 Cristina Rodilla1,2
Cristina Rodilla1,2 Gonzalo Núñez-Moreno1,2,3
Gonzalo Núñez-Moreno1,2,3 Yolanda Benitez1,3
Yolanda Benitez1,3 Raquel Romero1,2,3
Raquel Romero1,2,3 Lidia Fernández-Caballero1,2
Lidia Fernández-Caballero1,2 Pablo Mínguez1,2,3Marta Corton1,2*
Pablo Mínguez1,2,3Marta Corton1,2* Carmen Ayuso1,2*
Carmen Ayuso1,2*- 1Department of Genetics and Genomics, Instituto de Investigación Sanitaria-Fundación Jiménez Díaz University Hospital, Universidad Autónoma de Madrid (IIS-FJD, UAM), Madrid, Spain
- 2Center for Biomedical Network Research on Rare Diseases (CIBERER), Instituto de Salud Carlos III, Madrid, Spain
- 3Bioinformatics Unit, Instituto de Investigación Sanitaria-Fundación Jiménez Díaz University Hospital, Universidad Autónoma de Madrid (IIS-FJD, UAM), Madrid, Spain
Introduction: Long-read sequencing (LRS) enables accurate structural variant detection and variant phasing. When a molecular diagnosis is suspected, target enrichment can reduce the cost and duration of sequencing.
Methods: LRS was conducted in five inherited retinal dystrophy (IRD) patients harboring a monoallelic variant in RPE65 that remained uncharacterized after clinical exome sequencing (CES). CRISPR-Cas9 guide RNA probes were designed to target a 31 kb region, including the entire RPE65 locus. The DNA was sequenced on a MinION platform. Short-read ×30 whole-genome sequencing (WGS) was performed for five patients to validate nanopore results.
Results: The nanopore sequencing process yielded a median of 271 reads within the targeted region, with a mean depth of 109 and a median read size of 8 kb. All variants identified by CES have been detected using this approach, and no additional RPE65 gene causative variants were found. Nanopore variant detection demonstrated performance akin to short-read WGS at similar coverage levels, although exhibiting increased false positive calls at lower coverage.
Discussion: In this study, we explore the advantages of using a targeted approach together with long-read sequencing to identify variants associated with IRD. The results underscore the utility of targeted long reads for characterizing patients affected by rare diseases when first-tier diagnostic tests are non-conclusive.
Introduction
Inherited retinal diseases (IRDs) are a group of clinically and genetically heterogeneous rare disorders characterized by a neurodegenerative process in retina cells, primarily of photoreceptors. IRDs cause progressive vision loss and may differ in the age of onset and severity of symptoms. The retinal pigment epithelium-specific 65 kDa (RPE65) gene is located in chromosome 1 and codifies for an enzyme implicated in the regeneration of the 11-cis-retinal chromophore during phototransduction in both rod and cone photoreceptors (Jacobson et al., 2007). Loss-of-function biallelic RPE65 variants have been associated with two types of IRDs: Leber’s congenital amaurosis (LCA) and retinitis pigmentosa (RP).
In 2021, we reported RPE65 biallelic variants to be the molecular cause of 2.6% of non-syndromic IRD in Spanish families (Lopez-Rodriguez et al., 2021). This prevalence is similar to the 2% reported in the Italian IRD cohort (Karali et al., 2022), the 2.3% in the Russian population (Stepanova et al., 2023), and higher than the 0.8% prevalence reported in a Chinese cohort (S. Li et al., 2020).
Individuals with biallelic RPE65 variants are the first IRD cohort to undergo treatment with an approved gene-based therapy known as Voretigene Neparvovec (Patel et al., 2018). Molecular characterization of new RPE65 patients would mean access to disease treatment options for these individuals.
Genome sequencing allows the identification of many genetic variants, including single-nucleotide variants (SNVs) in coding and non-coding regions, as well as structural variants (SVs) that are often missed by other molecular analyses. Long-read sequencing (LRS) can generate reads of 10–100 kb in size, which can encompass a complete SV in a single continuous read, allowing to understand its size, position and frequency in the population (Logsdon, Vollger and Eichler, 2020). The most widespread long-read technologies are single-molecule real-time (SMRT) sequencing by Pacific Biosciences (PacBio) (Rhoads and Au, 2015) and nanopore-based sequencing developed by Oxford Nanopore Technologies (ONT) (Jain et al., 2016). The latter represents a scalable and accessible option for smaller budgets.
Whole-genome approaches generate large quantities of data that require large storage capacity and lead to the possibility of secondary genetic findings. Targeted genomic approaches are a better alternative to identifying these variants when a clinically distinct condition is suspected (Khan et al., 2020).
Here, we describe the application of CRISPR-Cas9-mediated enrichment combined with ONT sequencing to the RPE65 locus in undiagnosed IRD patients who are carriers of a heterozygous previously identified variant.
Material and methods
Patient selection and clinical diagnosis
Patients were recruited from the Fundación Jiménez Díaz (FJD) University Hospital (Madrid, Spain) inherited retinal dystrophy (IRD) cohort of 5,123 families from 1990 to February 2024 (March 2024 updated from Perea-Romero et al., 2021). Informed consent was obtained from all study subjects. This study adhered to the tenets of the Declaration of Helsinki and was approved by the FJD Research Ethics Committee (Approval No.: PIC172-20_FJD). For this study, we selected five patients who carried a heterozygous variant in RPE65 identified after genetic testing by clinical exome sequencing (CES), as previously described (Perea-Romero et al., 2021). None of these patients carried an additional known pathogenic variant in 280 other genes associated with nonsyndromic and syndromic IRD or 18 candidate genes from the studied panels (Supplementary Table 1). Additionally, WGS data from these patients are being analyzed with an extended panel without relevant diagnostic progress.
Clinical diagnosis was based on ophthalmology findings and a specific questionnaire. The diagnosis of retinitis pigmentosa (RP) was assigned when the first symptoms were poor night vision and/or peripheral vision loss, as previously described in Perea-Romero et al., 2021. Leber’s congenital amaurosis (LCA) was diagnosed based on severe visual impairment in the first year of life, nystagmus, and non-recordable electroretinography responses.
