 Hongmei Huang
Hongmei Huang Yue Qian
Yue Qian Chenlu Yang
Chenlu Yang- Beijing Children’s Hospital, Capital Medical University, National Center for Children’s Health, Beijing, China
Background: Tousled-like kinase 2 (TLK2) gene variant-related neurodevelopmental disorder was recently described. The haploinsufficiency of TLK2 was considered the most likely underlying disease mechanism, leading to a consistent neurodevelopmental phenotype. So far, only four studies, conducted on 49 patients from North America and Europe, have been reported.
Case presentation: In this study, we reported a Chinese family with a TLK2-related neuropsychiatric phenotype. The proband, a boy aged 2 years and 6 months, presented with temper tantrums, mood lability, aggressiveness, congenital astigmatism, and distinctive facial dysmorphism. Whole-exome sequencing identified a novel heterozygous variation in TLK2 gene (c.49dupG, p. E17Gfs*10) in them. His father carried the same TLK2 gene variant and exhibited anxiety and irritability. The parental grandparents and other family members had no such variation. Moreover, the proband was found to have global developmental delay, autism-like symptoms, and mild elevated homo-vanillic acid (HVA) and 2,3-dihydroxy-2-methylbutyric acid levels tested in urine.
Conclusion: Herein, we identified a novel TLK2 variant from a Chinese family and reported a new neuropsychiatric phenotype. This study also expanded the genotype profile of the newly defined TLK2-related neurodevelopmental disorder.
Introduction
Neurodevelopmental disorders (NDDs) are genetically heterogeneous diseases. Studies have shown that the genetic etiology of neurodevelopmental disorders can be found in up to 47% of patients with NDD (Woods et al., 2022). Tousled-like kinase 2 (TLK2) is located at 17q23.2, contains 22 exons, and encodes a ubiquitous 717-aa protein, which plays an important role in DNA replication, cell cycle checkpoint, recovery, and chromatin remodeling (Mortuza et al., 2018). The TLK2 gene variant was first described to be a candidate gene of NDDs in a cohort study in 2016 (Lelieveld et al., 2016). Following the initial publication, four additional reports were published, associating this gene with neurodevelopmental disorder (Reijnders et al., 2018; Topf et al., 2020; Pavinato et al., 2022; Woods et al., 2022). One study described a patient with a homozygous TLK2 variant, leading to an autosomal recessive severe neurodevelopmental disorder (Topf et al., 2020). Reijnders et al. identified a large number of individuals (38 probands and 2 affected mothers) with heterozygous mutations in TLK2 gene showed similar clinical characteristics, including mild-to-moderate intellectual developmental disorder, behavioral problems, gastrointestinal disorders, and typical facial dysmorphism (Reijnders et al., 2018). Recently, another two studies reported pathogenic TLK2 variants in eight patients with global developmental delay and also described that some novel phenotypes include sleep disturbance, non-differentiation of lateral semi-circular canals, vesico-ureteric reflux, and bilateral periauricular skin tags (Woods et al., 2022).
Totally, 39 TLK2 gene variants in 49 patients were reported to exhibit TLK2-related neurodevelopmental disorders in four studies. All cases were identified from North America and Europe, and more than two-thirds of them, from two countries, namely, the Netherlands and the UK (1, 4-6). With an estimated prevalence of 1/566 (17/9,625) of TLK2 variants in probands recruited to the NDD cohort study, it is expected that a larger number of individuals with TLK2 variants is present worldwide (Reijnders et al., 2018).
Here, we presented a Chinese family with TLK2-related neurodevelopmental disorder. A novel heterozygous variant (c.49dupG, p. E17Gfs*10) in TLK2 gene, was identified in a 2-year-old boy with mild global developmental delay, autism-like symptoms, and moderate-to-severe behavior problems. His father also carries this TLK2 variant and presented temper tantrums, mood lability, and aggressiveness. They also have congenital astigmatism and distinctive facial dysmorphism. The boy’s grandparents and other family members have no such variation.
Case presentation
The proband was a boy aged 2 years and 6 months with behavioral problems. He was recommended to our Pediatric Developmental Behavior Clinic because he presented temper tantrums, mood lability, self-injurious behavior, and aggressiveness toward others at home. He was full-term born to nonconsanguineous parents by selective C-section delivery, G1P1, with birth weight 3300 g (75–90th percentile), and birth height 50.0 cm (50–75th percentile). There were no other medical issues during pregnancy. It was observed that he was able to sit alone at 6 months, to walk alone at 13 months, and to speak the first meaningful words after 2 years of age.
