 Bingxuan Yu
Bingxuan Yu Jing Chen
Jing Chen Shuo Yang
Shuo Yang He Wang1,2
He Wang1,2 Yuanyuan Xiao
Yuanyuan Xiao Shanling Liu
Shanling Liu- 1Department of Medical Genetics/Prenatal Diagnostic Center, West China Second University Hospital, Sichuan University, Chengdu, China
- 2Key Laboratory of Birth Defects and Related Diseases of Women and Children (Sichuan University), Ministry of Education, Chengdu, China
Background: Developmental delay in children under 5 years old, which occurs globally with an incidence of 10%–15%, is caused by multiple factors including genetics, prenatal conditions, perinatal complications, postnatal influences, social factors, and nutritional deficiencies. Gene variants such as EFNB1, MECP2 and TRAPPC9 play a significant role in protein deformation and downregulation of nuclear factor κB (NF-κB) activity.
Methods: A 3-year-old girl, who exhibits poor gross motor skills, personal-social development, auditory language, hand-eye coordination, and visual performance, was diagnosed with global developmental delay. Trio whole exome sequencing was conducted to identify the genetic etiology of her condition. The identified genetic etiology was then validated through Sanger sequencing and quantitative polymerase chain reaction (qPCR).
Results: Genetic analysis revealed that the patient had compound heterozygous variants in the TRAPPC9 gene. These include a c.1928del frameshift variant inherited from the unaffected father and a deletion in exon 12 inherited from the unaffected mother. According to the American College of Medical Genetics (ACMG) guidelines, these variants were classified as “likely pathogenic”.
Conclusion: The study revealed that compound heterozygous TRAPPC9 gene variants cause developmental delay in a Chinese girl. These variants have been classified as having significant pathogenic effect according to the ACMG criteria, suggesting a recessive genetic pattern and highlighting the importance of prenatal testing for future offspring. Furthermore, our findings expand the genotype spectrum of the TRAPPC9 gene, and provide more comprehensive information regarding genetic counseling for children experiencing developmental delay.
1 Introduction
Development delay (DD) includes developmental disabilities, failure and degeneration. The prevalence of DD in children under 5 years old has been reported to range from 10% to 15%, while the incidence of global developmental delay (GDD) ranges from 1% to 3% (Choo et al., 2019). The causes of DD in young children are multifactorial and result from a combination of prenatal factors (such as genetic disorders, cerebral dysgenesis, vascular occlusion, etc.), perinatal factors (such as preterm birth, perinatal asphyxia, bilirubin-induced neurological dysfunction, etc.), or postnatal events (such as meningitis, hypernatremia, hyponatremia, etc.), along with social, nutritional, and other factors (Choo et al., 2019). Several clinical studies have reported gene variants, including EFNB1, MECP2, ATRX, NAA10, ANKRD11, ZNF699, TRAPPC9 and others associated with DD as significant causes of congenital growth retardation (Yan et al., 2019; Alvarez-Mora et al., 2021; Biela et al., 2022). Early intervention is essential for optimizing the developmental course of children with DD. If detected later, the opportunity for early intervention is lost, leading to adverse outcomes such as learning difficulties and behavioral problems. Therefore, it is important to refine the genetic spectrum of this disorder (Shannon and Anderson, 2008).
The gene we were concerned with, TRAFFICKING PROTEIN PARTICLE COMPLEX, SUBUNIT 9 (TRAPPC9, OMIM:611966), is associated with DD. Johannes Krämer et al. reported that all of 43 cases of TRAPPC9 variants, all showed intellectual disability (ID) and DD (Krämer et al., 2021). Other reports, including one consanguineous family in Pakistan and two consanguineous families in Iran, exhibited DD associated with TRAPPC9 variants (Yousefipour et al., 2021; Asif et al., 2022). The process by which TRAPPC9 genetic variants cause DD has not yet been elucidated, but it may be related to functional alterations in the transport protein particle complex subunit 9. This protein belongs to transporter particle II (TRAPPII) and plays an important role in Golgi vesicle tethering and Golgi transport in mammalian cells, thus influencing both cell development and individual development. At the same time, the clinical phenotype associated with TRAPPC9 variants is linked to a decrease in NF-κB activation, which is a transcription factor that plays a critical role in activating genes associated with various cellular processes, including inflammation, immune response, cell proliferation, and apoptosis (Mbimba et al., 2018; Aslanger et al., 2022). Variants in TRAPPC9 are also associated with autism spectrum disorder (ASD). A whole exome analysis of two siblings in Turkey reported that TRAPPC9 variants are not only linked to ID, but also to ASD and repetitive physical behaviors, such as clapping behavior (Bolat et al., 2022). The same procedure was applied to two Thai siblings with ASD and ID, and TRAPPC9 heterozygous variants were reported through whole exome sequencing (Hnoonual et al., 2019). The explanation of the mechanism mainly focuses on the deformation of related proteins and the downregulation of NF-κB activity (Wilton et al., 2020).
