 Artem Borovikov1*
Artem Borovikov1* Andrey Marakhonov1
Andrey Marakhonov1 Aysylu Murtazina1
Aysylu Murtazina1 Kseniya Davydenko1
Kseniya Davydenko1 Alexandra Filatova1
Alexandra Filatova1 Nailya Galeeva1Varvara Kadnikova1Natalya Ogorodova1Daria Gorodilova1
Nailya Galeeva1Varvara Kadnikova1Natalya Ogorodova1Daria Gorodilova1 Ilya Kanivets2,3Denis Pyankov2Konstantin Zherdev4,5Aleksandr Petel’guzov4Pavel Zubkov4Alexander Polyakov1
Ilya Kanivets2,3Denis Pyankov2Konstantin Zherdev4,5Aleksandr Petel’guzov4Pavel Zubkov4Alexander Polyakov1 Olga Shchagina1
Olga Shchagina1 Mikhail Skoblov1
Mikhail Skoblov1- 1Research Centre for Medical Genetics, Moscow, Russia
- 2Genomed, Moscow, Russia
- 3Federal State Budgetary Educational Institution, Further Professional Education, Russian Medical Academy of Continuous Professional Education, Ministry of Healthcare of the Russian Federation, Moscow, Russia
- 4National Medical Research Center of Children’s Health, Moscow, Russia
- 5Department of Pediatric Surgery and Urology-Andrology, I. M. Sechenov First Moscow State Medical University, Moscow, Russia
Multiple osteochondromas (MO) is a rare autosomal dominant skeletal disorder characterized by the development of multiple benign tumors known as osteochondromas. The condition is predominantly caused by loss-of-function variants in the EXT1 or EXT2 genes, facilitating relatively precise clinical diagnosis through established diagnostic criteria. Despite this, a notable percentage of MO cases (10%–20%) remains unresolved after sequencing coding regions and copy number analysis of both genes. In our study, we identified mosaic structural variants in two patients who initially yielded negative results on standard genetic analysis for MO. Specifically, mosaic deletions affecting exons 8–11 and exons 2–11 in the EXT1 gene were detected. RNA analysis was performed in one case, while both cases underwent genome sequencing. To date, only six mosaic copy number variations have been reported in association with MO, representing a minority among known variants in both genes. Our report provides a detailed analysis of these findings, highlighting the significance of advanced genetic testing techniques in detecting mosaic variants in the EXT1/2 genes.
1 Introduction
Multiple osteochondromas (MO), ICD-11: LD24.20, is a rare autosomal dominant skeletal disorder characterized by the formation of multiple benign tumors known as osteochondromas (Bovée, 2008; International Classification of Diseases, Eleventh Revision, 2019/2021). The disease is primarily caused by heterozygous loss-of-function variants in the EXT1 or EXT2 genes (Ahn et al., 1995; Stickens et al., 1996). Around 10%–20% of MO cases remain undiagnosed even after investigating the single-nucleotide variants (SNVs) within the coding regions and conducting copy number variation (CNV) analysis in the EXT1 and EXT2 genes (Ciavarella et al., 2013; Santos et al., 2018; Caino et al., 2022; Güneş et al., 2023). In our previous recent investigation involving a cohort of 244 families initially diagnosed with MO, we observed that 3.6% of these cases could be attributed to loss-of-function mutations in the PTPN11 gene (Borovikov et al., 2024). However, this finding requires confirmation by further cohort studies.
Other potential causes of undiagnosed cases could include deep intronic variants or variants in regulatory regions of the EXT1/2 genes, which are not routinely analyzed, and no such variants have been described to date (Stenson et al., 2003). Additionally, mosaic variants may contribute to disease pathology, as evidenced by six reported cases of mosaic CNVs (Szuhai et al., 2010; Sarrión et al., 2013; Oliver et al., 2019).
We identified mosaic structural variants in two patients whose routine genetic tests yielded negative findings, utilizing RNA analysis in one case and genome sequencing in both cases. In this report, we provide a detailed description of these two cases.
2 Materials and methods
2.1 Clinical data
The diagnosis of MO was based on typical clinical and/or radiographic findings (Bovée, 2008). Clinical examinations and scale classification perform according to the scale of severity of classification proposed by Istituto Ortopedico Rizzoli (IOR) (Mordenti et al., 2021).
