 Lan Zhong1,2†
Lan Zhong1,2† Wenxiang Wang1,2,3†
Wenxiang Wang1,2,3† Yuanqiong Duan1,2†Liang Song1,2Zhanghuan Li3Kaixuan Yang4
Yuanqiong Duan1,2†Liang Song1,2Zhanghuan Li3Kaixuan Yang4 Qintong Li2*‡
Qintong Li2*‡ Rutie Yin1,2*‡
Rutie Yin1,2*‡- 1Department of Obstetrics and Gynecology, West China Second University Hospital, Sichuan University, Chengdu, Sichuan, China
- 2Key Laboratory of Birth Defects and Related Diseases of Women and Children, Ministry of Education, West China Second University Hospital, Chengdu, Sichuan, China
- 3Department of Gynecologic Oncology, Central Hospital of Xinxiang, The Fourth Affiliated Hospital of Xinxiang Medical University, Xinxiang, Henan, China
- 4Department of Pathology, West China Second University Hospital, Sichuan University, Chengdu, Sichuan, China
Background: Lynch syndrome (LS) is an autosomal dominant inherited disorder caused by mutations in mismatch repair genes. Genetic counseling is crucial for the prevention and treatment of LS, as individuals with these mutations have an increased lifetime risk of developing multiple cancers. MutS Homolog 2 (MSH2) is a protein-coding gene that plays a key role in LS. A significant number of LS cases are linked to harmful heterozygous mutations in the MSH2 gene.
Case Presentation: The proband was a 50-year-old endometrial dedifferentiated carcinoma patient with a dMMR/MSI-H tumor negative for MSH2/MSH6 expression by immunohistochemistry. Genetic counseling and tumor gene testing were conducted using next-generation sequencing (NGS) technology, which revealed a previously unknown germline MSH2 gene nonsense mutation NM_000251.2:exon2.354T>A (p.Y118*), leading to a diagnosis of LS. Further analysis of this variant in five family members of the patient confirmed its presence in all individuals, with one family member being diagnosed with colorectal cancer (CRC) at the age of 43. The proband received postoperative chemoradiotherapy and achieved a disease-free survival of 2 years, with ongoing follow-up.
Conclusion: This study provides evidence that the MSH2 nonsense mutation c.354T>A is a highly likely pathogenic mutation and is responsible for typical LS-associated endometrial carcinoma. It emphasizes the importance of genetic counseling for proband family members to facilitate early diagnosis of LS-related carcinoma.
1 Introduction
Lynch syndrome (LS) is an autosomal dominant inherited disorder associated with germline mutations in mismatch repair (MMR) genes, including MLH1, MSH2, MSH6, PMS2, and EPCAM (Zhao et al., 2022). The MMR system corrects base substitution and insertion–deletion mismatches during DNA replication, leading to microsatellite instability (MSI) (Riedinger et al., 2024). The loss of MMR function is detrimental to genome integrity and sets the stage for cancer development. Clinically, MSI testing—performed by polymerase chain reaction (PCR) analysis—and/or immunohistochemistry (IHC) staining is regularly used in colorectal cancer (CRC) to check MMR deficiency (MMR-D). Because of this, MSI high (MSI-H) and MMR-D are the main genetic signs of LS-related tumors (Fanale et al., 2022). Apart from the increased risk of CRC, LS patients are also susceptible to other primary tumors affecting organs such as the endometrium, ovary, stomach, small intestine, hepatobiliary system, urologic tract, and skin. Women with LS have an approximately 40%–60% chance of presenting a sentinel endometrial carcinoma (EC), which is the most common extraintestinal sentinel cancer in LS patients (Lu et al., 2005; Tafe et al., 2014). Patients with LS-related EC have a 25% estimated risk of developing a second malignancy within 10 years and a 50% risk within 15 years after the initial EC diagnosis (Lynch et al., 1977). Therefore, it is crucial for gynecological oncologists to identify LS among patients with EC.