DNA extraction
High-molecular-weight genomic DNA (gDNA) was extracted from frozen blood cells using the EZ1 Advanced XL extraction system (QIAGEN) according to the manufacturer’s instructions. DNA concentration was assessed using Qubit double-stranded DNA assays (Thermo Fisher Scientific). The DNA quality was analyzed spectrophotometrically (NanoDrop ND-1000, Thermo Scientific), and integrity was ensured by gel electrophoresis.
Short-read sequencing
The five selected patients were analyzed as a first-tier analysis using Clinical Exome Solution (CES, developed by Sophia Genetics, Boston, MA, United States). Libraries were prepared following the manufacturer's instructions and sequenced on a NextSeq500 platform (Illumina) (Martin-Merida et al., 2019).
The whole-genome sequencing (WGS) library was prepared from 1 µg of gDNA using NEB Next® Ultra™ DNA Library Preparation Kit and sequenced paired-end (2 × 150 bp) in an Illumina Novaseq6000 sequencing platform at ×30 coverage. Raw sequencing reads were aligned to the GRCh37/hg19 assembly using the BWA Av 0.7.15 with default parameters.
Guide RNA design
The Alt-R™ CRISPR-Cas9 System (Integrated DNA Technologies, IDT) was selected to design and assemble the CRISPR RNAs (crRNA) and the trans-activating crRNAs (tracrRNA) into functional guide RNAs (gRNAs). crRNA sequences were identified using the CHOPCHOP tool (available at chopchop.cbu.uib.no; Labun et al., 2019).
The crRNA sequences were designed following the Oxford Nanopore Technologies (ONT) guidelines following a “tiling” approach as recommended for regions of interest larger than 20 kb. Thus, pairs of probes were designed for targeting in 10 kb overlapping blocks to achieve uniform coverage along a 31 kb region (chr1:68,423,822-68,454,954; GRCh38/hg38) that included the full-size RPE65 locus (chr1:68,428,822-68,449,954; GRCh38/hg38) and flanking 5 kb expanded regions on each end. A total of six gRNAs could be designed, and the theoretically best-performing gRNAs were selected based on the following criteria: i) a GC content between 30% and 80%; ii) a self-complementarity equal to 0; iii) off-target mismatches lower than 10; iv) a theoretic cut efficiency ≥0.2. In addition, the final selection of probes was made based on the complementary strand where the cut will be performed, the availability of a guide pair within a 10 kb distance in the other strand, the formation of overlapping clusters with other probes, and the best overall parameters predicted by CHOPCHOP. Finally, four gRNA pairs were selected, targeting both positive and negative strands (Table 1; Figure 1A), including two cutting upstream and downstream RPE65 loci, and six within intragenic sequences, mostly in intronic regions.
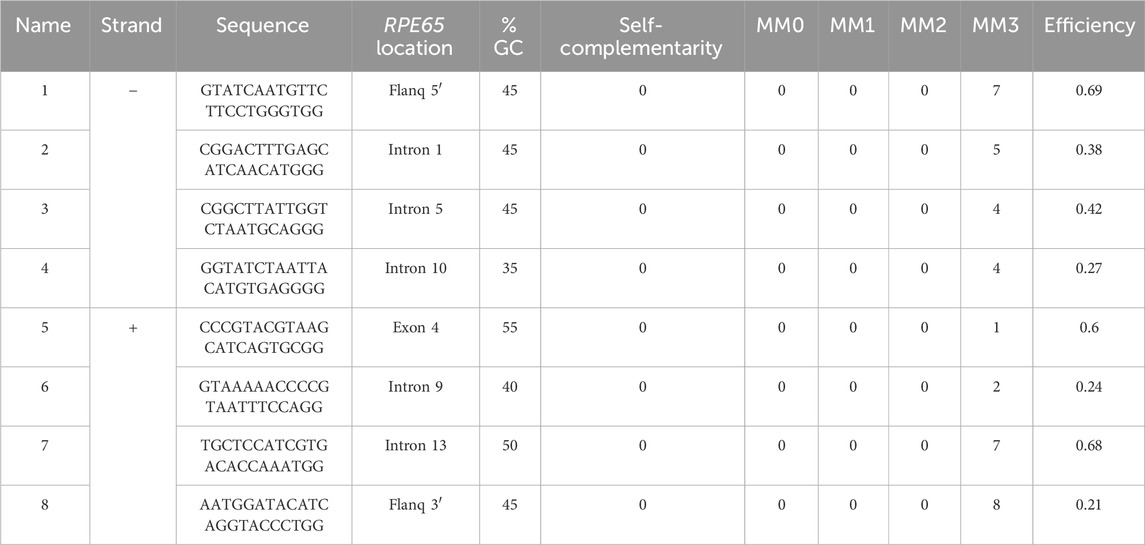
Table 1. Location and characteristics of the selected gRNAs for CRISPR-Cas9-mediated targeted capture of the RPE65 locus. MM: off-target mismatches.
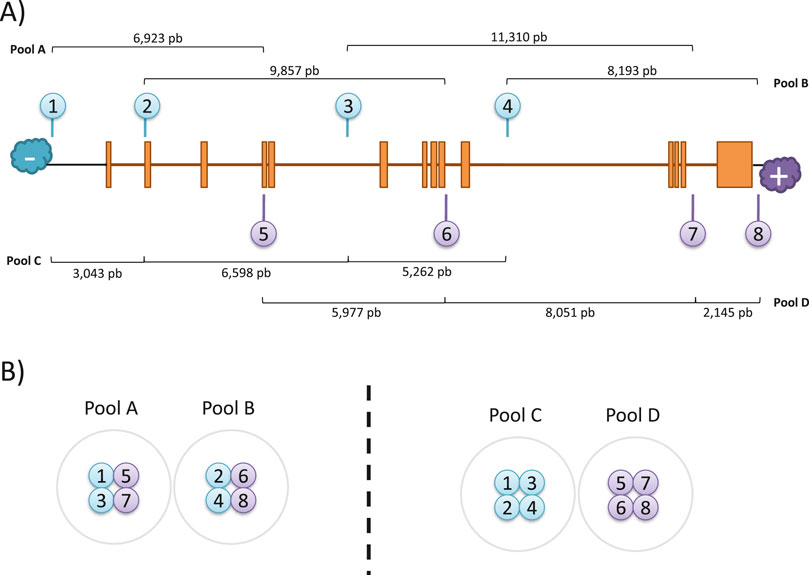
Figure 1. CRISPR-Cas9 enrichment design. (A) Schematic representation of gRNAs location in the target region of RPE65. The distances between expected cuts were calculated for each pool. (B) Arrangement of gRNAs for library preparation.
