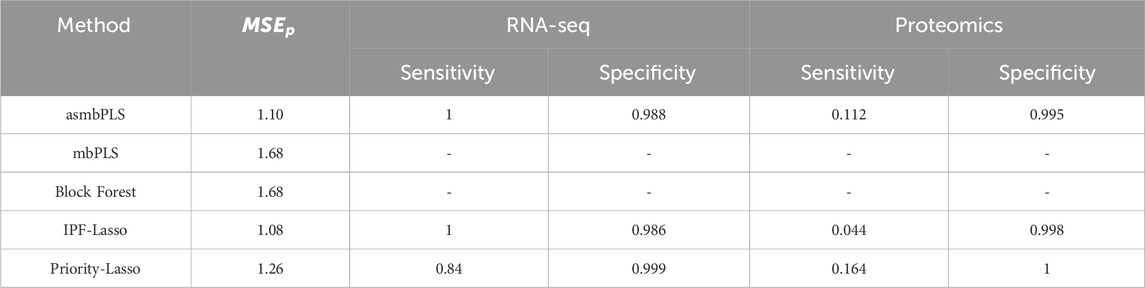
Table 5. Simulation results comparison regarding prediction and feature selection for simulation study 2. The results are the average of the 50 batches.
3.3 Application to melanoma patients data
In our study, we utilized data from Aitchison (1982), encompassing progression-free survival (PFS) interval/status, clinical variables, and diverse omics data gathered from 167 melanoma patients. We focused on three blocks of data: one low-dimensional block comprising clinical covariates, and two high-dimensional blocks consisting of microbiome data and proteomics data. The outcome of interest is the progression-free survival interval, with patients experiencing unobserved events (those without disease progression or death detection) considered right-censored. Among the available clinical variables, we selected 17 clinical variables, age and BMI are continuous with all the other variables are binary: sex, tumor response status, types of treatment, substage of disease in patients with late-stage disease, dietary fiber intake level, lactate dehydrogenase (LDH) level, probiotics use at baseline, antibiotic use at baseline, metformin use at baseline, steroid use at baseline, statin use at baseline, proton pump inhibitor (PPI) use at baseline, beta-blocker use at baseline, other (non-beta-blocker) hypertensive medication use at baseline, whether or not the patient received system therapy prior to baseline. The microbiome block and the proteomics block contain 225 microbial features and 6,773 proteins, respectively.
After filtering the subjects with missing values and without survival data, we obtained 89 samples (55 events) for the downstream implementation. The mean imputation was conducted first for the censored survival time, we then applied the AFT model to determine whether there is an association between PFS and any of the features in the three blocks. Since we have no prior information about the association, we implemented a univariate analysis for each feature to determine whether any of the features are predictive of PFS. To this end, we fitted the simple linear regression model with imputed log-survival PFS as our outcome and each feature as our predictor one at a time. The obtained p-values in each block were then adjusted using the false discovery rate (FDR). Specifically, at a significance level of 5%, no microbial features and proteins are significant. For clinical variables, only tumor response status and substage of disease are significant. With this information, we applied asmbPLS and the other methods in the real data and compared them in three aspects: 1) The fit of the model to data; 2) Prediction error of the model; 3) Feature selection of the model.
To measure the fit of the model to data, we used the MSE of fit to compare different methods:
where
where
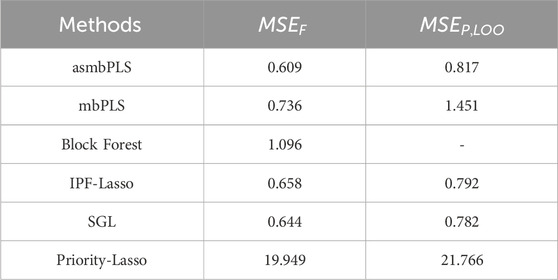
Table 6. Comparison of the model fitting and prediction performance for different methods using the melanoma patient data.