2.2 DNA analysis
DNA was extracted from whole-blood samples using a Wizard® Genomic DNA Purification Kit (Promega, United States). Sanger sequencing was carried out using the ABI PRISM 3500 Genetic Analyzer (Applied Biosystems, Foster City, CA, United States). A custom NGS panel, consisting of three genes, NM_000127.3 (EXT1), NM_207122.2 (EXT2), and NM_002834.5 (PTPN11), was used for sequencing on an Ion S5 (Thermo Fisher Scientific, Waltham, MA, United States) or MiSeq (Illumina, United States) sequencer. CNV analysis was carried out using the SALSA MLPA Probemix P215-B4 EXT (MRC Holland). Genome sequencing (GS) was performed using a DNBSEQ-G400 instrument in a pair-ended mode (2 × 150 bp) with an average on-target coverage of 30× with a DNBSEQ™ True PCR-Free kit (BGI, Beijing, China) for library preparation (GenoMed Ltd., Moscow, Russia). WGS findings were confirmed by Sanger sequencing.
2.3 Bioinformatics analysis
Bioinformatics analysis was performed using an in-house software pipeline described earlier with modifications (Rudenskaya et al., 2019). In brief, it included quality control of raw reads (FastQC tool v0.11.5), followed by read mapping to the hg19 human genome assembly (minimap2 v2.24-r1122), sorting of the alignments, and marking duplicates (Picard Toolkit v2.18.14). Base recalibration and variant calling were performed with GATK3.8. Variant annotation was done using ANNOVAR (v2018Apr16). CNV and SV analysis was performed using the Manta tool (v1.6.0).
2.4 RNA analysis
mRNA analysis was performed on primary cultured fibroblasts (PMBCs) isolated from the patient. Total RNA was extracted using the ExtractRNA reagent (Evrogen, Russia) according to the manufacturer’s recommendations. Reverse transcription was carried out using the ImProm-II™ Reverse Transcription System (Promega, United States). EXT1 and EXT2 mRNAs were divided into six overlapping loci each. The mRNA exonic structure was analyzed by PCR with locus-specific primers. Isoforms of EXT1/EXT2 were compared with mRNA isoforms from healthy donors. The PCR products were analyzed by gel electrophoresis, followed by Sanger sequencing. All loci were examined additionally by NGS. Libraries were prepared and sequenced on an Ion Torrent S5 system (with coverage >200,000). The raw sequencing data were processed with a custom pipeline based on open-source bioinformatics tools HISAT2, SAMtools, and SAJR. Splice junctions were visualized using a Sashimi plot in the IGV browser.
3 Case reports
3.1 Case 1
The first proband was an 11-year-old male child who exhibited dense bony formations of varying severities on the chest and extremity bones. The initial protrusion was discovered at the age of 3 years in the distal part of the popliteal fossa. Subsequently, the patient’s relatives observed the appearance of similar formations in the shoulders, hips, ribs, and forearm. Radiography of the knee, hip joints, and upper extremities at the age of 8–11 years revealed multiple osteochondromas in the popliteal fossa, distal part of the ulna, and around the hip joint (Figure 1A). The proband underwent several surgeries.