CRISPR-Cas9 enrichment
Library preparation was carried out following the ONT Cas9 Sequencing Kit (SQK-CS9109) protocol. The tiling approach requires preparing separate libraries with two sets of probes until the final steps. Probes were mixed in equimolar quantities to prepare the pools. From a pool, 1 µL was aliquoted and mixed with tracrRNA. The mix was incubated at 95°C for 5 min. Then, ribonucleoprotein complexes (RNPs) were formed by adding Alt-R™ S. p. Cas9 Nuclease (IDT) and incubating for 30 min.
Pool mixes were prepared in two ways to perform the RPE65 enrichment library. The first way was the “bricklayer” approach (Library A-B, Figure 1B), where overlapping pairs were separated between pools, and the second way was the “highway” approach (Library C-D) where probes were separated according to the direction of the cut.
On account of using the tiling approach, >1,000 ng of gDNA was used for each experiment (>500 ng for each pool of RNPs). The sample was divided into two equimolar reactions for each RNP pool; a minimum of 1.5 µg of DNA for each pool was ensured to be prepared. Following purification, pools were combined into a single library for sequencing. DNA dephosphorylation, cleaving, dA-tailing, adapter ligation, and purification of target DNA were performed according to the protocol.
Nanopore sequencing
Sequencing was carried out on a MinION flow cell (R9.4.1 FLO-MIN106). The MinION sequencer was run for 24 h using MinKNOW software (version 21.06.10). Basecalling was performed using Guppy (version 5.0.13) with the super-high accuracy model. Read filtering was established at Qscore 9, and filtering by read length was disabled.
Sequencing analysis
Fastq files from short-read sequencing were aligned to the GRCh38/hg38 genome assembly using BWA software (H. Li and Durbin, 2009). SNVs and indels (small insertions and deletions) were called using the HaplotypeCaller function in the GATK genomic analysis toolkit (McKenna et al., 2010). Copy-number variants (CNVs) were called using the commercial SOPHiA DDM platform (Sophia Genetics) for CES, and SVs were called using Manta (Chen et al., 2016) for WGS analysis. Variant annotation was performed using an in-house pipeline, NextVariantFJD, available at https://github.com/TBLabFJD/NextVariantFJD (Romero et al., 2022) that includes the variant effect predictor (VEP) (McLaren et al., 2016) for SNVs and indels and AnnotSV (Geoffroy et al., 2018) for SVs, plus additional custom information.
Fastq files from nanopore sequencing were aligned to the GRCh38/hg38 genome assembly using minimap2 (Li, 2018). SNVs and indels were called using PEPPER (PEPPER-Margin-DeepVariant, Shafin et al., 2021) and annotated using the NextVariantFJD pipeline. SVs were called and annotated following the wf-human-variation workflow from EPI2ME Labs, available at https://github.com/epi2me-labs/wf-human-variation, which performs Sniffles2 (Smolka et al., 2024) as the SV caller and SnpEff (Cingolani et al., 2012) for the annotation.
To compare quality statistics from different sequencing methods, we generated two bed files containing 1) the full gene for WGS and nanopore sequencing, and 2) the coding regions of the canonical transcript (NM_000329.3) as described in NCBI for CES sequencing. Mosdepth (Pedersen and Quinlan, 2018) was used to obtain depth and coverage statistics using bam files and the corresponding bed file. The number of reads and N50 reads were calculated using NanoStat (De Coster and Rademakers, 2023) from bam files restricted to the regions in the custom bed files. The variant files, vcfs, were compared using tabix to extract the region of interest and merged using a custom Python script.
Sequencing data were first tested for previously identified variants as a control step of the reading quality achieved in the experiments. To select possible causative variants, the following criteria were applied following the American College of Medical Genetics and Genomics (ACMG) guidelines (Richards et al., 2015). Disease associations reported in the Human Gene Mutation Database (Professional 2022.1), the Leiden Open Variation Database (https://databases.lovd.nl/shared/genes/RPE65), and ClinVar were explored, along with a review of relevant literature. Allele frequency <0.001 in general population databases such as GnomAD v4.1.0 (Genome Aggregation Database; http://gnomad.broadinstitute.org/), Kaviar (Known VARiants) and CSVS (Collaborative Spanish Variant Server). To assess variant impact on protein function, in silico analysis was performed using 20 pathogenicity assessment tools (including CADD, SIFT, Provean, etc.) and splicing predictors (SpliceAI, Ada score, Rf score, MaxEntScan).
Results
Selective enrichment of RPE65 locus sequencing
Our objective was to create a simple and effective method that facilitates fast and accurate characterization of RPE65-related IRD patients. The selected study cohort encompassed previously uncharacterized heterozygous patients with associated RPE65 conditions, LCA, or RP (Table 2).