Physical examination revealed his height was 95 cm (within the 50 to 75 centiles for his age), weight was 15 kg (within the 75 to 90 centiles for his age), and head circumference was 48.5 cm (within the 25 to 50 centiles for his age). Distinctive facial features were noticed including low-setting ear, flat nasal bridge with a broad nasal tip, and downturned mouth. The Gesell developmental diagnosis scale was used to assess the proband’s development quotient (DQ): adaptability DQ 46, gross motor DQ 64, fine motor DQ 47, sociality DQ 51, and language DQ 60. The result of the S-M social living ability test score was 7. Positive results were found in the Clancy Behavior Scale (CBS) and Modified Checklist for Autism in Toddlers (M-CHAT). The combined communication and social interaction score was 17 according to the Autism Diagnostic Observation Schedule (ADOS), which exceeded ADOS cutoff scores for autism spectrum disorder (ASD).
He exhibited poor communication and social interactions in an unfamiliar environment, as well as autism-like behavioral traits, hand-flapping movement, echolalia, and repetitive language. In addition, he also presented irritability, mood lability, and hyperactive behavior in the behavior observation room. Global developmental delay and ASD were considered by clinical professionals in accordance with the DSM-5 and clinical manifestations.
The proband showed elevated homo-vanillic acid and 2,3-dihydroxy-2-methylbutyric acid levels in urine. Other biochemical parameters were within the normal range, including the serum levels of neuron-specific enolase, lactic acid, cysteine, very long-chain fatty acids, amino acids, blood lipids, cholesterol, and hormones including TSH, FT4, LH, and FSH. No significant abnormalities were detected via EEG and head MRI. Congenital hyperopia in the right eye was found during the eye examination. There was no abnormal finding in hearing examination, echocardiography, abdominal ultrasound, and gastrointestinal ultrasound.
Furthermore, the proband’s father presented temper tantrums and mood lability, which affected his work and family to some extent. He reported marginal learning difficulty in his childhood but could not remember the developmental history in detail. He also presented distinctive facial features, prominent nasal bridge, and a broad nasal tip. The mother of the proband showed normal physical growth and mental development. No psychiatric or physical disorders were reported in maternal and paternal families.
Cytogenetics showed a normal male karyotype (46, XY), testing for Fragile X, and Leigh syndrome and other mitochondrial diseases were negative; microarray analysis detected no clinically significant imbalance. We performed whole-exome sequencing and the copy number variation test. A novel frameshift variant (c.49dupG, p. E17Gfs*10) was identified in the TLK2 gene of the proband, which may cause loss of gene function, and his father was also found to carry this de novo variant (Figure 1). However, the grandparents and mother of this proband did not have the variant (Figure 1). According to ACMG guidelines, the variation is preliminarily determined as likely pathogenic variation (PVS1 + PM2) (Richards et al., 2015). This mutation had not been reported in the literature and ClinVar database. (Figure 2).
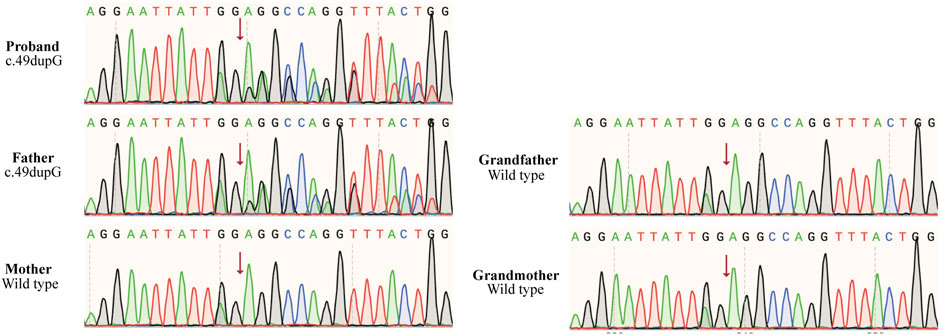
Figure 1. Sanger sequencing chromatograms of mutation c.49dupG (p.E17Gfs*10) in TLK2 gene, and the position is indicated by an arrow. The proband and his father were heterozygous of the mutated gene, while his mother and grandparents were normal.