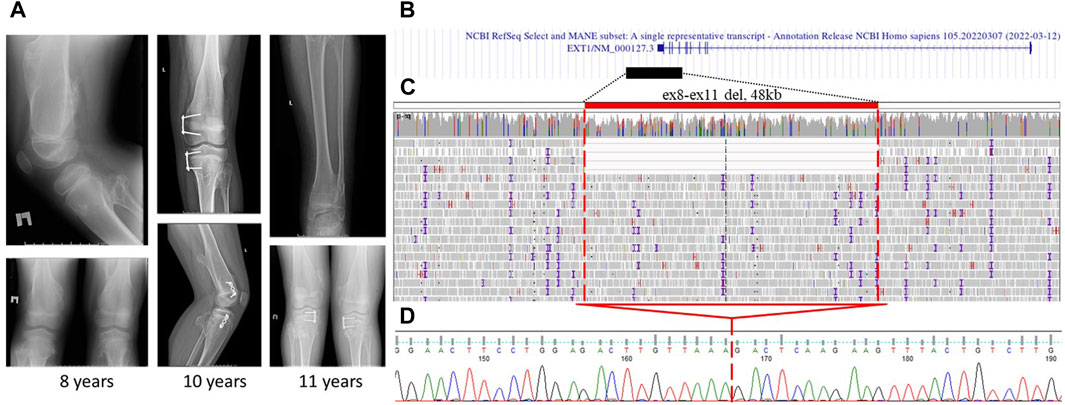
Figure 1. Case 1: (A) X-ray images of proband 1 at the age of 8, 10, and 11 years. Multiple osteochondromas around the knee and on the femur are shown. (B) Alignment of the deletion of exons 8–11 in the EXT1 gene in the UCSC browser was found. (C) GS data on mosaic gross deletion from case 1 visualized in the IGV browser. (D) Chromatogram showing the region encompassing the breakpoints of the deletion, with a red line marking the borders of the deletion.
Clinical examination showed a normal facial appearance and psychomotor development. However, the patient presented with hallux valgus deformity of the feet, weight-bearing imbalance when resting on the left leg, and tenderness during active movements of the left knee and hip joints. Palpation revealed dense formations on the posterior surface of the proximal part of the left lower leg, around the left scapula, wrist, and ribs. Additionally, there was ulnar deviation of both hands due to deformities in both radial bones. According to the IOR score, his clinical severity was categorized as IIB.
Targeted sequencing of the exon-coding regions by direct Sanger sequencing and MLPA of the EXT1 and EXT2 loci yielded negative results. The next step involved conducting RT-PCR on EXT1/2 genes to analyze the exon–exon structure and gene expression. This revealed a borderline unequal expression of alleles (45% for the reference allele G and 55% for the alternative allele A) at the common SNP (rs17439693) in exon 6 of the EXT1 gene and the absence of splicing aberrations in both genes. Notably, this SNP was the only heterozygous variant in the coding regions of the EXT1/2 genes. Following negative RNA analysis results, direct sequencing of the 5′untranslated and promoter region also did not reveal any variants. Subsequent GS was performed, which identified a mosaic deletion of chr8:118780297-118828217del (4/40 = 10.0% of the total read count) involving exons 8–11 of the EXT1 gene (Figures 1B, C). This finding was further confirmed by Sanger sequencing (Figure 1D).
3.2 Case 2
In another proband, the first osteochondroma was detected at 3.5 years in the left forearm area. Over 1.5 years of observation, additional small osteochondromas appeared in other parts of the body. Surgical interventions were not performed; however, at the time of examination, surgical removal of the osteochondroma in the forearm area was planned. A clinical examination conducted at the age of 5 years revealed dense formations in the distal region of the left forearm, in the area of the left knee joint, and small formations in the area of the right humerus. The OIR score was IA, which is appropriate severity for patients of such young age. After negative results of gene panel sequencing and MLPA, GS was performed and revealed mosaic gross deletion of chr8:118798015_118893064del affecting exons 2–11 in the EXT1 gene (5/43 = 11.6% of the total read count) (Figures 2A, B). Deletion was confirmed by Sanger sequencing (Figure 2C).