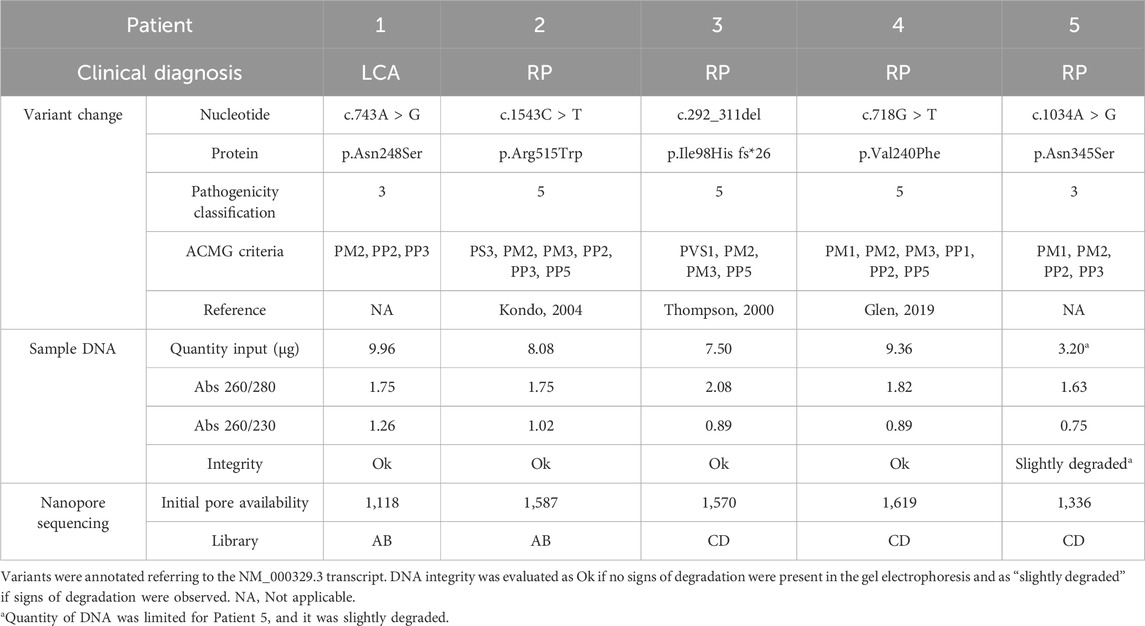
Table 2. Summary of patient information and library preparation for each experiment.
To comprehensively sequence the entire genomic loci of RPE65 using long reads, we selected eight specific crRNAs to perform nanopore sequencing. The crRNAs were designed to be located less than 10 kb away from each other, with pairs cutting on opposite strands at a mean distance of 9 kb, to ensure complete coverage of the region (Figure 1A).
Focusing on the library protocols, the sequencing depth achieved using the AB library was 1.78 times greater than that obtained with the CD library (Figure 2; Supplementary Table 2). The mean read size (N50) was also greater for the AB library, a result attributed to the experimental design.
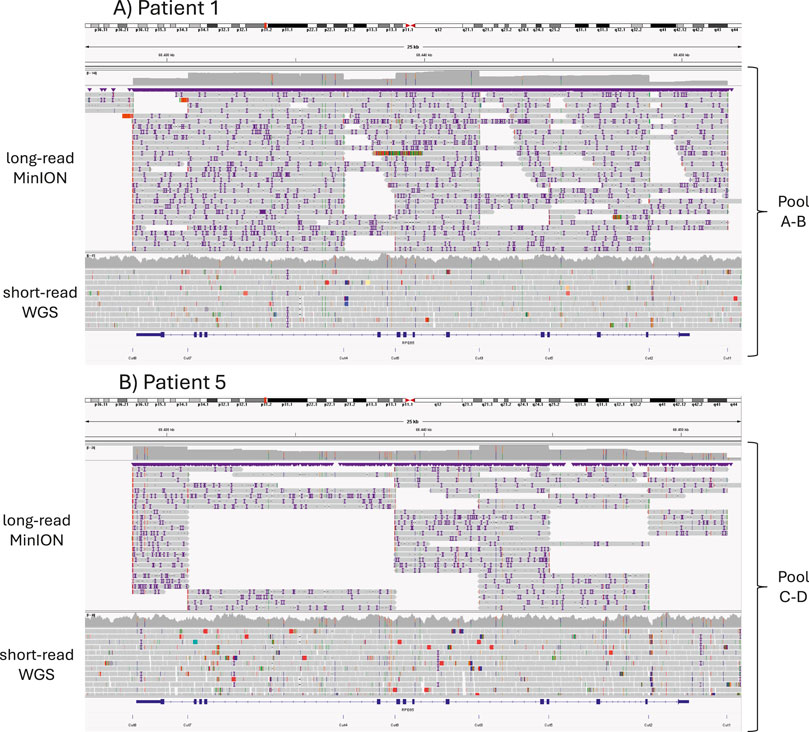
Figure 2. Alignment of long-read MinION sequencing reads using CRISPR-Cas9 enrichment (top) and short-read Illumina WGS reads (bottom); the expected CRISPR-Cas9 cuts are shown below. (A) Results from Patient 1, for whom the library was prepared using pools A and B. (B) Results from Patient 5 using pools C and D.
Variant detection
Patients were previously studied with CES and eight coding variants were identified within the patient samples. Using nanopore enrichment sequencing, we successfully identified all genetic variations that had been identified using CES (Supplementary Table 3).
No structural variant in the RPE65 locus was detected in the studied patients. However, a total of 71 SNVs and indels were detected in the studied patients using nanopore: 12.68% (9/71) were coding variants, 83.09% (59/71) were deep intronic variants, and 4.23% (3/71) were located in the 3′UTR region. Seven variants presented an allele frequency of less than 0.1% and of them, only the five previously reported variants were predicted to cause a deleterious effect with supporting in silico predictions. The remaining variants (88%, 66/71) have not been associated with pathogenicity, and more than half (42/66) were considered polymorphisms due to having an allele frequency higher than 1% in GnomAD.
Short-read WGS detected 69 variants, and 98.55% (68/69) were also detected using CRISPR-Cas9 nanopore sequencing. However, three variants were exclusively detected with nanopore in patients with WGS studies. These variants were reported to have low quality (ranging from 4 to 10), and for this reason, they were considered false positives. False positive detection was increased in experiments with lower coverage (Patient 5). There was also one indel variant exclusively detected in WGS for Patient 5. This variant implied a one nucleotide duplication and was detected in four of 19 reads for the long-read experiment, although the variant caller did not call it.
Comparison of targeted long-read data with short-read data
To further evaluate the different methodologies used, we have compared the sequencing data obtained with each approach.