We reported a patient presented with GDD and other clinical symptoms Through WES, we identified novel compound heterozygous variants in the TRAPPC9 gene, consisting of a heterozygous deletion of exon 12 and a frameshift variant (c.1928del, p.Y643Sfs*3). Our findings enhance the understanding of disease-causing variants in TRAPPC9 and provides valuable information for molecular diagnostics.
2 Materials and methods
2.1 Patients and clinical data
The present study focused on a 3-year-old female with GDD and other clinical symptoms from a Chinese family with no recorded family history. The family pedigree is illustrated in Figure 1. The child was admitted to West China Second Hospital at the age of three for medical evaluation and treatment. The findings of the brain magnetic resonance imaging (MRI) indicated abnormalities. The detailed clinical information of proband can be seen in Table 1. To investigate the genetic basis of the condition, Trio-WES was employed to analyze the family’s genetic profile. Subsequently, Sanger sequencing was used for validating variants initially detected through Trio-WES.
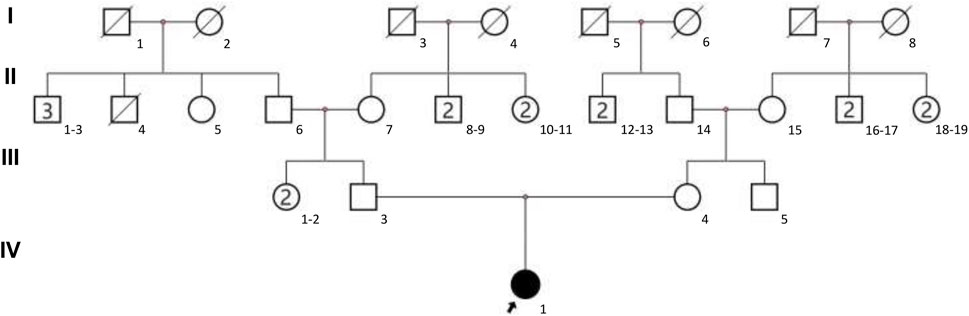
Figure 1. The family pedigree was depicted, with the proband designated by an arrow.
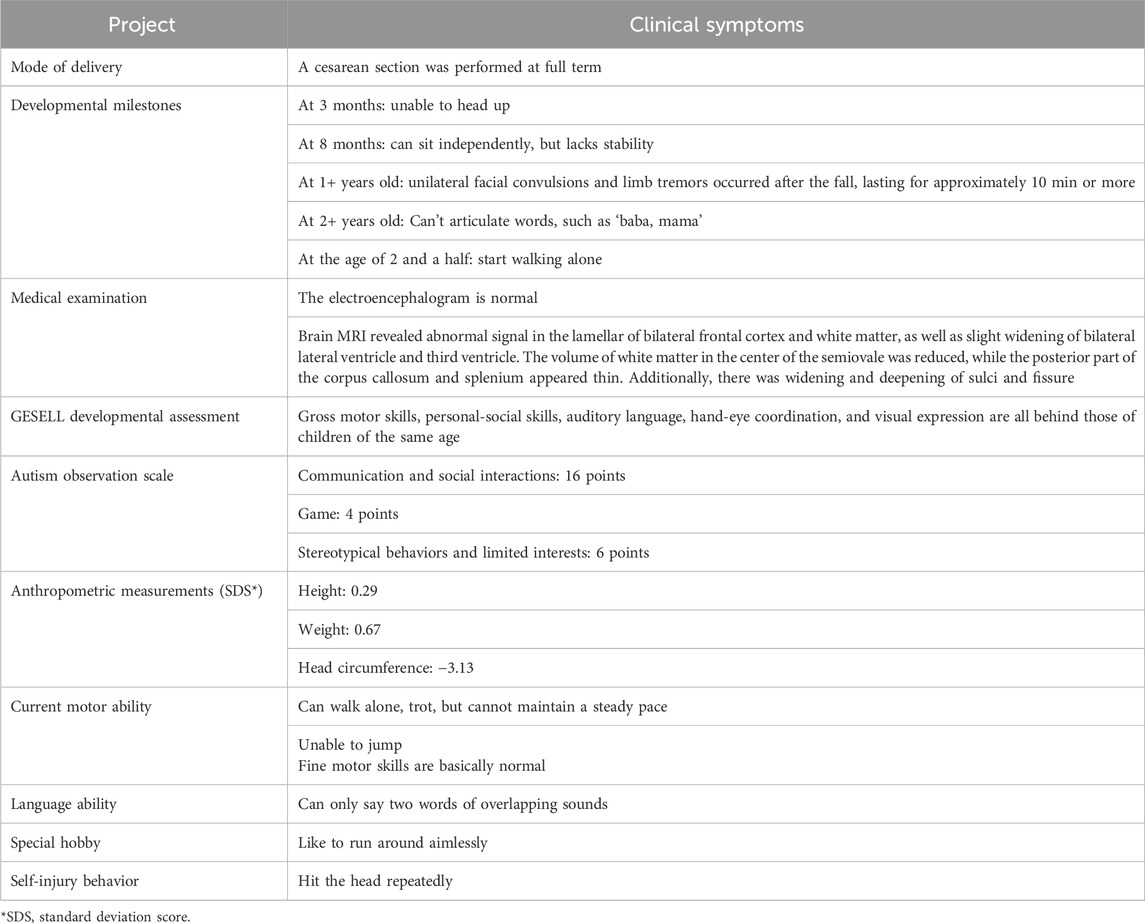
Table 1. The proband’s primary clinical symptoms.