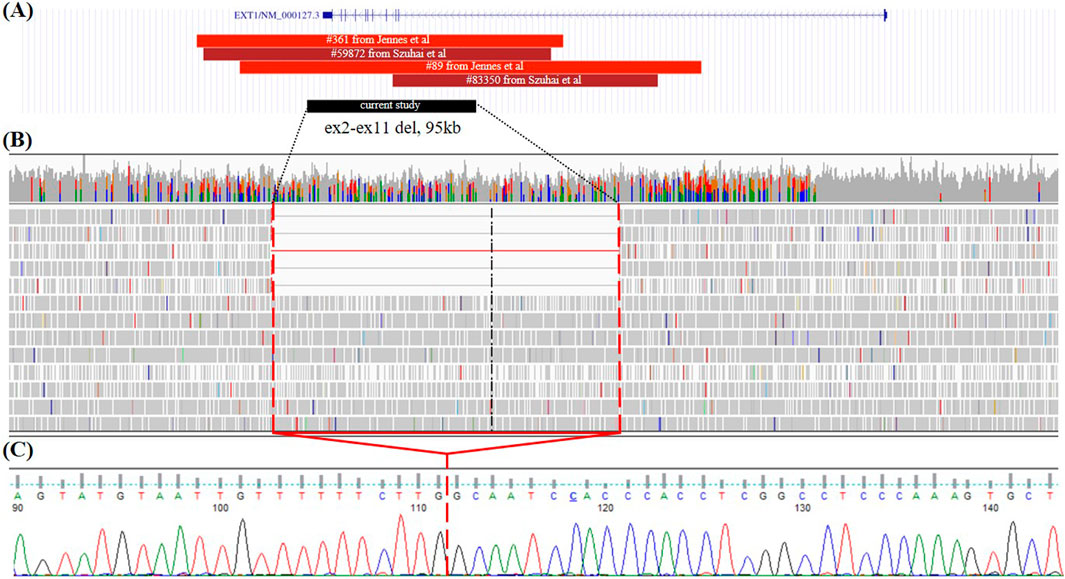
Figure 2. Case 2. (A) Alignment of all deletion of exons 2–11 in the EXT1 gene with known breakpoint borders on the UCSC browser from the literature and case 2 from the current study. Red—germline CNVs; dark red—mosaic CNVs; black—deletion found in the present study. (B) GS data on mosaic gross deletion from case 2 visualized in the IGV browser. (C) Chromatogram showing the region encompassing the breakpoints of the deletion, with a red line marking the borders of the deletion.
Both families were recommended to regularly monitor the size of the osteochondromas and bone deformities in order to take timely and appropriate orthopedic and surgical measures for correction.
4 Discussion
We presented two cases of mosaic partial deletion of the EXT1 gene. To the best of our knowledge, to date, only six cases with mosaic variants in the EXT1 and EXT2 genes are reported in three publications (Szuhai et al., 2010; Sarrión et al., 2013; Oliver et al., 2019). The most frequent (three out of six cases) are mosaic deletions of exons 2–11 of the EXT1 gene, one mosaic deletion of exons 2–3 in the EXT1 gene, and deletion of exon 2 of the EXT2 gene (Szuhai et al., 2010; Sarrión et al., 2013). The last of previously described mosaic variants was a deletion affecting exon 1 of the EXT1 gene and last exons of the SAMD12 gene; this deletion led to a fusion between two gene transcripts and was detected by RNA-seq (Oliver et al., 2019).
One of our patients had a recurrent mosaic deletion of exons 2–11 of the EXT1 gene, while another individual had a novel mosaic deletion of exons 8–11 of the same gene. Both children had disease onset at the age of 3–3.5 years when the first osteochondroma was discovered. One of them had more than 10 osteochondromas at the age of 11 years, with deformation of radial bones that correspond to the average moderate severity of MO at this age. It is interesting to note that despite the low percentage of mosaicism observed in all cases, including ours, the clinical presentation of patients does not differ significantly in severity from non-mosaic cases (Szuhai et al., 2010; Sarrión et al., 2013; Oliver et al., 2019). Both of our probands underwent standard management for MO, which includes periodic evaluation by orthopedic specialists and surgical intervention based on functional limitations or symptomatic presentation caused by osteochondromas. One patient underwent multiple surgical procedures, whereas the younger proband did not require surgical intervention.
Mosaic variants are also intriguing in the context of the two-hit hypothesis of MO pathogenesis (Bovée et al., 1999). According to this hypothesis, probands may harbor an additional somatic loss-of-function variant in the EXT1 gene, contributing to osteochondroma formation. Furthermore, it is plausible that the percentage of mosaic mutant alleles in osteochondromas could be higher, potentially explaining similarities in disease progression between mosaic and germline variants. Unfortunately, neither we nor other researchers had access to tumor samples to investigate the level of mosaicism of the identified variants or explore potential second somatic variants.