For asmbPLS, 1 microbial taxon, 1 protein, and 2 clinical variables were selected. And block weights for microbiome block, proteomics block, and clinical blocks were 0.069, 0.021, and 0.997, respectively. Due to the nature of asmbPLS, at least one feature was selected for each block. Nevertheless, the block weights for microbiome and proteomics data were notably less than that of the clinical block. This suggests that clinical features could be more relevant to the outcome, which was validated by the univariate analysis. The two clinical variables selected by asmbPLS were tumor response status and substage of disease, which were the only two significant clinical variables. Furthermore, all features identified by asmbPLS within the microbiome and proteomics blocks were significant prior to FDR adjustment. Notably, within the microbiome block, Ruminococcus lactaris stands out. It is worth highlighting that elevated levels of R. lactaris are associated with extended PFS and exhibit a tendency towards reduced systemic inflammation, as evidenced in a B cell-lymphoma patient group (Stein-Thoeringer et al., 2023). Our study corroborates these findings, indicating that increased levels of R. lactaris correspond to prolonged PFS among melanoma patients. Moreover, in the proteomics block, the selected feature is NADH oxidase, which plays a pivotal role in regulating growth and transcription in melanoma cells (SS et al., 2002).
For IPF-Lasso, 0 microbial feature, 0 protein, and 2 clinical variables were selected. The variable substage of disease, which was highly significant before and after FDR adjustment, was not selected by IPF-Lasso. Conversely, the lactate dehydrogenase (LDH) level, which was not significant prior to FDR adjustment, was selected. For SGL, 0 microbial feature, 0 protein, and 3 clinical variables were selected. In addition to the two significant variables, LDH level was also selected by SGL and assigned with a much higher weight than substage of disease. For priority-Lasso, only tumor response status was selected.
In addition to the three aspects discussed above, we explored the utility of the super score, which integrated information from all data blocks (e.g., microbiome, proteomics, and clinical data) and was uniquely derived using asmbPLS and mbPLS, as the predictor for the outcome. While asmbPLS selected a limited number of features, it prioritized those with the strongest and most relevant contributions to the outcome, enabling the super score to capture the most critical information from each block. Specifically, an optimal cut-point on super score was determined to define the two groups using the maximally selected rank statistics (Lausen and Schumacher, 1992) as implemented in the R package survminer (Kassambara et al., 2017), and the p-value was calculated based on a log-rank test between the resulting groups. As seen in Figure 4, super score is significantly associated with progression-free survival time. Patients with higher super scores (blue group) seem to have much higher survival probability. In other words, once we have a new sample with its corresponding microbiome, proteomics, and clinical data, asmbPLS can calculate the super score and then assign the sample to the high or low risk group. This information could be instrumental in designing personalized treatment tailored to each patient.
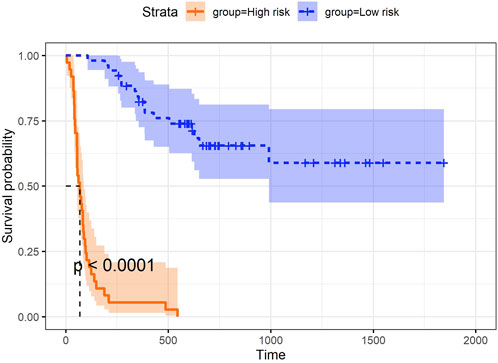
Figure 4. Prediction of PFS using super score from asmbPLS.
Furthermore, we have done the additional analysis without using the most significant clinical block. In this test, only microbiome block and proteomics are included as predictors. The same pre-determined quantile combinations set are used for microbiome and proteomics blocks. Based on the results of CV, the optimal number of PLS components is 1, combination (0.999, 0.9999) was selected for the first PLS component, indicating that 1 microbial taxon and 1 protein were selected. These two features are still the most significant feature in each block even though they are not significant after p-value adjustment. For the other three Lasso-based methods, 0 microbial taxon and 0 protein was selected. Table 7 lists the results for comparison of model fit and CV. With the fact that no predictor is relevant, all the methods show relatively higher
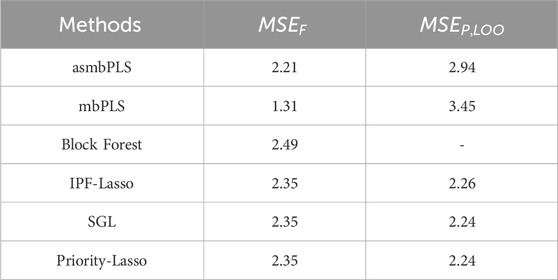
Table 7. Comparison of the model fitting and prediction performance for different methods using the melanoma patient data (without clinical block).
