 Rong Hua Wang1†
Rong Hua Wang1† Ke Wu
Ke Wu- 1Department of Laboratory Medicine, Quzhou Maternal and Child Healthcare Hospital, Quzhou, Zhejiang, China
- 2Laboratory of Prenatal Diagnosis Center, Quzhou Maternal and Child Healthcare Hospital, Quzhou, Zhejiang, China
A dicentric chromosome is an abnormal chromosome with two centromeres on the same chromosome. It has been reported that dicentric chromosomes are specific biomarkers of radiation exposure, but dicentric chromosomes are rarely identified in newborns with multiple congenital anomalies. At 16 weeks of gestation, a 39-year-old pregnant woman (gravida 2, para 1) was referred to the prenatal diagnosis center for genetic counseling. The fetal ultrasonography indicated multiple anomalies. Subsequently, amniocentesis was performed, and the G-banding karyotype analysis showed a rare type of mosaicism. The C-banding karyotype analysis indicated a pseudo-dicentric chromosome X [psu dic (X; 18) (p11.2; p11.2)]. A single-nucleotide polymorphism array (SNP array) revealed three pathogenic copy number variations (CNVs). After genetic counseling, the parents chose to terminate this pregnancy. This study provides new evidence for a better understanding of the diagnosis of dicentric chromosomes and emphasizes on the importance of genetic counseling.
Introduction
A dicentric chromosome is an abnormal chromosome with two centromeres on the same chromosome. The occurrence of dicentric chromosomes in cancer cells is a well-recognized event (Mackinnon and Campbell, 2011). It has been reported that dicentric chromosomes are specific biomarkers of radiation exposure (Jeong et al., 2022). However, they are rarely identified in newborns with multiple congenital anomalies. Most dicentric chromosomes are known to form through chromosomal inversions. Dicentric chromosomes