Nanopore sequencing obtained 271 mean reads, while short-read technologies produced more than 7 times as many for CES and 17 times as many for WGS (Table 3). Short reads had a standard size of 150 bp, while nanopore long reads reached more than 8,000 bp.
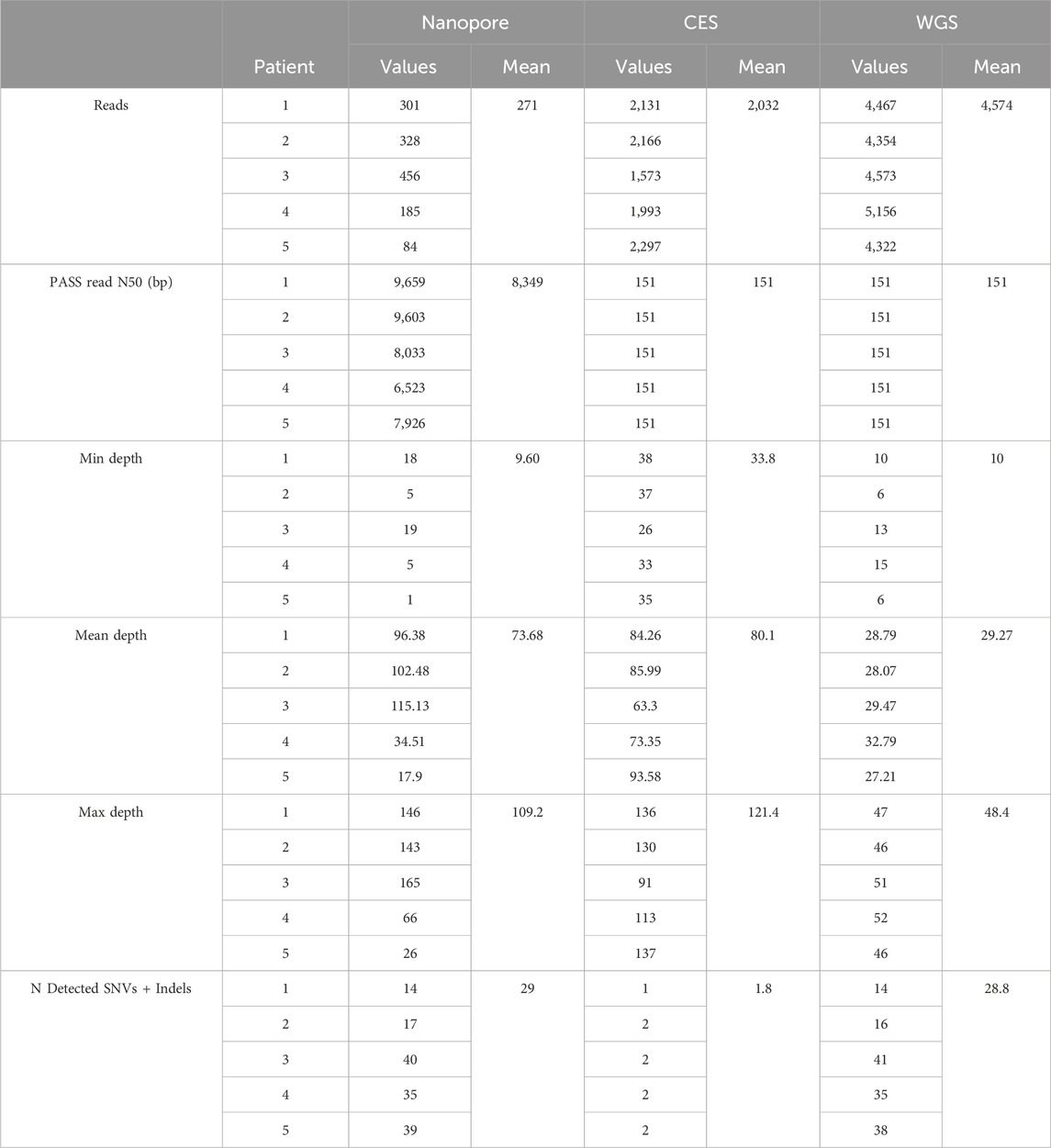
Table 3. Sequencing statistics of studied patients. N detected SNVs + indels: number of detected SNVs and indels.
Minimum coverage in the region was around 30 for CES and dropped to 10 and 9.6 reads for WGS and nanopore, respectively. Mean coverage obtained in the RPE65 locus varied between experiments, ranging from 18 to ×115 between nanopore experiments. CES studies had a similar mean coverage to nanopore, and WGS depth was close to 30. Nanopore reached a maximum of ×109, with one experiment having up to ×165 (Table 3).
On average, 81% of the RPE65 locus had coverage of at least ×20 using the nanopore method. More than ×60 mean nanopore coverage was reached in at least 56% of the RPE65 region, which was more than obtained with WGS (Figure 3). Although the minimum depth obtained with nanopore sequencing was similar to WGS, the resulting depth reaches higher values using a targeted approach (Table 3).
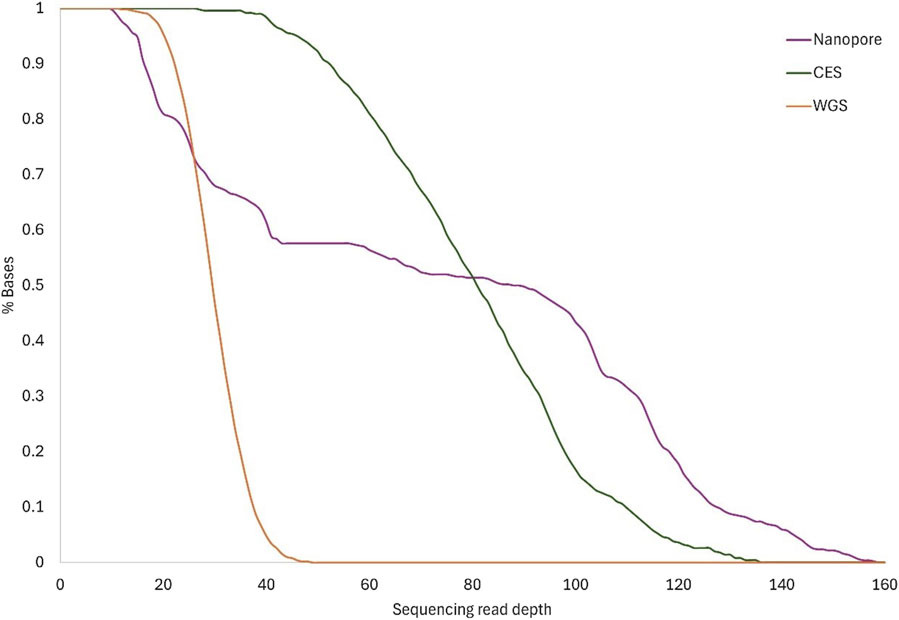
Figure 3. Sequence coverage. The lines show the cumulative read depth as a percentage of total bases for each method: Nanopore, CES, and WGS. Lines show the mean coverage for the same method between the different samples.