3.4 Application to TCGA-LUSC data
In addition, we applied TCGA-LUSC data to further assess the feature selection capability of asmbPLS. Lung squamous cell carcinoma is a type of non-small cell lung cancer (NSCLC). Among NSCLC, adenocarcinoma is the most common, followed by squamous cell carcinoma of the lung, especially in women (Sabbula et al., 2023). The analysis included gene expression, miRNA and survival data. Subjects with missing gene, miRNA or survival data were excluded, resulting in a final dataset of 362 subjects (91 events) with 45,362 genes and 1,409 miRNAs. Similarly, mean imputation was performed for the censored survival time, and asmbPLS was then applied on the dataset. For comparison, IPF-Lasso was also conducted. SGL and Priority-Lasso were excluded from the analysis as they generated error messages during the model fitting process. We included the first three PLS components for asmbPLS and 5-fold CV was used to determine the optimal quantile combination for model fitting. The pre-determined quantile combinations set were
Regarding feature selection, 1 gene (ENSG00000286152) and 2 miRNAs (has-mir-610 and has-mir-7977) were selected by asmbPLS, with block weights of 0.194 for the gene block and 0.981 for the miRNA block, indicating that the selected miRNAs might be more relevant. For the selected gene, ENSG00000286152, does not currently have an associated gene symbol in available databases. However, for selected miRNAs, miR-610 was found to suppress lung cancer cell proliferation and invasion by targeting GJA3 expression (Jin et al., 2014), while exosomal miR-7977 has been identified as a novel biomarker for patients with lung adenocarcinoma and may function as a tumor suppressor in lung cancer (Chen et al., 2020). No feature was selected by IPF-Lasso.
4 Discussion and conclusion
In this paper, we developed asmbPLS algorithm to identify the most significant features of the multi-omics data gathered from the same set of samples. Subsequently, these selected features are harnessed for outcome prediction. Different from conventional smbPLS, asmbPLS is flexible in determining the penalty factor for different omics data in different PLS components. With some prior knowledge of omics data, the pre-decided quantile set can be provided to each block. Then, the best quantile combination can be chosen in a completely data-driven manner. In addition, using the quantile makes the interpretation more straightforward, block with selected quantile = 0.95 indicates that only the top 5% features are relevant to the outcome. asmbPLS works with continuous predictor variables and continuous outcomes, binary variables can be transformed to 0/1 to meet the requirement. And for categorical variables with more than 2 levels, the one-hot encoding can be one strategy, where the categorical variable with G levels can be transformed into G - 1 dummy variables. asmbPLS is implemented in the R package asmbPLS available on our GitHub (https://github.com/RunzhiZ/asmbPLS).
Simulation study 1 has demonstrated the superior predictive performance of asmbPLS compared to other methods, particularly in scenarios characterized by higher censoring rates, especially when dealing with fewer relevant features such as
The performance of asmbPLS is further validated through its application to melanoma patient data. Notably, asmbPLS retained features from both the microbiome and proteomics blocks, even in cases where no significant feature was identified (after p-value adjustment) within these blocks. The calculated block weights of 0.069, 0.021, and 0.997 validate the limited contributions of the microbiome and proteomics blocks. Furthermore, it is noteworthy that despite the retention of features, the top features selected by asmbPLS from the microbiome and proteomics blocks exhibit meaningful biological significance. In addition, the exploration of the super score in melanoma patient data has demonstrated that super score of asmbPLS is an effective predictor for classifying different survival groups. This practice holds significant potential, enabling the assignment of patients into high-risk or low-risk groups, thereby facilitating the development of personalized treatment plans. In addition to the melanoma dataset, we applied asmbPLS to the TCGA-LUSC dataset, which includes gene expression, miRNA, and survival data, to further evaluate its feature selection capability in lung cancer. asmbPLS successfully identified 1 gene and 2 miRNAs as key features, with miRNAs appearing to have higher relevance based on block weights. This further underscores asmbPLS’s ability to select biologically meaningful features, outperforming other methods like IPF-Lasso, which failed to select any features in this dataset.