All reported mosaic cases of MO are structural variants, particularly deletions, with no instances of mosaic SNVs reported to date. Among eight reported mosaic CNVs, including our own findings, four CNVs are deletions affecting exons 2–11 of the EXT1 gene (Szuhai et al., 2010; Sarrión et al., 2013; Oliver et al., 2019). Although germline deletions of exons 2–11 have been noted in various cohort studies, in our cohort study, the deletion of exons 2–11 was counted for 4 cases out of 10 pathogenic CNVs in the EXT1 gene (Vink et al., 2004; Sarrión et al., 2013; Li et al., 2018; Borovikov et al., 2024). Breakpoint details are available for two germline deletions and two mosaic deletions affecting exons 2–11 of the EXT1 gene (Figure 2A) (Szuhai et al., 2010; Jennes et al., 2011). The deletion of exons 2–11 of the EXT1 gene identified in our study (case 2) is the smallest, spanning 95,049 base pairs. Furthermore, each deletion displays distinct breakpoints, suggesting an absence of hotspot regions for breakpoint junctions. Multiple sites for breakpoints due to the size of intron 1 (272,882 bp) of the EXT1 gene (312,457 bp) could correspond to the higher prevalence of deletion events affecting exons 2–11 among all germline and mosaic CNVs.
In both cases where CNVs were identified, we observed the absence of long homologous sequences or closely located repeats in breakpoint regions of the same class. The deletion spanning exons 8–11 exhibited a 2-base microhomology, while the deletion spanning exons 2–11 displayed a 3-base microhomology. These findings suggest that non-homologous end joining (NHEJ) is the most plausible mechanism for both deletions as NHEJ typically requires short homologous sequences, typically 1–4 base pairs in length. Comparatively, another study investigating CNVs in the EXT1/2 genes reported that 6 out of 10 CNVs likely originate from NHEJ (Jennes et al., 2011).
In both cases analyzed in our study, we observed a mosaic deletion with an approximate allele frequency of 10%. In both cases, retrospective analysis of the MLPA profile could be regarded as suggestive for a mosaic deletion, but the decrease in signals was too subtle (0.71–0.9 for various probes within affected exons) to detect with the routinely used parameters (dosage quotient for a normal copy number between 0.80 < DQ < 1.20). In case 1, mosaic deletion was initially detected through RNA analysis in fibroblast culture, revealing allelic imbalance (45:55) in the EXT1 gene. This distribution of alleles corresponded with the count of deletion-supporting reads in the GS data, thereby validating the effect of the deletion at the RNA level. However, the observed difference in allele expression revealed by RNA analysis may not have been adequately discerned solely due to the presence of a mosaic deletion, highlighting the crucial role of GS data in this validation process.
Routine diagnostic methods often struggle to detect such variants, and even RNA analysis may not always be conclusive. Additionally, genome sequencing has limitations, particularly in achieving sufficient coverage for accurately identifying variants with low mosaic frequencies. This underscores the need for caution in interpreting our findings as they raise intriguing questions about the pathogenic threshold of mosaic variants, particularly given their rarity in most hereditary diseases. Future investigations on this subject would greatly benefit from access to more patient samples exhibiting similar characteristics.
In conclusion, our study contributes two novel cases of mosaic partial deletions of the EXT1 gene, adding to a limited pool of reported mosaic variants in MO. Despite the rarity of mosaic events, the clinical severity of these cases remains comparable to non-mosaic cases. The predominance of deletion events in mosaic variants underscores the distinct genetic landscape of MO, highlighting the need for further investigation into the genetic mechanisms underlying this condition. Future research with larger cohorts and access to tumor samples could provide deeper insights into the prevalence and impact of mosaic events in MO pathogenesis and clinical outcomes.
Data availability statement
The datasets for this article are not publicly available due to concerns regarding participant/patient anonymity. Requests to access the datasets should be directed to the corresponding author.
Ethics statement
The studies involving humans were approved by the Ethics Committee of the Research Centre for Medical Genetics. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants’ legal guardians/next of kin. Written informed consent was obtained from the individual(s), and minor(s)’ legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
Author contributions
AB: conceptualization, data curation, formal analysis, investigation, methodology, visualization, writing–original draft, and writing–review and editing. AnM: formal analysis, funding acquisition, investigation, supervision, visualization, and writing–review and editing. AyM: writing–original draft and writing–review and editing. KD: formal analysis, investigation, and writing–review and editing. AF: investigation and writing–review and editing. NG: investigation and writing–review and editing. VK: investigation and writing–review and editing. NO: investigation and writing–review and editing. DG: investigation and writing–review and editing. IK: investigation and writing–review and editing. DP: investigation and writing–review and editing. KZ: investigation and writing–review and editing. AP’g: investigation and writing–review and editing. PZ: investigation and writing–review and editing. AP: investigation and writing–review and editing. OS: data curation, investigation, supervision, and writing–review and editing. MS: data curation, funding acquisition, investigation, supervision, and writing–review and editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This study was supported by the state assignment of the Ministry of Science and Higher Education of the Russian Federation.