mainly (up to 80%) happen between acrocentric chromosomes (Stimpson et al., 2010). Robertsonian translocations (ROBs) involving acrocentric chromosomes (13, 14, 15, 21, and 22) most frequently generate dicentric chromosomes (Chiatante et al., 2017). In this study, we present the first description of a dicentric chromosome (X; 18).
Materials and methods
A 39-year-old pregnant woman (gravida 2, para 1) was referred to the center of prenatal diagnosis at Yiwu Maternity and Children Hospital for genetic counseling. At 16 weeks of gestation, the fetal ultrasonography indicated ventricular septal defect (VSD), pulmonary stenosis, cystic hygroma colli (CHC), choroid plexus cyst, and bilateral hydronephrosis (Figure 1).
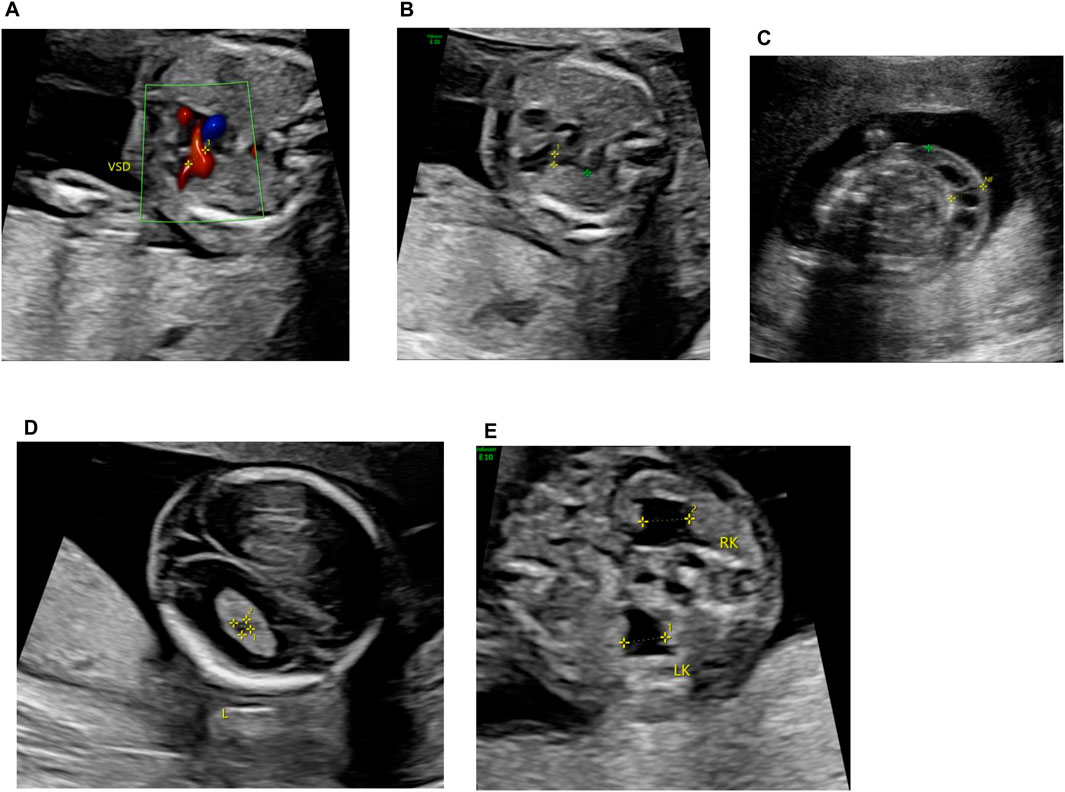
Figure 1. Fetal ultrasonography (at 16 weeks of gestation). (A) VSD, (B) pulmonary stenosis, (C) CCH, (D) choroid plexus cyst, and (E) bilateral hydronephrosis.
The parents signed an informed consent for genetic analysis and amniocentesis. Subsequently, the amniocentesis was performed, and the fetal sample detection was performed by single-nucleotide polymorphism array (SNP array) analysis, G-banding karyotype analysis with a band resolution of 400 bands, and C-banding karyotype analysis.
Results
G-banding and C-banding karyotype analysis
The G-banding karyotype analysis revealed a rare type of mosaicism 45, X, psu dic (X; 18) (p11.2; p11.2) [31]/45,X [26] (Figure 2). The C-banding analysis showed a dicentric chromosome X (Figure 3).
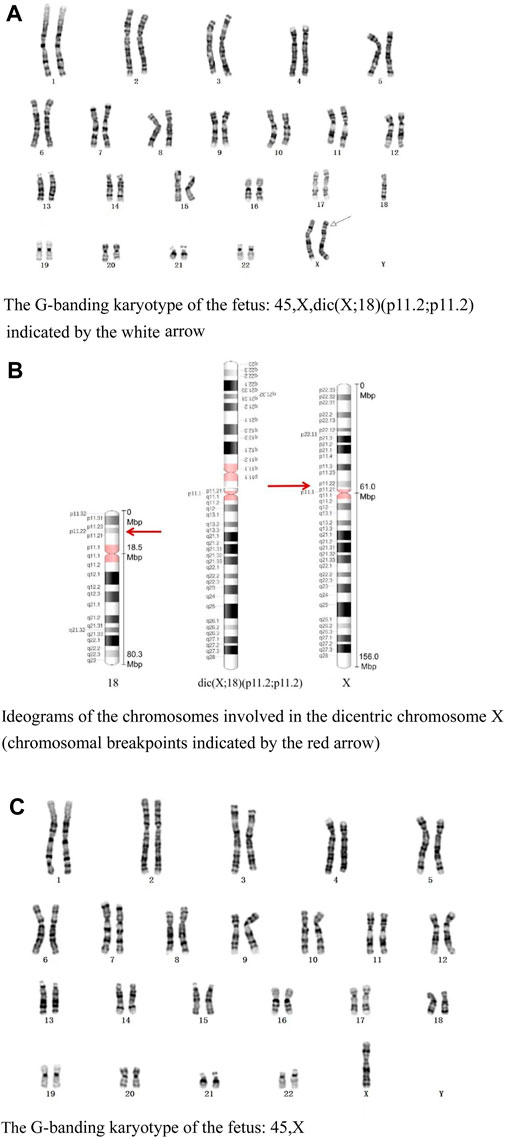
Figure 2. (A) G-banding karyotype of the fetus: 45,X,dic (X; 18) (p11.2; p11.2) indicated by the white arrow. (B) Ideograms of the chromosomes involved in the dicentric chromosome X (chromosomal breakpoints are indicated by the red arrow). (C) G-banding karyotype of the fetus: 45, X.