A notable limitation of asmbPLS emerges when there is no real relevant feature in the predictor blocks. In such cases, the inclusion of a certain number of features within each block might sacrifice the predictive performance of asmbPLS, although enhancing our understanding of the relative importance of these features. In addition, to perform the model fitting in asmbPLS, each subject must have the same number of predictors and outcomes with no missing data. If any predictors contain missing values, the affected samples must be removed before model fitting, which can reduce the effective sample size and potentially bias the results. This limitation may be particularly problematic in datasets with a high proportion of missing data, as it can lead to a loss of statistical power and hinder the model’s ability to generalize. Future enhancements could include integrating imputation methods to better manage missing data. Furthermore, while mean imputation offers computational convenience for handling right censored survival data, it has the potential to introduce bias, especially in cases where the censoring mechanism is not random. In future extensions of this work, it’s worth to explore the impact of different imputation methods, such as reweighting and multiple imputation (Datta et al., 2007), on the prediction performance of asmbPLS.
In summary, by integrating multi-omics data and continuous phenotypes, asmbPLS can identify the most relevant features across various omics layers and utilizes these selected features for prediction. asmbPLS delivers competitive performance in prediction, feature selection, and computational efficiency compared with other state-of-the-art methods. We anticipate asmbPLS to be a valuable tool for multi-omics research.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://github.com/RunzhiZ/Runzhi_Susmita_asmbPLS_2024.
Author contributions
RZ: Formal Analysis, Investigation, Methodology, Software, Validation, Writing–original draft, Writing–review and editing. SD: Conceptualization, Funding acquisition, Methodology, Resources, Supervision, Writing–original draft, Writing–review and editing.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. Research reported in this publication is partially supported by the National Center for Advancing Translational Sciences of the National Institutes of Health under University of Florida and Florida State University Clinical and Translational Science Awards TL1TR001428.
Acknowledgments
We appreciate the staff at the University of Florida Research Computing Center for providing computational resources and adequate technological support for carrying out the bioinformatics analysis.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2024.1444054/full#supplementary-material
References
Aitchison, J. (1982). The statistical analysis of compositional data. J. R. Stat. Soc. Ser. B Methodol. 44 (2), 139–160. doi:10.1111/j.2517-6161.1982.tb01195.x
Bell, J. (2004). Predicting disease using genomics. Nature 429 (6990), 453–456. doi:10.1038/nature02624
Blekherman, G., Laubenbacher, R., Cortes, D. F., Mendes, P., Torti, F. M., Akman, S., et al. (2011). Bioinformatics tools for cancer metabolomics. Metabolomics 7 (3), 329–343. doi:10.1007/s11306-010-0270-3
Boulesteix, A.-L., De Bin, R., Jiang, X., and Fuchs, M. (2017). IPF-LASSO: integrative L1-penalized regression with penalty factors for prediction based on multi-omics data. Comput. Math. methods Med. 2017, 7691937. doi:10.1155/2017/7691937
Calderón-González, K. G., Hernández-Monge, J., Herrera-Aguirre, M. E., and Luna-Arias, J. P. (2016). “Bioinformatics tools for proteomics data interpretation. Modern Proteomics–Sample Preparation,” in Analysis and practical applications, 281–341.
Chen, J., and Li, H. (2013). Variable selection for sparse Dirichlet-multinomial regression with an application to microbiome data analysis. Ann. Appl. statistics 7 (1). doi:10.1214/12-AOAS592
Chen, L., Cao, P., Huang, C., Wu, Q., Chen, S., and Chen, F. (2020). Serum exosomal miR-7977 as a novel biomarker for lung adenocarcinoma. J. Cell. Biochem. 121 (5-6), 3382–3391. doi:10.1002/jcb.29612
Chen, Y., and Blaser, M. J. (2007). Inverse associations of Helicobacter pylori with asthma and allergy. Archives Intern. Med. 167 (8), 821–827. doi:10.1001/archinte.167.8.821
Chung, R.-H., and Kang, C.-Y. (2019). A multi-omics data simulator for complex disease studies and its application to evaluate multi-omics data analysis methods for disease classification. GigaScience 8 (5), giz045. doi:10.1093/gigascience/giz045