The prognosis for Lynch syndrome treatment heavily relies on early diagnosis. The screening criteria for identifying Lynch syndrome-associated endometrial carcinoma (LS-EC) have gradually improved. The Amsterdam Ⅱ and Bethesda Guideline, which are based on family history, are widely used clinical criteria to screen for LS in CRC. Extrapolation of both tools to EC has shown a specificity of 61% and 49%, respectively (Syngal et al., 2000). However, universal screening for MMR is recommended for all women diagnosed with EC to identify those with underlying LS (Dillon et al., 2017; Kahn et al., 2019). The newer prediction tools based on molecular screening methods, including MMR-immunohistochemical staining (IHC), microsatellite instability (MSI) testing, and gene sequencing, are widely used to increase the accuracy of screening for LS (Barnetson et al., 2006; Chen et al., 2006) A combination of molecular methods should be used for screening for LS among EC patients (Crosbie et al., 2019). In addition, high-throughput technologies and computational prediction tools, such as deep mutational scanning or multiplexed assays of variant effect (MAVE) technologies, have been developed for assessing variants of unknown significance in Lynch syndrome (Abildgaard et al., 2023). Mutations in MSH2 or MLH1 are the most frequently observed genetic mutations in LS patients, accounting for 40% and 50% of LS cases, respectively. Among LS-EC, the mutation rate for MSH2 genes is 50%–66%, 24%–40% for MLH1, and 10%–13% for MSH6 (Bonadona et al., 2011).
In this study, we identified an EC patient who was diagnosed with LS based on family history and loss of MSH2 protein in IHC analysis. We conducted a genetic analysis using peripheral blood samples to identify the specific germline mutation. Utilizing second-generation high-throughput sequencing (NGS) technology on the Illumina platform, we analyzed variants, including single-nucleotide variations and small insertions/deletions, in the complete sequences and junction sequences of exons and introns of genes associated with LS in the proband. Through this analysis, we discovered a previously unknown mutation in the MSH2 gene.
2 Case report
Our proband was a 50-year-old Chinese woman (G3P1A2, BMI:23.5 kg/m2) who presented with abnormal vaginal bleeding. She had previously undergone fractional curettage through a hysteroscope in another hospital. Pathological results after consultation with pathology experts in our hospital revealed poorly differentiated cancer in endometrium tissue. We performed a radical hysterectomy (Querleu-Morrow, type B), a salpingo-oophorectomy, pelvic lymph node dissection, and a para-aorta lymph node biopsy. Postoperative pathological results revealed that the dedifferentiated carcinoma of the endometrium invaded less than one-half of the uterine muscle, the carcinoma had not invaded the cervical canal, the surgical margins were negative, and no metastatic carcinoma was found in lymph nodes.
Based on the histopathological examination of the sample obtained at surgery, endometrial dedifferentiated carcinoma stage I A was diagnosed. IHC staining demonstrated positive for ER, PR, vimentin, Ki67, MLH1, and PMS2 proteins and negative for MSH2 and MSH6 proteins (Figure 1). Due to a family history of Lynch-related malignancies (Figure 2), the patient met the Amsterdam II criteria and was clinically diagnosed with LS. Therefore, we advised her to undergo genetic counseling and testing. According to The Cancer Genome Atlas (TCGA) molecular subgroups in EC, the molecular classification of this patient was mismatching repair (MMR)-deficient (MMR-d) (Kandoth et al., 2013). Then, in order to identify the germline mutation, we used NGS technology based on the Illumina platform to analyze variants (including single-nucleotide variation and small insertion/deletion) in the complete sequences and the junction sequence of exons and introns of genes related to LS in the proband. Germline testing revealed an NM_000251.2:exon2:c.354T>A (p.Y118*) mutation of the MSH2 gene, which is a nonsense mutation, resulting in the mutation of the 118th amino acid of the gene coding protein from tyrosine to the termination codon. This mutation is expected to lead to functional damage or inactivation of the protein due to premature protein truncation or nonsense-mediated mRNA decay. NGS was performed to verify the mutation. The functional and clinical significance of this mutation has not been reported in the literature. Genetic analyses of this variant were conducted on five members of this pedigree (Figure 3). All of them were positive for the same MSH2 gene mutation. Among them, one member (III-15) was phenotypically affected with LS and had been diagnosed with CRC at the age of 43. Four members (IV20, IV21, IV23, and IV24) had been phenotypically free from LS until the screening (Figure 2).