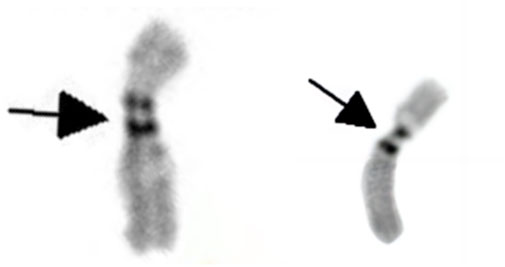
Figure 3. C-banding karyotype of dic (X; 18) (p11.2; p11.2) indicating two centromeres (as shown by the black arrow).
Chromosomal microarray analysis
The chromosomal microarray analysis (CMA) with an SNP array (Affymetrix CytoScan 750K Array, Santa Clara, California) revealed that the fetus had three pathogenic (Figure 4) CNVs: arr [GRCh37]18p11.32p11.21 (136,228_15181208)×1.32, arr [GRCh37]Xp22.33p11.22 (168,552_52154982) × 1, and arr [GRCh37]Xp11.22q28 (52705315_155233098) × 1.72. The dosage-sensitive genes in CNVs are listed in Table 1.
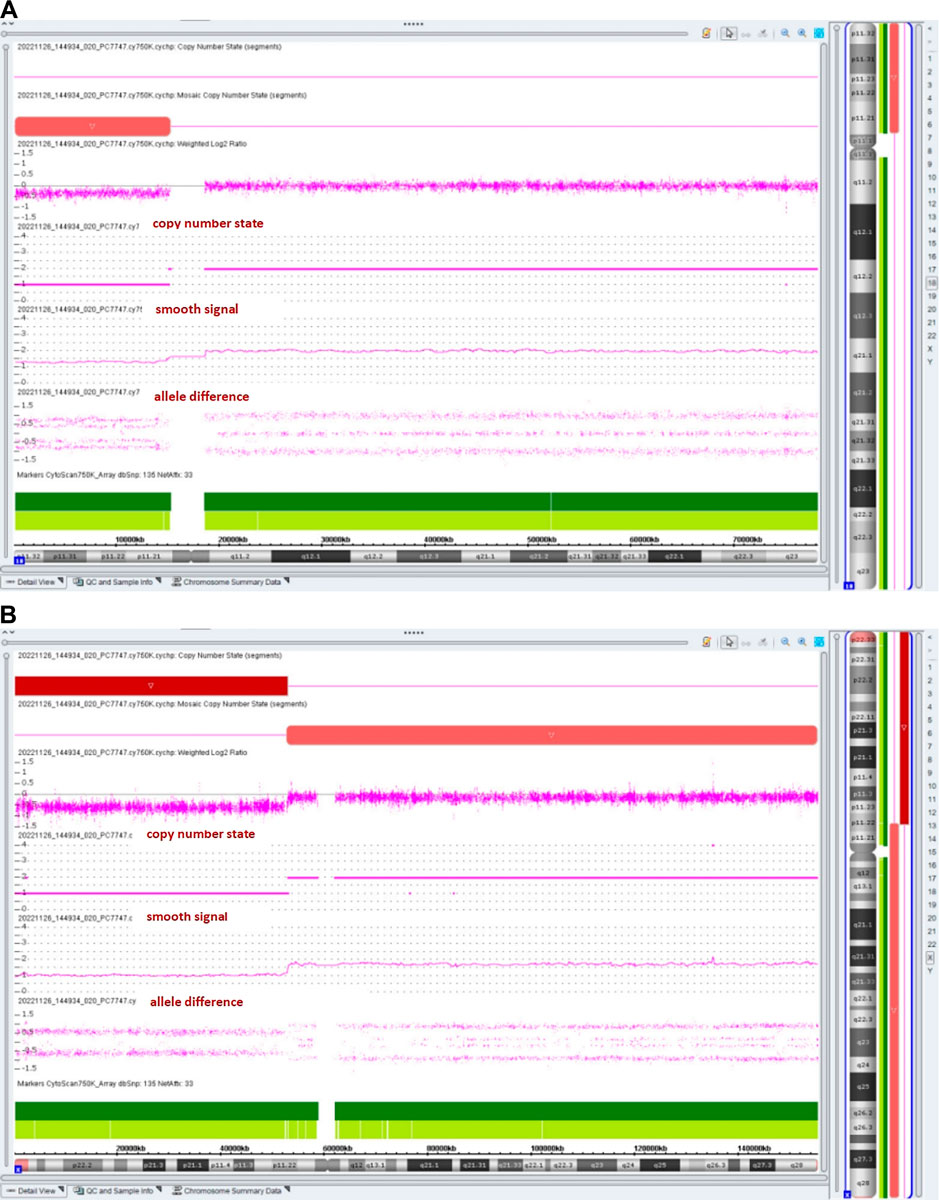
Figure 4. Results of the SNP array analysis (the “smooth signal” representing a copy number, and the “allele difference” with two rows representing haploid regions, with three rows representing diploid regions, and with four rows representing mosaic regions).