Acknowledgments
The authors thank the families of the individuals who participated in this study. The authors also thank the Russian multiple osteochondromas patient organization “ECHO” for their unwavering support and assistance.
Conflict of interest
IK and DP were employed by LTD Genomed.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Ahn, J., Lüdecke, H. J., Lindow, S., Horton, W. A., Lee, B., Wagner, M. J., et al. (1995). Cloning of the putative tumour suppressor gene for hereditary multiple exostoses (EXT1). Nat. Genet. 11 (2 11), 137–143. doi:10.1038/ng1095-137
Borovikov, A., Galeeva, N., Marakhonov, A., Murtazina, A., Kadnikova, V., Davydenko, K., et al. (2024). The missing piece of the puzzle: unveiling the role of PTPN11 gene in multiple osteochondromas in a large cohort study. Hum. Mutat. 2024, 1–13. doi:10.1155/2024/8849348
Bovée, J. V. M. G. (2008). Multiple osteochondromas. Orphanet J. Rare Dis. 3, 3. doi:10.1186/1750-1172-3-3
Bovée, J. V. M. G., Cleton-Jansen, A. M., Wuyts, W., Caethoven, G., Taminiau, A. H. M., Bakker, E., et al. (1999). EXT-mutation analysis and loss of heterozygosity in sporadic and hereditary osteochondromas and secondary chondrosarcomas. Am. J. Hum. Genet. 65, 689–698. doi:10.1086/302532
Caino, S., Cubilla, M. A., Alba, R., Obregón, M. G., Fano, V., Gómez, A., et al. (2022). Clinical and genetic analysis of multiple osteochondromas in a cohort of Argentine patients. Genes 13, 2063. doi:10.3390/GENES13112063
Ciavarella, M., Coco, M., Baorda, F., Stanziale, P., Chetta, M., Bisceglia, L., et al. (2013). 20 novel point mutations and one large deletion in EXT1 and EXT2 genes: report of diagnostic screening in a large Italian cohort of patients affected by hereditary multiple exostosis. Gene 515, 339–348. doi:10.1016/J.GENE.2012.11.055
Güneş, N., Alkaya, D. U., Toylu, A., Özüdoğru, P., Sunamak, E. Ç., Şeker, A., et al. (2023). Phenotypic and molecular spectrum of a Turkish cohort with hereditary multiple osteochondromas. Turkish archives Pediatr. 58, 376–381. doi:10.5152/TURKARCHPEDIATR.2023.23011
International Classification of Diseases, Eleventh Revision (ICD-11) (2019/2021). Geneva, Switzerland: World Health Organization. Available at: https://icd.who.int/browse11.