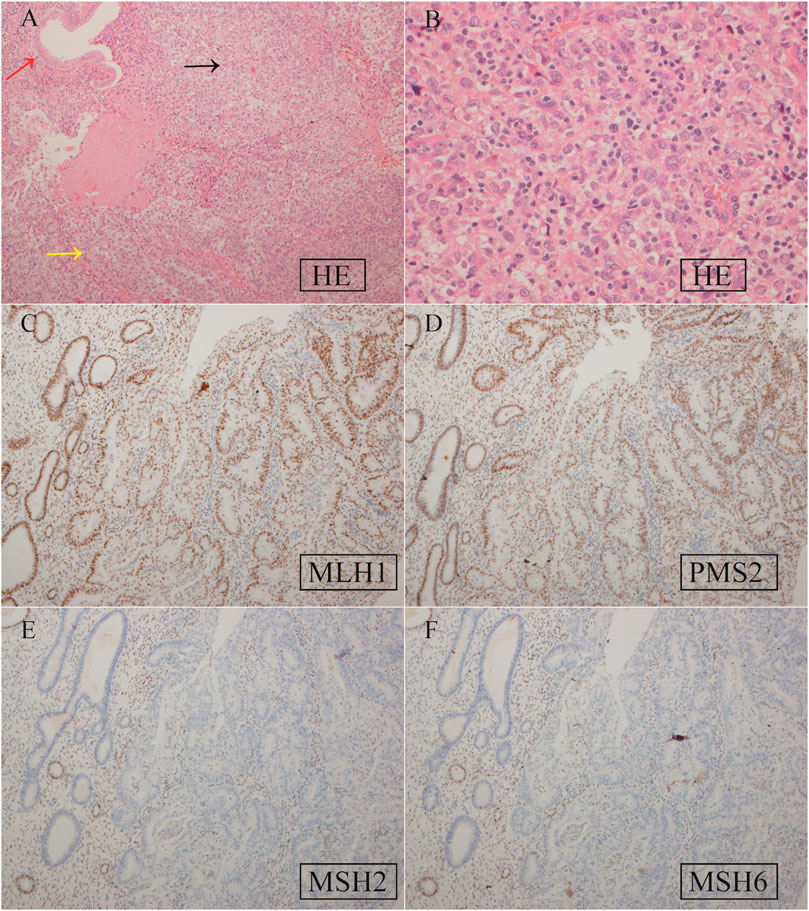
Figure 1. Hematoxylin–eosin (HE) staining (A, B) and IHC staining (C–F) of EC tissue specimens. (A) HE staining. The red, yellow, and black arrows represent normal endometrial glands and well-differentiated and dedifferentiated EC, respectively. Magnification ×200; (B) HE staining for dedifferentiated EC. ×400; (C) positive MLH1 expression, ×100; (D) positive PMS2 expression, ×100; (E) negative MSH2 expression, ×100; (F) negative MSH6 expression, ×100.