Table 1. List of the dosage-sensitive genes in CNVs.
Pregnancy outcome
Parental karyotypes and CMA were normal. The pregnant woman was informed of these results. After genetic counseling, this family decided to terminate this pregnancy at 20 weeks of gestation.
Discussion
Chromosome 18p deletion syndrome (OMIM#146390), also known as monosomy 18p, is a rare type of chromosomal syndrome with phenotypic heterogeneity. The clinical manifestations of 18p deletion syndrome include cardiac abnormalities (VSD, pulmonary stenosis, tetralogy of Fallot, and mild aortic valve abnormality), neurologic abnormalities (seizures, hypotonia, and holoprosencephaly), short stature, intellectual disability, holoprosencephaly, hypoplastic pituitary stalk, septo-optic dysplasia, isolated scoliosis, facial dysmorphism, genitourinary abnormalities, gastrointestinal abnormalities, hearing loss, pituitary abnormalities, and ophthalmologic abnormalities (Sebold et al., 2015).
The prevalence is estimated to be about 1:50,000 in live-born infants (Hasi-Zogaj et al., 2015). Over 100 individuals with 18p deletion syndrome have been reported (Kocaaga and Yimenicioglu, 2022), but rare cases (about 10 cases) have been described in prenatal diagnosis. It has been reported that fetuses with a pure 18p deletion present with severe hydronephrosis (Jin et al., 2021), holoprosencephaly (Yin et al., 2017), tetralogy of Fallot (Yi et al., 2014), reduced head circumference (Fogu et al., 2014), increased nuchal translucency (Monosomy, 2009), craniofacial abnormalities, and premaxillary agenesis (Chen et al., 2013). The CMA result of our case showed arr [GRCh37]18p11.32p11.21 (136,228_15181208)×1.32, with a smooth signal of 1.32 and a top allele difference row value of 0.66, indicating that approximately 68% of fetal cells had a 15.0-Mb deletion in the 18p11.32-p11.21 region, which overlapped the 18p deletion syndrome and contained a total of 58 OMIM genes, including a critical dosage-sensitive gene TGIF1 (OMIM*602630). The heterozygous mutations/deletions of the TGIF1 gene were associated with autosomal dominant holoprosencephaly 4 (OMIM#142946). This fetus did not show holoprosencephaly. Fetal ultrasonography indicated VSD and bilateral hydronephrosis, which have been reported in literature. The clinical phenotypes and severity may vary with the proportion and distribution of abnormal cells in various tissues.
This case was also a rare variant of Turner syndrome (TS). Vorsanova et al. (2021) reported that Turner syndrome mosaicism occurs in 1.9% of girls with neurodevelopmental disorders and congenital anomalies. The CMA result showed arr [GRCh37]Xp11.22q28 (52705315_155233098)×1.72 (smooth signal was 1.72, and the value of the top allele difference row was 0.86) indicate that approximately 28% of fetal cells had a 102.5-Mb deletion in the Xp11.22-q28 region. The CMA result also showed that the fetus had a 51.9-Mb heterozygous deletion in the Xp22.33-p11.22 region, which contained 264 OMIM genes such as SHOX (OMIM*312865). Two copies of the short-stature homeobox (SHOX) genes are located in the pseudoautosomal region 1 (PAR1) of Xp22.33 and Yp11.3, respectively. SHOX plays a particularly important role in short-stature conditions and bone development. SHOX has a clear haploinsufficiency effect. SHOX deficiency was associated with Léri–Weill dyschondrosteosis (OMIM#127300) and isolated short stature (OMIM#300582). In the absence of a family history of short stature, SHOX haploinsufficiency-related diseases were rarely diagnosed before late childhood (Ramachandrappa et al., 2018). Li et al. (2023) reported that only 62.5% (5/8) of fetuses with SHOX haploinsufficiency presented with short, long bones (Li et al., 2023). So it is understandable that the ultrasound examination indicated the normal length of the fetal long bones at 20 weeks of gestation.