Colaprico, A., Silva, T. C., Olsen, C., Garofano, L., Cava, C., Garolini, D., et al. (2016). TCGAbiolinks: an R/Bioconductor package for integrative analysis of TCGA data. Nucleic acids Res. 44 (8), e71. doi:10.1093/nar/gkv1507
Datta, S. (2005). Estimating the mean life time using right censored data. Stat. Methodol. 2 (1), 65–69. doi:10.1016/j.stamet.2004.11.003
Datta, S., Le-Rademacher, J., and Datta, S. (2007). Predicting patient survival from microarray data by accelerated failure time modeling using partial least squares and LASSO. Biometrics 63 (1), 259–271. doi:10.1111/j.1541-0420.2006.00660.x
Duijf, P. H., Nanayakkara, D., Nones, K., Srihari, S., Kalimutho, M., and Khanna, K. K. (2019). Mechanisms of genomic instability in breast cancer. Trends Mol. Med. 25 (7), 595–611. doi:10.1016/j.molmed.2019.04.004
Ejtahed, H.-S., Angoorani, P., Soroush, A.-R., Hasani-Ranjbar, S., Siadat, S.-D., and Larijani, B. (2020). Gut microbiota-derived metabolites in obesity: a systematic review. Biosci. microbiota, food health 39 (3), 65–76. doi:10.12938/bmfh.2019-026
Geladi, P., and Kowalski, B. R. (1986). Partial least-squares regression: a tutorial. Anal. Chim. acta 185, 1–17. doi:10.1016/0003-2670(86)80028-9
Haffner, M. C., Zwart, W., Roudier, M. P., True, L. D., Nelson, W. G., Epstein, J. I., et al. (2021). Genomic and phenotypic heterogeneity in prostate cancer. Nat. Rev. Urol. 18 (2), 79–92. doi:10.1038/s41585-020-00400-w
Hasin, Y., Seldin, M., and Lusis, A. (2017). Multi-omics approaches to disease. Genome Biol. 18 (1), 83–15. doi:10.1186/s13059-017-1215-1
Hornung, R., and Wright, M. N. (2019). Block forests: random forests for blocks of clinical and omics covariate data. BMC Bioinforma. 20, 1–17. doi:10.1186/s12859-019-2942-y
Jin, J., Li, C., You, J., and Zhang, B. (2014). miR-610 suppresses lung cancer cell proliferation and invasion by targeting GJA3. Zhonghua Zhong liu za zhi Chin. J. Oncol. 36 (6), 405–411.
Kamboh, M. I. (2022). Genomics and functional genomics of Alzheimer’s disease. Neurotherapeutics 19 (1), 152–172. doi:10.1007/s13311-021-01152-0
Kassambara, A., Kosinski, M., Biecek, P., and Fabian, S. (2017). Package ‘survminer’. Drawing Survival Curves using ‘ggplot2’(R package version 03 1).
Klau, S., Jurinovic, V., Hornung, R., Herold, T., and Boulesteix, A.-L. (2018). Priority-Lasso: a simple hierarchical approach to the prediction of clinical outcome using multi-omics data. BMC Bioinforma. 19, 1–14. doi:10.1186/s12859-018-2344-6
Lausen, B., and Schumacher, M. (1992). Maximally selected rank statistics. Biometrics 48, 73–85. doi:10.2307/2532740
Lee-Sarwar, K. A., Lasky-Su, J., Kelly, R. S., Litonjua, A. A., and Weiss, S. T. (2020). Gut microbial-derived metabolomics of asthma. Metabolites 10 (3), 97. doi:10.3390/metabo10030097
Li, W., Zhang, S., Liu, C.-C., and Zhou, X. J. (2012). Identifying multi-layer gene regulatory modules from multi-dimensional genomic data. Bioinformatics 28 (19), 2458–2466. doi:10.1093/bioinformatics/bts476
Lin, Z., He, Y., Qiu, C., Yu, Q., Huang, H., Zhang, Y., et al. (2022). A multi-omics signature to predict the prognosis of invasive ductal carcinoma of the breast. Comput. Biol. Med. 151, 106291. doi:10.1016/j.compbiomed.2022.106291
Louis, P., Hold, G. L., and Flint, H. J. (2014). The gut microbiota, bacterial metabolites and colorectal cancer. Nat. Rev. Microbiol. 12 (10), 661–672. doi:10.1038/nrmicro3344
Morton, S. U., Quiat, D., Seidman, J. G., and Seidman, C. E. (2022). Genomic frontiers in congenital heart disease. Nat. Rev. Cardiol. 19 (1), 26–42. doi:10.1038/s41569-021-00587-4
Mosimann, J. E. (1962). On the compound multinomial distribution, the multivariate β-distribution, and correlations among proportions. Biometrika 49 (1/2), 65–82. doi:10.2307/2333468
Patterson, E., Ryan, P. M., Cryan, J. F., Dinan, T. G., Ross, R. P., Fitzgerald, G. F., et al. (2016). Gut microbiota, obesity and diabetes. Postgrad. Med. J. 92 (1087), 286–300. doi:10.1136/postgradmedj-2015-133285
Ribeiro, I. P., Esteves, L., Caramelo, F., Carreira, I. M., and Melo, J. B. (2022). Integrated multi-omics signature predicts survival in head and neck cancer. Cells 11 (16), 2536. doi:10.3390/cells11162536
Roumpeka, D. D., Wallace, R. J., Escalettes, F., Fotheringham, I., and Watson, M. (2017). A review of bioinformatics tools for bio-prospecting from metagenomic sequence data. Front. Genet. 8, 23. doi:10.3389/fgene.2017.00023
Sabbula, B. R., Gasalberti, D. P., and Anjum, F. (2023). “Squamous cell lung cancer,” in StatPearls (Island, FL: StatPearls Publishing).