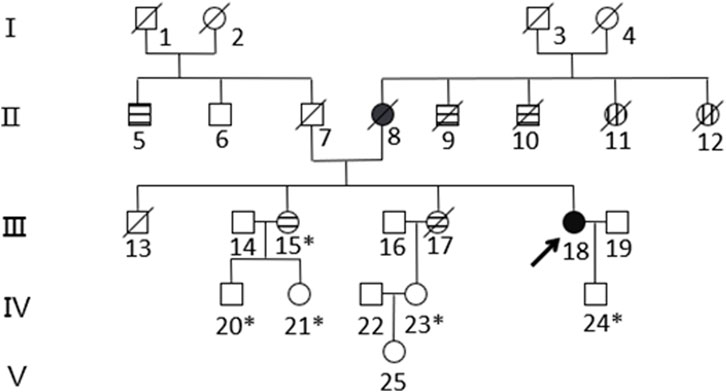
Figure 2. Pedigree of the family with MSH2 c.354T>A. The arrow (→) indicates the proband. Squares and circles denote men and women, respectively. Roman numbers indicate generations. Black solids represent EC. Patients 8 and 18 suffered from EC at the ages of 57 and 50, respectively. Horizontal stripes represent CRC. Patient 5 suffered rectal cancer at the age of 74. Patients 9 and 10 suffered bowel cancer at the ages of 30 and 70, respectively. Patients 15 and 17 suffered ascending colon cancer at the ages of 43 and 34, respectively. Vertical stripes represent unknown cancer. Patients 11 and 12 suffered unknown cancer in their 30s. A backslash (/) represents people who have died. An asterisk (*) indicates MSH2 c.354T>A carriers. The remaining family members have not been tested for MSH2 mutations.

Figure 3. Sequence chromatogram of the proband’s family analyzed by NGS. (A) Heterozygous (Het) mutations were observed in the proband. (B–F) The same Het mutations were observed in the proband’s five family members. The green stripes show the MSH2 c.354T>A (p.Y118*) mutation.
3 Discussion
The proband, a 50-year-old woman, initially presented with endometrial dedifferentiated carcinoma. Her mother had EC, while her two uncles and two sisters had CRC, spanning two generations. One of the proband’s uncles (II-9) and two sisters (III-15, III-17) were diagnosed at ages 43 and 34, respectively, in accordance with the Amsterdam II criteria. This pedigree exhibited typical Lynch syndrome (LS)-related cancers, including colorectal, uterine, and an unknown carcinoma.
Genetic testing of MMR genes has been widely used for LS diagnosis. In this report, we demonstrated that the c.354T>A mutation resulted in a nonsense mutation in the MSH2 gene. The International Society for Gastrointestinal Hereditary Tumors (InSiGHT) database (http://www.insight-group.org) currently lists 9,061 reported MSH2 mutations. The most common types are frameshift mutations and nonsense mutations, which lead to truncated proteins, accounting for 49% and 19% of all MSH2 mutations, respectively (Peltomäki and Vasen, 2004).
The MMR proteins form heterodimers, with MLH1 pairing with PMS2 and MSH2 pairing with MSH6 (Crosbie et al., 2019). These proteins are unstable in their unpaired state, and while MLH1 and MSH2 can form stable heterodimers with other proteins, PMS2 and MSH6 can only dimerize with MLH1 and MSH2, respectively. Mutation of MSH2 usually results in IHC loss of both MSH2 and MSH6. In this study, we confirmed the absence of MSH2 and MSH6 expression, indicating that MSH2 is the primary event, and recommended germline testing for LS-associated mutations. Genetic testing of MMR genes was performed to aid the diagnosis of LS. Based on NGS results, a point mutation was identified in MSH2 (c.354T>A, p.Y118*), located in exon 2. This mutation is a nonsense mutation, leading to the alteration of the 118th amino acid from tyrosine to a termination codon. It is expected that this mutation will result in functional damage or inactivation of the protein through premature protein truncation or nonsense-mediated mRNA decay.
EC is pathogenetically divisible into type I and type II tumors. Type I is low-grade endometrioid adenocarcinoma, and type II is high-grade endometrioid adenocarcinoma and most non-endometrioid carcinomas (Bokhman, 1983). LS-related ECs often exhibit a more diverse histology, including both endometrioid and non-endometrioid types, such as clear cell carcinoma, endometrioid serous carcinoma, undifferentiated carcinoma, and carcinosarcoma (Broaddus et al., 2006). The MSH2 gene mutation appears to be more frequently associated with a non-endometrioid histology (Broaddus et al., 2006; Carcangiu et al., 2010). However, MSH2 loss is relatively rarely reported in endometrial undifferentiated carcinoma. Zhou et al. (2020) reported that one of three dedifferentiated endometrioid carcinomas with neuroendocrine differentiation showed a loss of MSH2/PMS6 expression, which is consistent with our findings. These reports suggest a correlation between MSH2 loss and endometrial dedifferentiated carcinoma.