The conventional C-band karyotype (Zhu et al., 2019) is still an effective complementary method to identify chromosomal heteromorphisms and is also essential for diagnosis of chromosomal disorders. The pseudo dicentric chromosome 45, X, psu dic (X; 18) (p11.2; p11.2) indicated by the C-band karyotype is an unbalanced X-autosome translocation. Some unbalanced X-autosome translocations have been reported to be preferentially inactivated, which could spread X chromosome inactivation (XCI) into the autosomal chromosome regions and inactivate autosomal genes (Favilla et al., 2021). We could not measure the extent of the spreading into the translocated 18. However, the spreading of inactivation into chromosome 18 could not be excluded. In this case, most probably, some genes from the fragment of chromosome 18p11.1-qter attached to chromosome X would be silenced. The spreading of X inactivation into translocated autosomes has been previously observed over large distances.
The X inactive specific transcript (XIST) gene is located in Xq13.2. XIST mediates the X-inactivation center (XIC). It has been found that the deletion of Xist is associated with preferential upregulation of XCI escape genes compared to XCI subjective genes, which underlies the variability in Xist deficiency phenotypes between different cell lineages and tissues (Yang et al., 2022). So these variable phenotypes may depend on the extent of silenced autosomal genes and genomic imbalances. The fetus was diagnosed with both extremely rare variant Turner syndrome mosaicism and chromosome 18p deletion syndrome mosaicism. The fetus may present with certain phenotypes related to the “silence” of genes in chromosome 18p11.1-qter.
Based on the origin of the centromere, dicentric chromosomes could be categorized into three types (Therman et al., 1986): 1) heterologous dicentric chromosomes derived from non-homologous chromosomes; 2) homologous dicentric chromosomes derived from two homologous chromosomes; and 3) isodicentric chromosomes derived from homologous chromosomes with two identical arms. According to the above categories, this case should belong to type 1, and the derivative dicentric X chromosome is asymmetrical. Individuals with dicentric chromosomes are rarely observed in humans, and we review previously published cases with dic (X; autosomes), which had detailed clinical information (Table 2). They presented with various phenotypes resulting from the extent of the monosomy or trisomy of the concerned segments. Dicentric chromosomes are prone to malsegregation during mitosis, resulting in anaphase bridges and chromosome breakage (Anbumani and Mohankumar, 2015). Most dicentric chromosomes are known to form through chromosomal inversions. Chromosomal inversions are rotations within the chromosomal arm due to chromosomal breakages or intra-chromosomal recombination.

Table 2. Summary of previously published dic (X; autosomes) cases.
As we have seen, the levels of mosaicism performed by conventional karyotype and CMA analyses are different. We know that the procedures for conventional cytogenetic testing and CMA are different. Conventional cytogenetic testing needs a key process of the short-term culture of cells derived from a specimen for generating karyotypes. During the period of the culture, the fetal cells with abnormal karyotypes may not grow well (Hao et al., 2020). So the number of fetal cells with abnormal karyotypes may not be counted. However, the procedures of CMA do not require cultured amniotic fluid cells; they just need direct extraction of cellular DNA (Hao et al., 2020). We consider it an “understandable” discrepancy between the cytogenetic karyotype and CMA results.
In this report, we describe a phenotypically abnormal fetus with mosaic karyotype 45, X, psu dic (X; 18) (p11.2; p11.2)[31]/45,X [26]. This is a rare type of pseudo-dicentric chromosome X that has not been previously reported. Our case report added descriptive information on this fetus and provided a useful reference for genetic counseling.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material; further inquiries can be directed to the corresponding author.
Ethics statement
The studies involving humans were approved by the Ethical Committee of Quzhou Maternity and Child Healthcare Hospital. The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author contributions
RW: writing–original draft and writing–review and editing. KW: data curation and writing–review and editing. XH: formal analysis, funding acquisition, and writing–review and editing.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This research was supported by grants from projects of Science and Technology of Quzhou (2023ZD084).