Discussion
In this study, we demonstrated that the implementation of Cas9-mediated long-read sequencing can explore the RPE65 locus in depth for IRD patients.
Biallelic variants in the RPE65 gene are the molecular cause of 2.6% of IRD families in Spain (Lopez-Rodriguez et al., 2021). To fully characterize any potential RPE65 patient, we have applied this technology to determine if a second allele was present in the remaining five monoallelic RPE65-uncharacterized IRD families in our cohort (0.26%, 5/5123 uncharacterized IRD families). The importance of investigating monoallelic patients in the context of a recessive disorder lies in the potential of identifying a second hit in the same gene, potentially leading to a definitive molecular diagnosis for the individual. This holds particular significance for the RPE65 gene because therapeutic intervention is already available (Fischer et al., 2024). By performing this analysis, we can rule out biallelic RPE65 single-nucleotide variants as the molecular cause of IRD in the studied cohort, thereby refining our understanding of the genetic mechanisms contributing to the disorder.
Previously detected variants were identified in all studied patients with this new method, even at a ×17 mean read depth. Although the bricklayer strategy (library AB) obtained longer reads and more sequencing depth throughout the region, differences could not be attributed exclusively to library preparation because the two protocols were not compared in the same samples, and the number of samples was small. In addition, sequencing yield might have been affected for Patient 5 due to low integrity DNA and limited quantity. Almost all variants (98.55%) detected by second generation sequencing were also identified with long-read nanopore sequencing.
Nanopore sequencing using R9 chemistry has higher error rates (Q20 accuracy) than short-read NGS sequencing (Wang et al., 2021) or similar third-generation sequencing technologies such as PacBio (Lang et al., 2020). In our hands, only three variants were exclusively detected in nanopore sequences. However, the most recent R10 nanopore chemistry achieves an F1-score, accuracy, and false-discovery rate closer to Illumina sequencing (Kolmogorov et al., 2023; Ni et al., 2023). Although the latest ONT basecaller, Dorado, outperforms the Guppy basecaller (Kuśmirek, 2023), it could not be directly tested in our study because raw data were unavailable. Additionally, ONT reads are reported to reach higher contiguity assemblies (Lang et al., 2020). A standard method such as Sanger sequencing is recommended for variant confirmation when no previous technology is available to validate the variant results.
When assessing the cost-effectiveness of a new technology, it is essential to look beyond the price of reagents (library kits and flowcell) and consider the broader financial implications. Enrichment sequencing using nanopore technology costs approximately 900€ per sample. This is more expensive than NGS techniques such as CES and WGS, which cost between 200€ and 600€. However, these prices do not consider direct labor costs and sequencing analysis, which greatly increase the overall price because NGS libraries take at least 3 days to prepare and comprehend the analysis of thousands of variants. Nanopore enrichment sequencing only takes 6 h to prepare and a day to complete sequencing. Furthermore, variants can be analyzed from the start of the sequencing, and because it is an enrichment approach, only an average of 28 variants need to be interpreted. For this reason, the time saved through nanopore technology translates directly into cost savings and increased productivity.
CRISPR-Cas9 enrichment using nanopore sequencing limits the analysis scope to the selected targeted region. Any variants or structural rearrangement breakpoints localized further from the end of the region would not be captured even if they affect the function of the gene.
Alternative targeted enrichment techniques are PCR-dependent (McClinton et al., 2023), which has length limitations, may introduce artifacts, or produce allele-biased amplification. Another amplification-free enrichment technique is ReadFish adaptive sampling (Payne et al., 2021), which is recommended for larger targets of >3 Mb. Moreover, adaptive sampling requires library reload on a MinION flow cell (Nakamichi et al., 2023) or a run in a more powerful and expensive device using a PromethION flow cell (Deserranno et al., 2023), limiting implementation due to sample availability and cost constraints. Lastly, low-coverage long-read WGS might be effective for characterizing structural variants (Lavrichenko, Johansson, and Jonassen, 2021), but the detection of single-nucleotide variants could be affected.
In conclusion, nanopore sequencing could be considered a robust and feasible system for finding or implementing a second allele strategy in monoallelic recessive cases, with good sensitivity due to the fact that it was able to detect previously identified variants. Additionally, in the context of patient care, using such advanced technologies can significantly improve patients’ clinical management by enabling faster and more accurate molecular diagnosis. This leads to personalized treatment options and genetic counselling, ultimately improving overall healthcare quality and resulting in better health outcomes.
Data availability statement
The original contributions presented in the study are publicly available. This data can be found here: European Genome-phenome Archive (EGA) repository, accession numbers: EGAS50000000596 and EGAD50000000847.
Ethics statement
The studies involving humans were approved by the Comité de Ética de la Investigación de la Fundación Jiménez Díaz. The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study.