Compared with the general population, individuals with LS and MMR gene germline mutations have a significantly higher lifetime risk of developing CRC, EC, and other malignancies such as gastric and ovarian cancers (Bonadona et al., 2011; Engel et al., 2012). It is recommended that interventions to reduce the risk of these cancers in affected family members be implemented. In this particular family, genetic analysis was performed on five members of the pedigree (III15, IV20, IV21, IV23, and IV24), and all of them tested positive for the MSH2 gene mutation. Among them, individual III-15 developed colorectal cancer at the age of 43, while the other four members (IV20, IV21, IV23, and IV24) have not shown any LS-related symptoms so far, possibly due to their young age (16 years, 24 years, 28 years, and 26 years, respectively). This identified mutation is considered “pathogenic” and is likely responsible for the manifestation of LS. Prompt management strategies are crucial for these individuals, particularly for subject III-15, who had already been diagnosed with colon cancer at the age of 43.
There are currently no standardized protocols for the surveillance of MMR pathogenic variant carriers because there are insufficient data to demonstrate clinical benefits. Many reports suggested that the combination of TVS and endometrial biopsy may enhance the efficacy of surveillance (Gerritzen et al., 2009; Auranen and Joutsiniemi, 2011; Tzortzatos et al., 2015; Gambini et al., 2022). According to National Comprehensive Cancer Network (NCCN) guidelines for uterine neoplasms, a yearly endometrial biopsy is recommended for patients and family members with LS but without EC to assess for cancer (Abu-Rustum et al., 2023). Prophylactic hysterectomy/bilateral salpingo-oophorectomy is recommended after childbearing is complete. A retrospective cohort study by Schmeler et al. found that patients who underwent prophylactic gynecologic surgery had no occurrences of EC, ovarian cancer, or primary peritoneal cancer (Schmeler et al., 2006). However, 33% of the patients in the control group were diagnosed with EC. These findings suggest that risk-reducing surgery is an effective strategy for preventing EC in patients with LS. The timing and options of risk-reducing surgery can be individualized based on childbearing potential, comorbidities, menopause status, family history, and types of germline mutations (Seppälä et al., 2021; Abu-Rustum et al., 2023). The risk of LS-EC varies based on the types of germline pathogenic variants (PVs); MLH1 or MSH2 carriers might consider risk-reducing surgery at approximately 35 years when they no longer need fertility (Crosbie et al., 2019). For individuals like subject III-15, who has been diagnosed with CRC at the age of 43 and carries an MSH2 variant without the need for fertility, risk-reducing surgery is recommended. There is no uniform guideline for other young female MSH2 variant carriers, such as IV-21 (24 years old) and IV-23 (26 years old), due to limited research on gynecologic surveillance in LS. We recommended that the three carriers should raise awareness of “red flag symptoms,” including abnormal bleeding, weight loss, bloating, change in bowel habits, recurrent urinary symptoms, and abdominal discomfort (Funston et al., 2018). When they have fulfilled their reproductive goals, prophylactic hysterectomy/bilateral salpingo-oophorectomy can be considered.
For the monitoring of CRC, individuals with MLH1 or MSH2 germline mutations have a 52%–82% risk of CRC by the time they are 70 years old (Bonadona et al., 2011). A systematic literature review showed that an initial screening colonoscopy is recommended from the age of 20–25 years with reexamination every 1–2 years for carriers of MLHI or MSH2 gene mutations, or 10 years younger than the youngest age of the person diagnosed in the family (Lindor et al., 2006). In this pedigree, the youngest age of CRC diagnosis is 34 years (III-17), so we recommended the six carriers (including the proband) undergo colonoscopy every 1–2 years from the age of 24.