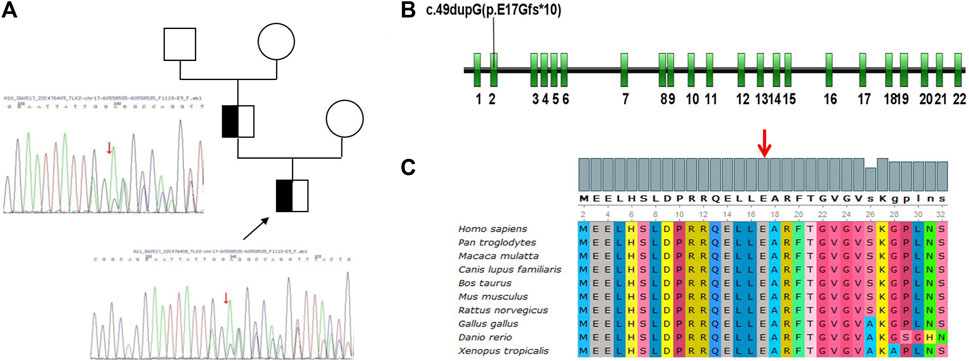
Figure 2. (A) Pedigrees of the two individuals issued from the Chinese families. The black arrow indicates the proband. The affected individuals with the homozygous TLK2 variant were indicated with a half-black square. Empty circles or squares indicated other non-carriers. (B) Exon models of TLK2 gene showing the location of the variant (c.49dupG, p. E17Gfs*10) in Exon 2. (C) Multi-species conservation alignment of TLK2 homologs. The red arrow indicates the site of the variant.
Discussion and conclusion
We reported a Chinese family with a novel heterozygous variant (c.49dupG, p. E17Gfs*10) in TLK2 gene. The proband, a 2-year-old boy, inherited this TLK2 variant from his father who carried the de novo TLK2 variant. These two affected individuals both presented prominent behavioral problems like temper tantrums, mood lability, and aggressiveness. Similar distinctive facial features, including an abnormal nasal bridge, broad nasal tip, and thin vermilion of the upper lip, were observed in them. The proband also displayed global developmental delay and autism-like symptoms, consistent with TLK2-related neurodevelopmental disorder in the available clinical literature (Woods et al., 2022). In addition, we observed mild elevated homo-vanillic acid (HVA) and 2,3-dihydroxy-2-methylbutyric acid levels in the urine of the proband. A high HVA level in urine, first reported in the affected individual with the TLK2 variant, may relate to the prominent behavioral problems in the proband.
This novel TLK2 variant (c.49dupG, p. E17Gfs*10) was predicted to result in nonsense-mediated mRNA decay, consistent with a loss of function and haploinsufficiency (Lambert et al., 2020). It was a novel variant that had not been reported before (Figure 3). It has been estimated that approximately 30% of inherited human diseases are due to the presence of premature termination codons or frameshifts that induce nonsense codons in mRNAs (Popp and Maquat, 2016). In the majority of reported cases, the disease is likely due to TLK2 haploinsufficiency as most of cases are heterozygous for loss-of-function alleles, which is in line with the strong constraint against loss-of-function variants in the gene (pLI = 1, GnomAD database) (Pavinato et al., 2022). The site was highly conserved in different species, which supported that the frameshift mutation within the N-terminal domain was a candidate for disease causation. TLK2 polypeptide is a predominantly nuclear protein composed of an N-terminal region harboring a nuclear localization signal, a middle region of helices predicted to contain three coiled coils (CC), and a C-terminal kinase domain (Mortuza et al., 2018). The N-terminal domain of TLK2 protein contains a nuclear localization signal, and mutants lacking the first 160 amino acids resulted in failing to localize to the nucleus (Mortuza et al., 2018). TLK2 activation requires dimerization through an N-terminal coiled coil motif, suggesting that inactive mutants could have a dominant negative effect (Mortuza et al., 2018). Thus, the predominant pathogenic mechanism of TLK2 mutations appears to be a reduction in its overall activity. Moreover, TLK2 variants were indicated to alter proximal interactions with many genes encoding proteins involved in chromatin remodeling that are associated with neurodevelopmental disorders (Pavinato et al., 2022).
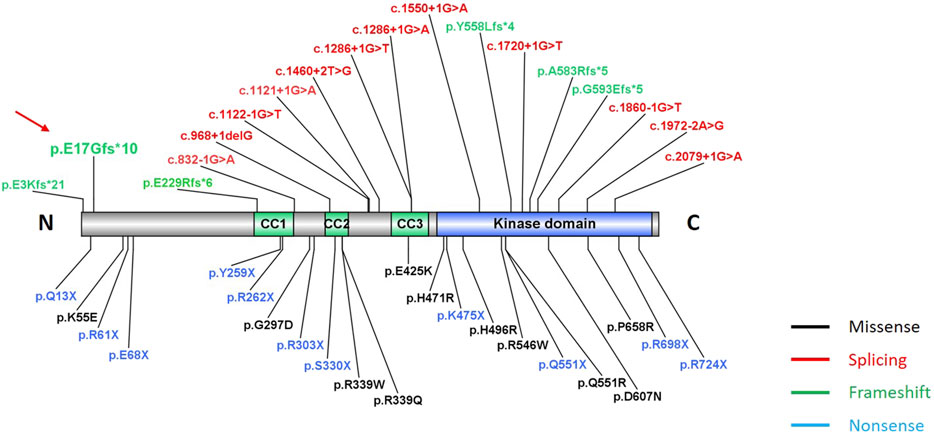
Figure 3. Location of pathogenic variants in TLK2 gene. Totally, 40 variants in our study and previous literature studies are showed here, including missense (black), splicing (red), frameshift (green), and nonsense (blue) variants. The red arrow indicates the novel frameshift variant in our study. The three green functional domains are coiled-coil domains (CC1, CC2, and CC3), and the blue domain is the kinase domain.