Author contributions
CR: conceptualization, data curation, investigation, methodology, validation, writing–original draft, writing–review and editing. GN-M: methodology, software, writing–review and editing. YB: validation, visualization, writing–original draft, writing–review and editing. RR: software, validation, writing–review and editing. LF-C: writing–review and editing. PM: conceptualization, supervision, writing–review and editing. MC: conceptualization, methodology, supervision, writing–review and editing. CA: supervision, writing–review and editing.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This work was supported by the Instituto de Salud Carlos III (ISCIII) of the Spanish Ministry of Health (Grants PI19/00321, PI20/00851 and PI22/00321) IIS-FJD BioBank (PT23/00114), Centro de Investigación Biomédica en Red Enfermedades Raras, ISCIII (CIBERER, 06/07/0036), and co-funded by the European Union, European Regional Development Fund (FEDER), Fundación Conchita Rábago, the Organización Nacional de Ciegos Españoles (ONCE), and the University Chair UAM-IIS-FJD of Genomic Medicine.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2024.1439153/full#supplementary-material
References
Chen, X., Schulz-Trieglaff, O., Shaw, R., Barnes, B., Schlesinger, F., Källberg, M., et al. (2016). Manta: rapid detection of structural variants and indels for germline and cancer sequencing applications. Bioinformatics 32 (8), 1220–1222. doi:10.1093/bioinformatics/btv710
Cingolani, P., Platts, A., Wang, Le L., Coon, M., Nguyen, T., Wang, L., et al. (2012). A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w 1118; iso-2; iso-3. Fly 6 (2), 80–92. doi:10.4161/fly.19695
De Coster, W., and Rademakers, R. (2023). NanoPack2: population-scale evaluation of long-read sequencing data. Bioinforma. Oxf. Engl. 39 (5), btad311. doi:10.1093/bioinformatics/btad311
Deserranno, K., Tilleman, L., Rubben, K., Deforce, D., and Van Nieuwerburgh, F. (2023). Targeted haplotyping in pharmacogenomics using Oxford nanopore technologies’ adaptive sampling. Front. Pharmacol. 14, 1286764. doi:10.3389/fphar.2023.1286764
Fischer, M. D., Simonelli, F., Sahni, J., Holz, F. G., Maier, R., Fasser, C., et al. (2024). Real-world safety and effectiveness of Voretigene Neparvovec: results up to 2 Years from the prospective, registry-based PERCEIVE study. Biomolecules 14 (1), 122. doi:10.3390/biom14010122
Geoffroy, V., Herenger, Y., Kress, A., Stoetzel, C., Piton, A., Dollfus, H., et al. (2018). AnnotSV: an integrated tool for structural variations annotation. Bioinformatics 34 (20), 3572–3574. doi:10.1093/bioinformatics/bty304
Jacobson, S. G., Aleman, T. S., Cideciyan, A. V., Heon, E., Golczak, M., Beltran, W. A., et al. (2007). Human cone photoreceptor dependence on RPE65 isomerase. Proc. Natl. Acad. Sci. 104 (38), 15123–15128. doi:10.1073/pnas.0706367104
Jain, M., Olsen, H. E., Paten, B., and Akeson, M. (2016). The Oxford nanopore MinION: delivery of nanopore sequencing to the genomics community. Genome Biol. 17 (1), 239. doi:10.1186/s13059-016-1103-0
Karali, M., Testa, F., Di Iorio, V., Torella, A., Zeuli, R., Scarpato, M., et al. (2022). Genetic epidemiology of inherited retinal diseases in a large patient cohort followed at a single center in Italy. Sci. Rep. 12 (1), 20815. doi:10.1038/s41598-022-24636-1
Khan, M., Cornelis, S. S., Pozo-Valero, M. D., Whelan, L., Runhart, E. H., Mishra, K., et al. (2020). Resolving the dark matter of ABCA4 for 1054 stargardt disease probands through integrated genomics and transcriptomics. Genet. Med. Official J. Am. Coll. Med. Genet. 22 (7), 1235–1246. doi:10.1038/s41436-020-0787-4
Kolmogorov, M., Billingsley, K. J., Mastoras, M., Meredith, M., Monlong, J., Lorig-Roach, R., et al. (2023). Scalable nanopore sequencing of human genomes provides a comprehensive view of haplotype-resolved variation and methylation. Nat. Methods 20 (10), 1483–1492. doi:10.1038/s41592-023-01993-x
Kuśmirek, W. (2023). Estimated nucleotide reconstruction quality symbols of basecalling tools for Oxford nanopore sequencing. Sensors 23 (15), 6787. doi:10.3390/s23156787
Labun, K., Montague, T. G., Krause, M., Torres Cleuren, Y. N., Tjeldnes, H., and Valen, E. (2019). CHOPCHOP v3: expanding the CRISPR web toolbox beyond genome editing. Nucleic Acids Res. 47 (W1), W171–W174. doi:10.1093/nar/gkz365
Lang, D., Zhang, S., Ren, P., Liang, F., Sun, Z., Meng, G., et al. (2020). Comparison of the two up-to-date sequencing technologies for genome assembly: HiFi reads of pacific Biosciences sequel II system and ultralong reads of Oxford nanopore. GigaScience 9 (12), giaa123. doi:10.1093/gigascience/giaa123
Lavrichenko, K., Johansson, S., and Jonassen, I. (2021). Comprehensive characterization of copy number variation (CNV) called from array, long- and short-read data. BMC Genomics 22 (1), 826. doi:10.1186/s12864-021-08082-3
Li, H. (2018). Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34 (18), 3094–3100. doi:10.1093/bioinformatics/bty191
Li, H., and Durbin, R. (2009). Fast and accurate short read alignment with burrows-wheeler transform. Bioinforma. Oxf. Engl. 25 (14), 1754–1760. doi:10.1093/bioinformatics/btp324
Li, S., Xiao, X., Yi, Z., Sun, W., Wang, P., and Zhang, Q. (2020). RPE65 mutation frequency and phenotypic variation according to exome sequencing in a tertiary centre for genetic eye diseases in China. Acta Ophthalmol. 98 (2), e181–e190. doi:10.1111/aos.14181