4 Conclusion
We report a novel case of LS-related endometrial dedifferentiated carcinoma with dMMR/MSI-H by MSH2 NM_000251.2:exon2:c. 354T>A (p.Y118*), which is considered a likely pathogenetic mutation. We provided MSH2-specific genetic counseling to the proband and five other members of her family and confirmed the MSH2 c. 354T>A (p.Y118*) mutation. There are no publications of LS bearing this variant in the literature. Together with this observation obtained from an LS pedigree, we conclude that this variant may be a likely pathogenic variant of LS.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material; further inquiries can be directed to the corresponding authors.
Ethics statement
The studies involving humans were approved by the Ethics Committee of West China Second Hospital. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participant’s legal guardians/next of kin. Written informed consent was obtained from the individual (s) for the publication of any potentially identifiable images or data included in this article.
Author contributions
LZ: Writing–original draft, Conceptualization, Investigation. WW: Writing–original draft, Writing–review and editing. YD: Writing–review and editing. LS: Data curation, Investigation, Writing–review and editing. ZL: Writing–review and editing, Data curation, Formal analysis. KY: Writing–review and editing, Data curation, Methodology. QL: Conceptualization, Project administration, Writing–review and editing. RY: Funding acquisition, Project administration, Writing–review and editing.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. 1. The Key Project of Sichuan Provincial, Department of Science and Technology (19YFS0532) 2. The Project of Chengdu Science and Technology Administration (2021-YF05-01725-SN) 3. WU JIEPING Medical Foundation (320.6750.2021-02-129).
Acknowledgments
We thank the patient and her family. AmoyDx(Xiamen) Co., Ltd., provided the sequencing service for this research.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Abildgaard, A. B., Nielsen, S. V., Bernstein, I., Stein, A., Lindorff-Larsen, K., and Hartmann-Petersen, R. (2023). Lynch syndrome, molecular mechanisms and variant classification. Br. J. Cancer 128 (5), 726–734. doi:10.1038/s41416-022-02059-z
Abu-Rustum, N., Yashar, C., Arend, R., Barber, E., Bradley, K., Brooks, R., et al. (2023). Uterine neoplasms, version 1.2023, NCCN clinical practice guidelines in oncology. J. Natl. Compr. Cancer Netw. 21 (2), 181–209. doi:10.6004/jnccn.2023.0006
Auranen, A., and Joutsiniemi, T. (2011). A systematic review of gynecological cancer surveillance in women belonging to hereditary nonpolyposis colorectal cancer (Lynch syndrome) families. Acta Obstet. Gynecol. Scand. 90 (5), 437–444. doi:10.1111/j.1600-0412.2011.01091.x
Barnetson, R. A., Tenesa, A., Farrington, S. M., Nicholl, I. D., Cetnarskyj, R., Porteous, M. E., et al. (2006). Identification and survival of carriers of mutations in DNA mismatch-repair genes in colon cancer. N. Engl. J. Med. 354 (26), 2751–2763. doi:10.1056/NEJMoa053493
Bokhman, J. V. (1983). Two pathogenetic types of endometrial carcinoma. Gynecol. Oncol. 15 (1), 10–17. doi:10.1016/0090-8258(83)90111-7
Bonadona, V., Bonaïti, B., Olschwang, S., Grandjouan, S., Huiart, L., Longy, M., et al. (2011). Cancer risks associated with germline mutations in MLH1, MSH2, and MSH6 genes in Lynch syndrome. Jama 305 (22), 2304–2310. doi:10.1001/jama.2011.743