Acknowledgments
The authors are grateful to the patients and their families for their participation in this study, as well as to all the physicians for their assistance during the course of the medical treatment. They wish to thank the staff of Chigene (Beijing) Translational Medical Research Center Co. Ltd. for assisting with data analysis.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Anbumani, S., and Mohankumar, M. N. (2015). Nucleoplasmic bridges and tailed nuclei are signatures of radiation exposure in Oreochromis mossambicus using erythrocyte micronucleus cytome assay (EMNCA). Environ. Sci. Pollut. Res. Int. 22 (23), 18425–18436. doi:10.1007/s11356-015-5107-1
Chen, C. P., Huang, J. P., Chen, Y. Y., Chern, S. R., Wu, P. S., Su, J. W., et al. (2013). Chromosome 18p deletion syndrome presenting holoprosencephaly and premaxillary agenesis: prenatal diagnosis and aCGH characterization using uncultured amniocytes. Gene 527 (2), 636–641. doi:10.1016/j.gene.2013.06.081
Chiatante, G., Giannuzzi, G., Calabrese, F. M., Eichler, E. E., and Ventura, M. (2017). Centromere destiny in dicentric chromosomes: new insights from the evolution of human chromosome 2 ancestral centromeric region. Mol. Biol. Evol. 34 (7), 1669–1681. doi:10.1093/molbev/msx108
Eggermann, T., Mau, U., Klein-Vogler, U., Kendziorra, H., Mackensen-Haen, S., Sieverding, L., et al. (1998). Molecular and cytogenetic [correction of cytogenate] analysis of an X/autosomal translocation: 45,X,dic(X;17)(p22.2;p13). Clin. Genet. 53 (4), 293–297. doi:10.1111/j.1399-0004.1998.tb02699.x
Favilla, B. P., Meloni, V. A., Perez, A. B., Moretti-Ferreira, D., de Souza, D. H., Bellucco, F. T., et al. (2021). Spread of X-chromosome inactivation into autosomal regions in patients with unbalanced X-autosome translocations and its phenotypic effects. Am. J. Med. Genet. A 185 (8), 2295–2305. doi:10.1002/ajmg.a.62228
Fogu, G., Capobianco, G., Cambosu, F., Bandiera, P., Pirino, A., Moro, M. A., et al. (2014). Prenatal diagnosis and molecular cytogenetic characterisation of a de novo 18p deletion. J. Obstet. Gynaecol. 34 (2), 192–193. doi:10.3109/01443615.2013.834300
Hao, M., Li, L., Zhang, H., Liu, R., and Yu, Y. (2020). The difference between karyotype analysis and chromosome microarray for mosaicism of aneuploid chromosomes in prenatal diagnosis. J. Clin. Lab. Anal. 34 (12), e23514. doi:10.1002/jcla.23514
Hasi-Zogaj, M., Sebold, C., Heard, P., Carter, E., Soileau, B., Hill, A., et al. (2015). A review of 18p deletions. Am. J. Med. Genet. C Semin. Med. Genet. 169 (3), 251–264. doi:10.1002/ajmg.c.31445
Jeong, S. K., Oh, S. J., Kim, S. H., Jang, S., Kang, Y. R., Kim, H., et al. (2022). Dicentric chromosome assay using a deep learning-based automated system. Sci. Rep. 12 (1), 22097. doi:10.1038/s41598-022-25856-1
Jin, Q., Qiang, R., Cai, B., Wang, X., Zhen, S., et al. (2021). The genotype and phenotype of chromosome 18p deletion syndrome: case series. Med. Baltim. 100 (18), e25777. doi:10.1097/MD.0000000000025777
Kocaaga, A., and Yimenicioglu, S. (2022). Presentation of an infant with chromosome 18p deletion syndrome and asymmetric septal hypertrophy. Glob. Med. Genet. 9 (2), 179–181. doi:10.1055/s-0042-1743261
Li, L., Fu, F., Li, R., Jing, X., Yu, Q., Zhou, H., et al. (2023). Genetic analysis and sonography characteristics in fetus with SHOX haploinsufficiency. Genes (Basel) 14 (1), 140. doi:10.3390/genes14010140
Mackinnon, R. N., and Campbell, L. J. (2011). The role of dicentric chromosome formation and secondary centromere deletion in the evolution of myeloid malignancy. Genet. Res. Int. 2011, 643628. doi:10.4061/2011/643628