In addition, we identified, for the first time, HVA and 2,3-dihydroxy-2-methylbutyric acid levels were increased in the urine of the proband with TLK2 variants. It was not clear whether the high HVA and 2,3-dihydroxy-2-methylbutyric acid levels were involved in TLK2-related neurodevelopmental disorder. This might be one of causes for the severe behavioral disorder in the proband. HVA is the last metabolite of the neurotransmitter dopamine, and 2,3-dihydroxy-3-methylpentanoic acid is an intermediate metabolite of isoleucine in the human body (Gatarek and Kaluzna-Czaplinska, 2022). Isoleucine, a branched-chain amino acid, is an essential amino acid involved in biological functions of brain development and recently linked with autism (Anand et al., 2021). In addition, the elevated HVA level in urine may be resulted from the reduced dopamine β-hydroxylase enzyme activity, which is a very important enzyme in catecholamine metabolism and converts dopamine to norepinephrine in noradrenergic neurons and adrenergic neurons (Gevi et al., 2020; Gatarek and Kaluzna-Czaplinska, 2022). High levels of HVA indicate that oxidative stress caused by superoxide free radicals affects the sympathetic nervous system (Shaw, 2010; Fu et al., 2020). In the literature, a relationship between the severity of ASD symptoms and HVA levels has been observed. High HVA levels in the urine of children with ASD suggest some dopaminergic system disturbance, causing mood disorders, disorders of social relationships, aggression, and also repeated behaviors (Kaluzna-Czaplinska et al., 2010).
In summary, we described a Chinese family with a novel heterozygous TLK2 variant, leading to behavior disorders, global developmental delay, and typical facial dysmorphism. Our finding expanded the genotype of TLK2 variation in neurodevelopmental disorders and also reinforced the focus on the behavior problems in individuals with TLK2 variants. Extensive further studies are needed to explore the pathogenesis of variants in TLK2 gene.
Data availability statement
The data presented in the study are deposited in the China National Genebank (CNGB, https://db.cngb.org/cnsa/), accession number CNP0006065.
Ethics statement
The studies involving humans were approved by the ethics committees of Beijing Children’s Hospital (Beijing) (Approval number: [2021]-E-234-Y). The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants’ legal guardians/next of kin. Written informed consent was obtained from the participant/patient(s) for the publication of this case report.
Author contributions
HH: writing–original draft, writing–review and editing, conceptualization, data curation, and investigation. YQ: investigation and writing–review and editing. CY: formal analysis and writing–review and editing. SL: data curation and writing–review and editing.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Acknowledgments
The authors would like to thank the patient and his family for contributing information for this study.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
TLK2, Tousled-like kinase 2; HVA, homo-vanillic acid; NDDs, neurodevelopmental disorders; DQ, development quotient; TSH, thyroid-stimulating hormone; FT4, free thyroxine; LH, luteinizing hormone; FSH: follicle-stimulating hormone; EEG, electroencephalogram; MRI, magnetic resonance imaging; CBS, Clancy Behavior Scale; M-CHAT, Modified Checklist for Autism in Toddlers; ASD, autism spectrum disorder; CC, coiled coil.