Broaddus, R. R., Lynch, H. T., Chen, L. M., Daniels, M. S., Conrad, P., Munsell, M. F., et al. (2006). Pathologic features of endometrial carcinoma associated with HNPCC: a comparison with sporadic endometrial carcinoma. Cancer 106 (1), 87–94. doi:10.1002/cncr.21560
Carcangiu, M. L., Radice, P., Casalini, P., Bertario, L., Merola, M., and Sala, P. (2010). Lynch syndrome--related endometrial carcinomas show a high frequency of nonendometrioid types and of high FIGO grade endometrioid types. Int. J. Surg. Pathol. 18 (1), 21–26. doi:10.1177/1066896909332117
Chen, S., Wang, W., Lee, S., Nafa, K., Lee, J., Romans, K., et al. (2006). Prediction of germline mutations and cancer risk in the Lynch syndrome. Jama 296 (12), 1479–1487. doi:10.1001/jama.296.12.1479
Crosbie, E. J., Ryan, N. A. J., Arends, M. J., Bosse, T., Burn, J., Cornes, J. M., et al. (2019). The Manchester International Consensus Group recommendations for the management of gynecological cancers in Lynch syndrome. Genet. Med. 21 (10), 2390–2400. doi:10.1038/s41436-019-0489-y
Dillon, J. L., Gonzalez, J. L., DeMars, L., Bloch, K. J., and Tafe, L. J. (2017). Universal screening for Lynch syndrome in endometrial cancers: frequency of germline mutations and identification of patients with Lynch-like syndrome. Hum. Pathol. 70, 121–128. doi:10.1016/j.humpath.2017.10.022
Engel, C., Loeffler, M., Steinke, V., Rahner, N., Holinski-Feder, E., Dietmaier, W., et al. (2012). Risks of less common cancers in proven mutation carriers with lynch syndrome. J. Clin. Oncol. 30 (35), 4409–4415. doi:10.1200/jco.2012.43.2278
Fanale, D., Corsini, L. R., Brando, C., Dimino, A., Filorizzo, C., Magrin, L., et al. (2022). Impact of different selection approaches for identifying lynch syndrome-related colorectal cancer patients: unity is strength. Front. Oncol. 12, 827822. doi:10.3389/fonc.2022.827822
Funston, G., O'Flynn, H., Ryan, N. A. J., Hamilton, W., and Crosbie, E. J. (2018). Recognizing gynecological cancer in primary care: risk factors, red flags, and referrals. Adv. Ther. 35 (4), 577–589. doi:10.1007/s12325-018-0683-3
Gambini, D., Ferrero, S., and Kuhn, E. (2022). Lynch syndrome: from carcinogenesis to prevention interventions. Cancers (Basel) 14 (17), 4102. doi:10.3390/cancers14174102
Gerritzen, L. H., Hoogerbrugge, N., Oei, A. L., Nagengast, F. M., van Ham, M. A., Massuger, L. F., et al. (2009). Improvement of endometrial biopsy over transvaginal ultrasound alone for endometrial surveillance in women with Lynch syndrome. Fam. Cancer 8 (4), 391–397. doi:10.1007/s10689-009-9252-x
Kahn, R. M., Gordhandas, S., Maddy, B. P., Baltich Nelson, B., Askin, G., Christos, P. J., et al. (2019). Universal endometrial cancer tumor typing: how much has immunohistochemistry, microsatellite instability, and MLH1 methylation improved the diagnosis of Lynch syndrome across the population? Cancer 125 (18), 3172–3183. doi:10.1002/cncr.32203
Kandoth, C., Schultz, N., Cherniack, A. D., Akbani, R., Liu, Y., Shen, H., et al. (2013). Integrated genomic characterization of endometrial carcinoma. Nature 497 (7447), 67–73. doi:10.1038/nature12113
Lindor, N. M., Petersen, G. M., Hadley, D. W., Kinney, A. Y., Miesfeldt, S., Lu, K. H., et al. (2006). Recommendations for the care of individuals with an inherited predisposition to Lynch syndrome: a systematic review. Jama 296 (12), 1507–1517. doi:10.1001/jama.296.12.1507