Monosomy, S. W. (2009). Monosomy 18p presenting with holoprosencephaly and increased nuchal translucency in the first trimester: report of 2 cases. J. Ultrasound Med. 28 (8), 1077–1080. doi:10.7863/jum.2009.28.8.1077
Ramachandrappa, S., Kulkarni, A., Gandhi, H., Ellis, C., Hutt, R., Roberts, L., et al. (2018). SHOX haploinsufficiency presenting with isolated short long bones in the second and third trimester. Eur. J. Hum. Genet. 26 (3), 350–358. doi:10.1038/s41431-017-0080-4
Scheuerle, A., Zenger-Hain, J. L., Van Dyke, D. L., Ledbetter, D. H., Greenberg, F., and Shaffer, L. G. (1995). Replication banding and molecular studies of a mosaic, unbalanced dic(X;15)(Xpter-->Xq26.1::15p11-->15qter). Am. J. Med. Genet. 56 (4), 403–408. doi:10.1002/ajmg.1320560411
Sebold, C., Soileau, B., Heard, P., Carter, E., O'Donnell, L., Hale, D. E., et al. (2015). Whole arm deletions of 18p: medical and developmental effects. Am. J. Med. Genet. A 167A (2), 313–323. doi:10.1002/ajmg.a.36880
Stimpson, K. M., Song, I. Y., Jauch, A., Holtgreve-Grez, H., Hayden, K. E., Bridger, J. M., et al. (2010). Telomere disruption results in non-random formation of de novo dicentric chromosomes involving acrocentric human chromosomes. PLoS Genet. 6 (8), e1001061. doi:10.1371/journal.pgen.1001061
Therman, E., Trunca, C., Kuhn, E. M., and Sarto, G. E. (1986). Dicentric chromosomes and the inactivation of the centromere. Hum. Genet. 72 (3), 191–195. doi:10.1007/BF00291876
Vorsanova, S. G., Kolotii, A. D., Kurinnaia, O. S., Kravets, V. S., Demidova, I. A., Soloviev, I. V., et al. (2021). Turner’s syndrome mosaicism in girls with neurodevelopmental disorders: a cohort study and hypothesis. Mol. Cytogenet 14 (1), 9. doi:10.1186/s13039-021-00529-2
Yang, T., Ou, J., and Yildirim, E. (2022). Xist exerts gene-specific silencing during XCI maintenance and impacts lineage-specific cell differentiation and proliferation during hematopoiesis. Nat. Commun. 13 (1), 4464. doi:10.1038/s41467-022-32273-5
Yi, Z., Yingjun, X., Yongzhen, C., Liangying, Z., Meijiao, S., and Baojiang, C. (2014). Prenatal diagnosis of pure partial monosomy 18p associated with holoprosencephaly and congenital heart defects. Gene 533 (2), 565–569. doi:10.1016/j.gene.2013.09.027
Yin, Z., Zhang, K., Ni, B., Fan, X., and Wu, X. (2017). Prenatal diagnosis of monosomy 18p associated with holoprosencephaly: case report. J. Obstet. Gynaecol. 37 (6), 804–806. doi:10.1080/01443615.2017.1306836
Zhu, J. J., Qi, H., Cai, L. R., Wen, X. H., Zeng, W., Tang, G. D., et al. (2019). C-banding and AgNOR-staining were still effective complementary methods to indentify chromosomal heteromorphisms and some structural abnormalities in prenatal diagnosis. Mol. Cytogenet 12, 41. doi:10.1186/s13039-019-0453-1
Keywords: dicentric chromosome, C-banding karyotype, fetal ultrasonography, genetic counseling, chromosomal microarray analysis
Citation: Wang RH, Wu K and Hu XL (2024) Prenatal diagnosis of dicentric chromosome X mosaicism: a case report and review. Front. Genet. 15:1436469. doi: 10.3389/fgene.2024.1436469
Received: 22 May 2024; Accepted: 01 July 2024;
Published: 18 July 2024.
Edited by:
Paulo Ricardo Gazzola Zen, Federal University of Health Sciences of Porto Alegre, BrazilReviewed by:
Rafaella Mergener, Federal University of Health Sciences of Porto Alegre, BrazilPatricia Trevisan, University of Colorado, United States
Copyright © 2024 Wang, Wu and Hu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiao Ling Hu, NTQzMDg5MTc5QHFxLmNvbQ==
†These authors have contributed equally to this work