Logsdon, G. A., Vollger, M. R., and Eichler, E. E. (2020). Long-read human genome sequencing and its applications. Nat. Rev. Genet. 21 (10), 597–614. doi:10.1038/s41576-020-0236-x
Lopez-Rodriguez, R., Lantero, E., Blanco-Kelly, F., Avila-Fernandez, A., Merida, I. M., Pozo-Valero, M. del, et al. (2021). RPE65-Related retinal dystrophy: mutational and phenotypic spectrum in 45 affected patients. Exp. Eye Res. 212, 108761. doi:10.1016/j.exer.2021.108761
Martin-Merida, I., Avila-Fernandez, A., Pozo-Valero, M. D., Blanco-Kelly, F., Zurita, O., Perez-Carro, R., et al. (2019). Genomic landscape of sporadic retinitis pigmentosa: findings from 877 Spanish cases. Ophthalmology 126 (8), 1181–1188. doi:10.1016/j.ophtha.2019.03.018
McClinton, B., Crinnion, L. A., McKibbin, M., Mukherjee, R., Poulter, J. A., Smith, C. E. L., et al. (2023). Targeted nanopore sequencing enables complete characterisation of structural deletions initially identified using exon-based short-read sequencing strategies. Mol. Genet. and Genomic Med. 11 (6), e2164. doi:10.1002/mgg3.2164
McKenna, A., Hanna, M., Banks, E., Sivachenko, A., Cibulskis, K., Kernytsky, A., et al. (2010). The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20 (9), 1297–1303. doi:10.1101/gr.107524.110
McLaren, W., Gil, L., Hunt, S. E., Singh Riat, H., Ritchie, G. R. S., Thormann, A., et al. (2016). The ensembl variant effect predictor. Genome Biol. 17 (1), 122. doi:10.1186/s13059-016-0974-4
Nakamichi, K., Van Gelder, R. N., Chao, J. R., and Mustafi, D. (2023). Targeted adaptive long-read sequencing for discovery of complex phased variants in inherited retinal disease patients. Sci. Rep. 13 (1), 8535. doi:10.1038/s41598-023-35791-4
Ni, Y., Liu, X., Mengistie Simeneh, Z., Yang, M., and Li, R. (2023). Benchmarking of nanopore R10.4 and R9.4.1 flow cells in single-cell whole-genome amplification and whole-genome shotgun sequencing. Comput. Struct. Biotechnol. J. 21, 2352–2364. doi:10.1016/j.csbj.2023.03.038
Patel, U., Boucher, M., Léséleuc, L. de, and Visintini, S. (2018). Voretigene Neparvovec: an emerging gene therapy for the treatment of inherited blindness. CADTH Issues Emerg. Health Technol.
Payne, A., Holmes, N., Clarke, T., Munro, R., Debebe, B. J., and Loose, M. (2021). Readfish enables targeted nanopore sequencing of gigabase-sized genomes. Nat. Biotechnol. 39 (4), 442–450. doi:10.1038/s41587-020-00746-x
Pedersen, B. S., and Quinlan, A. R. (2018). Mosdepth: quick coverage calculation for genomes and exomes. Bioinforma. Oxf. Engl. 34 (5), 867–868. doi:10.1093/bioinformatics/btx699
Perea-Romero, I., Gordo, G., Iancu, I. F., Pozo-Valero, M. D., Almoguera, B., Blanco-Kelly, F., et al. (2021). Genetic landscape of 6089 inherited retinal dystrophies affected cases in Spain and their therapeutic and extended epidemiological implications. Sci. Rep. 11 (1), 1526. doi:10.1038/s41598-021-81093-y
Rhoads, A., and Au, K. F. (2015). PacBio sequencing and its applications. Genomics, Proteomics and Bioinforma. 13 (5), 278–289. doi:10.1016/j.gpb.2015.08.002
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of medical genetics and genomics and the association for molecular pathology. Genet. Med. 17 (5), 405–424. doi:10.1038/gim.2015.30
Romero, R., de la Fuente, L., Pozo-Valero, M. D., Riveiro-Álvarez, R., Trujillo-Tiebas, M. J., Martín-Mérida, I., et al. (2022). An evaluation of pipelines for DNA variant detection can guide a reanalysis protocol to increase the diagnostic ratio of genetic diseases. NPJ Genomic Med. 7 (1), 7. doi:10.1038/s41525-021-00278-6
Shafin, K., Pesout, T., Chang, P.-C., Nattestad, M., Kolesnikov, A., Goel, S., et al. (2021). Haplotype-aware variant calling with PEPPER-margin-DeepVariant enables high accuracy in nanopore long-reads. Nat. Methods 18 (11), 1322–1332. doi:10.1038/s41592-021-01299-w
Smolka, M., Paulin, L. F., Grochowski, C. M., Horner, D. W., Mahmoud, M., Behera, S., et al. (2024). Detection of mosaic and population-level structural variants with Sniffles2. Nat. Biotechnol. doi:10.1038/s41587-023-02024-y
Stepanova, A., Ogorodova, N., Kadyshev, V., Shchagina, O., Kutsev, S., and Polyakov, A. (2023). A molecular genetic analysis of RPE65-associated forms of inherited retinal degenerations in the Russian federation. Genes 14 (11), 2056. doi:10.3390/genes14112056
Keywords: RPE65 gene, Leber congenital amaurosis, nanopore sequencing, CRISPR, retinitis pigmentosa
Citation: Rodilla C, Núñez-Moreno G, Benitez Y, Romero R, Fernández-Caballero L, Mínguez P, Corton M and Ayuso C (2024) Cas9-targeted-based long-read sequencing for genetic screening of RPE65 locus. Front. Genet. 15:1439153. doi: 10.3389/fgene.2024.1439153
Received: 27 May 2024; Accepted: 06 August 2024;
Published: 14 October 2024.
Edited by:
Luisa Azevedo, University of Porto, PortugalReviewed by:
Kimberley Billingsley, National Institutes of Health (NIH), United StatesKandarp Joshi, Johns Hopkins All Children’s Hospital, United States
Copyright © 2024 Rodilla, Núñez-Moreno, Benitez, Romero, Fernández-Caballero, Mínguez, Corton and Ayuso. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Marta Corton, bWNvcnRvbkBxdWlyb25zYWx1ZC5lcw==; Carmen Ayuso, Y2F5dXNvQGZqZC5lcw==