References
Anand, N. S., Ji, Y., Wang, G., Hong, X., van der Rijn, M., Riley, A., et al. (2021). Maternal and cord plasma branched-chain amino acids and child risk of attention-deficit hyperactivity disorder: a prospective birth cohort study. J. Child. Psychol. Psychiatry 62 (7), 868–875. doi:10.1111/jcpp.13332
Fu, B., Chen, H., Yan, Z., Zhang, Z., Chen, J., Liu, T., et al. (2020). A simple ultrasensitive electrochemical sensor for simultaneous determination of homovanillic acid and vanillylmandelic acid in human urine based on MWCNTs-Pt nanoparticles as peroxidase mimics. J. Electroanal. Chem. 866, 114165. doi:10.1016/j.jelechem.2020.114165
Gatarek, P., and Kaluzna-Czaplinska, J. (2022). Effect of supplementation on levels of homovanillic and vanillylmandelic acids in children with autism spectrum disorders. Metabolites 12 (5), 423. doi:10.3390/metabo12050423
Gevi, F., Belardo, A., and Zolla, L. (2020). A metabolomics approach to investigate urine levels of neurotransmitters and related metabolites in autistic children. Biochim. Biophys. Acta Mol. Basis Dis. 1866 (10), 165859. doi:10.1016/j.bbadis.2020.165859
Kaluzna-Czaplinska, J., Socha, E., and Rynkowski, J. (2010). Determination of homovanillic acid and vanillylmandelic acid in urine of autistic children by gas chromatography/mass spectrometry. Med. Sci. Monit. 16 (9), CR445–50.
Lambert, J. M., Ashi, M. O., Srour, N., Delpy, L., and Sauliere, J. (2020). Mechanisms and regulation of nonsense-mediated mRNA decay and nonsense-associated altered splicing in lymphocytes. Int. J. Mol. Sci. 21 (4), 1335. doi:10.3390/ijms21041335
Lelieveld, S. H., Reijnders, M. R., Pfundt, R., Yntema, H. G., Kamsteeg, E. J., de Vries, P., et al. (2016). Meta-analysis of 2,104 trios provides support for 10 new genes for intellectual disability. Nat. Neurosci. 19 (9), 1194–1196. doi:10.1038/nn.4352
Mortuza, G. B., Hermida, D., Pedersen, A. K., Segura-Bayona, S., Lopez-Mendez, B., Redondo, P., et al. (2018). Molecular basis of tousled-like kinase 2 activation. Nat. Commun. 9 (1), 2535. doi:10.1038/s41467-018-04941-y
Pavinato, L., Villamor-Paya, M., Sanchiz-Calvo, M., Andreoli, C., Gay, M., Vilaseca, M., et al. (2022). Functional analysis of TLK2 variants and their proximal interactomes implicates impaired kinase activity and chromatin maintenance defects in their pathogenesis. J. Med. Genet. 59 (2), 170–179. doi:10.1136/jmedgenet-2020-107281
Popp, M. W., and Maquat, L. E. (2016). Leveraging rules of nonsense-mediated mRNA decay for genome engineering and personalized medicine. Cell. 165 (6), 1319–1322. doi:10.1016/j.cell.2016.05.053
Reijnders, M. R. F., Miller, K. A., Alvi, M., Goos, J. A. C., Lees, M. M., de Burca, A., et al. (2018). De novo and inherited loss-of-function variants in TLK2: clinical and genotype-phenotype evaluation of a distinct neurodevelopmental disorder. Am. J. Hum. Genet. 102 (6), 1195–1203. doi:10.1016/j.ajhg.2018.04.014
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genet. Med. 17 (5), 405–424. doi:10.1038/gim.2015.30
Shaw, W. (2010). Increased urinary excretion of a 3-(3-hydroxyphenyl)-3-hydroxypropionic acid (HPHPA), an abnormal phenylalanine metabolite of Clostridia spp. in the gastrointestinal tract, in urine samples from patients with autism and schizophrenia. Nutr. Neurosci. 13 (3), 135–143. doi:10.1179/147683010X12611460763968
Topf, A., Oktay, Y., Balaraju, S., Yilmaz, E., Sonmezler, E., Yis, U., et al. (2020). Severe neurodevelopmental disease caused by a homozygous TLK2 variant. Eur. J. Hum. Genet. 28 (3), 383–387. doi:10.1038/s41431-019-0519-x
Keywords: TLK2 gene, behavioral disorder, neurodevelopmental disorder, case report, heterozygous variant
Citation: Huang H, Qian Y, Yang C and Li S (2024) Case report: A novel TLK2 variant with a neuropsychiatric phenotype from a Chinese family. Front. Genet. 15:1419027. doi: 10.3389/fgene.2024.1419027
Received: 22 April 2024; Accepted: 10 July 2024;
Published: 04 September 2024.
Edited by:
Hong Ni, Children’s Hospital of Soochow University, ChinaReviewed by:
Shahid Mahmood Baig, National Institute for Biotechnology and Genetic Engineering, PakistanMohammad Abdullah Shboul, Jordan University of Science and Technology, Jordan
Copyright © 2024 Huang, Qian, Yang and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hongmei Huang, aHVhbmdob25nbWF5QGFsaXl1bi5jb20=