Simon, N., Friedman, J., Hastie, T., and Tibshirani, R. (2013). A sparse-group lasso. J. Comput. Graph. statistics 22 (2), 231–245. doi:10.1080/10618600.2012.681250
Spencer, C. N., McQuade, J. L., Gopalakrishnan, V., McCulloch, J. A., Vetizou, M., Cogdill, A. P., et al. (2021). Dietary fiber and probiotics influence the gut microbiome and melanoma immunotherapy response. Science 374 (6575), 1632–1640. doi:10.1126/science.aaz7015
SS, B., Kennedy, T. P., Sturrock, A. B., Huecksteadt, T. P., Quinn, M. T., Whorton, A. R., et al. (2002). An NAD (P) H oxidase regulates growth and transcription in melanoma cells. Am. J. Physiol. Cell Physiol. 282, C1212–C1224. doi:10.1152/ajpcell.00496.2001
Stein-Thoeringer, C. K., Saini, N. Y., Zamir, E., Blumenberg, V., Schubert, M.-L., Mor, U., et al. (2023). A non-antibiotic-disrupted gut microbiome is associated with clinical responses to CD19-CAR-T cell cancer immunotherapy. Nat. Med. 29 (4), 906–916. doi:10.1038/s41591-023-02234-6
Wadowska, K., Bil-Lula, I., Trembecki, Ł., and Śliwińska-Mossoń, M. (2020). Śliwińska-Mossoń M: genetic markers in lung cancer diagnosis: a review. Int. J. Mol. Sci. 21 (13), 4569. doi:10.3390/ijms21134569
Wangen, L., and Kowalski, B. (1989). A multiblock partial least squares algorithm for investigating complex chemical systems. J. Chemom. 3 (1), 3–20. doi:10.1002/cem.1180030104
Wlodarska, M., Luo, C., Kolde, R., d’Hennezel, E., Annand, J. W., Heim, C. E., et al. (2017). Indoleacrylic acid produced by commensal peptostreptococcus species suppresses inflammation. Cell host and microbe 22 (1), 25–37. doi:10.1016/j.chom.2017.06.007
Wold, S. (1987). PLS modeling with latent variables in two or more dimensions. Verlag nicht ermittelbar.
Yang, M., Yang, H., Ji, L., Hu, X., Tian, G., Wang, B., et al. (2022). A multi-omics machine learning framework in predicting the survival of colorectal cancer patients. Comput. Biol. Med. 146, 105516. doi:10.1016/j.compbiomed.2022.105516
Keywords: feature selection, mbPLS, multi-omics, PLS, prediction, survival
Citation: Zhang R and Datta S (2024) asmbPLS: biomarker identification and patient survival prediction with multi-omics data. Front. Genet. 15:1444054. doi: 10.3389/fgene.2024.1444054
Received: 05 June 2024; Accepted: 11 November 2024;
Published: 22 November 2024.
Edited by:
Paweł P. Łabaj, Jagiellonian University, PolandReviewed by:
Shaojun Yu, Northwestern University, United StatesZhiwei Zhou, Stanford University, United States
Copyright © 2024 Zhang and Datta. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Susmita Datta, c3VzbWl0YS5kYXR0YUB1ZmwuZWR1