Lu, K. H., Dinh, M., Kohlmann, W., Watson, P., Green, J., Syngal, S., et al. (2005). Gynecologic cancer as a “sentinel cancer” for women with hereditary nonpolyposis colorectal cancer syndrome. Obstet. Gynecol. 105 (3), 569–574. doi:10.1097/01.AOG.0000154885.44002.ae
Lynch, H. T., Harris, R. E., Lynch, P. M., Guirgis, H. A., Lynch, J. F., and Bardawil, W. A. (1977). Role of heredity in multiple primary cancer. Cancer 40 (4 Suppl. l), 1849–1854. doi:10.1002/1097-0142(197710)40:4+<1849::aid-cncr2820400813>3.0.co;2-u
Peltomäki, P., and Vasen, H. (2004). Mutations associated with HNPCC predisposition -- Update of ICG-HNPCC/INSiGHT mutation database. Dis. Markers 20 (4-5), 269–276. doi:10.1155/2004/305058
Riedinger, C. J., Esnakula, A., Haight, P. J., Suarez, A. A., Chen, W., Gillespie, J., et al. (2024). Characterization of mismatch-repair/microsatellite instability-discordant endometrial cancers. Cancer 130 (3), 385–399. doi:10.1002/cncr.35030
Schmeler, K. M., Lynch, H. T., Chen, L. M., Munsell, M. F., Soliman, P. T., Clark, M. B., et al. (2006). Prophylactic surgery to reduce the risk of gynecologic cancers in the Lynch syndrome. N. Engl. J. Med. 354 (3), 261–269. doi:10.1056/NEJMoa052627
Seppälä, T. T., Dominguez-Valentin, M., Crosbie, E. J., Engel, C., Aretz, S., Macrae, F., et al. (2021). Uptake of hysterectomy and bilateral salpingo-oophorectomy in carriers of pathogenic mismatch repair variants: a Prospective Lynch Syndrome Database report. Eur. J. Cancer 148, 124–133. doi:10.1016/j.ejca.2021.02.022
Syngal, S., Fox, E. A., Eng, C., Kolodner, R. D., and Garber, J. E. (2000). Sensitivity and specificity of clinical criteria for hereditary non-polyposis colorectal cancer associated mutations in MSH2 and MLH1. J. Med. Genet. 37 (9), 641–645. doi:10.1136/jmg.37.9.641
Tafe, L. J., Riggs, E. R., and Tsongalis, G. J. (2014). Lynch syndrome presenting as endometrial cancer. Clin. Chem. 60 (1), 111–121. doi:10.1373/clinchem.2013.206888
Tzortzatos, G., Andersson, E., Soller, M., Askmalm, M. S., Zagoras, T., Georgii-Hemming, P., et al. (2015). The gynecological surveillance of women with Lynch syndrome in Sweden. Gynecol. Oncol. 138 (3), 717–722. doi:10.1016/j.ygyno.2015.07.016
Zhao, S., Chen, L., Zang, Y., Liu, W., Liu, S., Teng, F., et al. (2022). Endometrial cancer in Lynch syndrome. Int. J. Cancer 150 (1), 7–17. doi:10.1002/ijc.33763
Keywords: Lynch syndrome, endometrial carcinoma, MSH2, nonsense mutation, case report
Citation: Zhong L, Wang W, Duan Y, Song L, Li Z, Yang K, Li Q and Yin R (2024) A new subtype of Lynch syndrome associated with MSH2 c.354T>A (p. Y118*) identified in a Chinese family: case report and literature review. Front. Genet. 15:1440179. doi: 10.3389/fgene.2024.1440179
Received: 29 May 2024; Accepted: 17 September 2024;
Published: 07 October 2024.
Edited by:
Eleonora Marchina, University of Brescia, ItalyReviewed by:
Daniele Fanale, Azienda Ospedaliera Universitaria Policlinico Paolo Giaccone, ItalyShirley V. Hodgson, St George’s, University of London, United Kingdom
Copyright © 2024 Zhong, Wang, Duan, Song, Li, Yang, Li and Yin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Qintong Li, bGlxaW50b25nQHNjdS5lZHUuY24=; Rutie Yin, eWlucnV0aWVAc2N1LmVkdS5jbg==
†These authors have contributed equally to this work and share first authorship
‡These authors have contributed equally to this work