Jennes, I., de Jong, D., Mees, K., Hogendoorn, P. C. W., Szuhai, K., and Wuyts, W. (2011). Breakpoint characterization of large deletions in EXT1 or EXT2 in 10 Multiple Osteochondromas families. BMC Med. Genet. 12, 85. doi:10.1186/1471-2350-12-85
Li, Y., Wang, J., Tang, J., Wang, Z., Han, B., Li, N., et al. (2018). Heterogeneous spectrum of EXT gene mutations in Chinese patients with hereditary multiple osteochondromas. Medicine 97, e12855. doi:10.1097/MD.0000000000012855
Mordenti, M., Gnoli, M., Boarini, M., Trisolino, G., Evangelista, A., Pedrini, E., et al. (2021). The Rizzoli Multiple Osteochondromas Classification revised: describing the phenotype to improve clinical practice. Am. J. Med. Genet. A 185, 3466–3475. doi:10.1002/AJMG.A.62470
Oliver, G. R., Blackburn, P. R., Ellingson, M. S., Conboy, E., Pinto e Vairo, F., Webley, M., et al. (2019). RNA-Seq detects a SAMD12-EXT1 fusion transcript and leads to the discovery of an EXT1 deletion in a child with multiple osteochondromas. Mol. Genet. Genomic Med. 7, e00560. doi:10.1002/MGG3.560
Rudenskaya, G. E., Marakhonov, A. V., Shchagina, O. A., Lozier, E. R., Dadali, E. L., Akimova, I. A., et al. (2019). Ataxia with oculomotor apraxia type 4 with PNKP common "Portuguese" and novel mutations in two Belarusian families. J. Pediatr. Genet. 8 (2), 58–62. doi:10.1055/s-0039-1684008
Santos, S. C. L., Rizzo, I. M. P. O., Takata, R. I., Speck-Martins, C. E., Brum, J. M., and Sollaci, C. (2018). Analysis of mutations in EXT1 and EXT2 in Brazilian patients with multiple osteochondromas. Mol. Genet. Genomic Med. 6, 382–392. doi:10.1002/MGG3.382
Sarrión, P., Sangorrin, A., Urreizti, R., Delgado, A., Artuch, R., Martorell, L., et al. (2013). Mutations in the EXT1 and EXT2 genes in Spanish patients with multiple osteochondromas. Sci. Rep. 3 (1 3), 1346–1347. doi:10.1038/srep01346
Stenson, P. D., Ball, E. V., Mort, M., Phillips, A. D., Shiel, J. A., Thomas, N. S. T., et al. (2003). Human gene mutation database (HGMD): 2003 update. Hum. Mutat. 21, 577–581. doi:10.1002/HUMU.10212
Stickens, D., Clines, G., Burbee, D., Ramos, P., Thomas, S., Hogue, D., et al. (1996). The EXT2 multiple exostoses gene defines a family of putative tumour suppressor genes. Nat. Genet. 14, 25–32. doi:10.1038/NG0996-25
Szuhai, K., Jennes, I., de Jong, D., Bovée, J. V., Wiweger, M., Wuyts, W., et al. (2010). Tiling resolution array-CGH shows that somatic mosaic deletion of the EXT gene is causative in EXT gene mutation negative multiple osteochondromas patients. Hum. Mutat. 32 (2), 2036–E2049. doi:10.1002/humu.21423
Vink, G. R., White, S. J., Gabelic, S., Hogendoorn, P. C. W., Breuning, M. H., and Bakker, E. (2004). Mutation screening of EXT1 and EXT2 by direct sequence analysis and MLPA in patients with multiple osteochondromas: splice site mutations and exonic deletions account for more than half of the mutations. Eur. J. Hum. Genet. 13 (4 13), 470–474. doi:10.1038/sj.ejhg.5201343
Keywords: multiple osteochondromas, exostosis, EXT1, EXT2, mosaic deletion, genome sequencing
Citation: Borovikov A, Marakhonov A, Murtazina A, Davydenko K, Filatova A, Galeeva N, Kadnikova V, Ogorodova N, Gorodilova D, Kanivets I, Pyankov D, Zherdev K, Petel’guzov A, Zubkov P, Polyakov A, Shchagina O and Skoblov M (2024) Cases report: Mosaic structural variants of the EXT1 gene in previously genetically unconfirmed multiple osteochondromas. Front. Genet. 15:1435493. doi: 10.3389/fgene.2024.1435493
Received: 20 May 2024; Accepted: 26 July 2024;
Published: 13 August 2024.
Edited by:
Karoly Szuhai, Leiden University Medical Center (LUMC), NetherlandsReviewed by:
Wim Wuyts, University of Antwerp, BelgiumPancras C. W. Hogendoorn, Leiden University Medical Center (LUMC), Netherlands
Copyright © 2024 Borovikov, Marakhonov, Murtazina, Davydenko, Filatova, Galeeva, Kadnikova, Ogorodova, Gorodilova, Kanivets, Pyankov, Zherdev, Petel’guzov, Zubkov, Polyakov, Shchagina and Skoblov. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Artem Borovikov, Ym9yb3Zpa292MzNAZ21haWwuY29t