The written informed consent of all subjects has been obtained and approved by the Medical Ethics Committee of West China Second Hospital of Sichuan University.
2.2 DNA extraction
The genomic DNA of this family member was extracted from the blood, according to the manufacturer’s instructions (QIAamp DNA Blood Minikit). The proband and her parents’ genomic DNA was tested for WES, with sequences captured using Nano WES Human Exome V1 (Berry Genomics).
2.3 Whole exome sequencing
To identify variants in the proband, this study utilized the Nano WES Human Exome V1 for sequence capture and library formation. The enriched libraries were sequenced on the Illumina NovaSeq6000 platform, yielding 150 bp reads. The sequencing reads were then aligned to the hg38/GRCh38 reference genome using the Burrows-Wheeler Aligner (BWA) software (v0.7.17). Post-alignment, local recalibration of reads was conducted using the GATK Indel Realigner, and base recalibration was performed using the GATK Base Recalibrator (broadinstitute.org/). Single Nucleotide Variants (SNVs) and small indels were identified using the GATK Unified Genotyper. Variants with minor allele frequencies less than 0.05 were filtered using databases such as gnomAD (V3.1) (http://gnomad.broadinstitute.org/), the 1000 Genomes Project (phase 3) (http://browser.1000genomes.org). These selected variants were then subjected to pathogenicity prediction using platforms such as PolyPhen2 (http://genetics.bwh.harvard.edu/pph2), SIFT (http://sift.jcvi.org), and CADD (V1.6) (https://cadd.gs.washington.edu). Furthermore, to pinpoint causative variants, additional resources were consulted, including the National Institutes of Health, the Online Mendelian Inheritance in Man (OMIM,20230828) database (http://www.omim.org), the Human Gene Mutation Database (http://www.hgmd.org), and the ClinVar (20230819) database (http://www.ncbi.nlm.nih.gov/clinvar). The SNVs obtained in this way were divided into five categories, including pathogenic, likely pathogenic, uncertain significance, likely benign and benign, according to theACMG guidelines (Richards et al., 2015).
2.4 Sanger sequencing
To validate candidate variants, we sequenced PCR products of TRAPPC9 c.1928del using an ABI 3500 genetic analyzer (Thermo Fisher Scientific) after amplifying each independent gDNA sample (both proband and her parents) with specific primer pairs 303 bp in length through polymerase chain reaction (PCR) amplification of each independent (Forward sequence: GAGCAGGAAGCATGAAGG; Reversed sequence: CTGTCTCCGAAGTTCTCTG).
2.5 Quantitative polymerase chain reaction (qPCR)
To validate the candidate variants selected by WES, qPCR was performed on gDNA from the proband and her parents using a SYBR Green qPCR master mix system and an Applied Biosystems 7500 real time PCR system. The copy number of TRAPPC9 in each sample’s exon 12 was assessed using specific primer pairs through the 2 [-Delta Delta C (T)] analysis method (Livak and Schmittgen, 2001). Experiments were repeated three times for each sample. The forward sequence was TAGATTTCCAGTGGGTTCAA, and the reversed sequence was TGTGCCTCAGACATTAAGTA.
3 Results
3.1 Compound heterozygous variants in TRAPPC9 gene were found in a patients with developmental delay
The WES revealed a frameshift variant in TRAPPC9 (NM 031466.8, c.1928del, p.Y643Sfs*3) from the paternal allele and a heterozygous deletion of TRAPPC9 exon 12 (NM 031466.8) from the maternal allele. The frameshift variant was further validated by Sanger sequencing (Figure 2A) and the heterozygous deletion of exon 12 was confirmed by qPCR (Figure 3B). These results were consistent with WES, confirming the presence of this novel heterozygous compound variants.
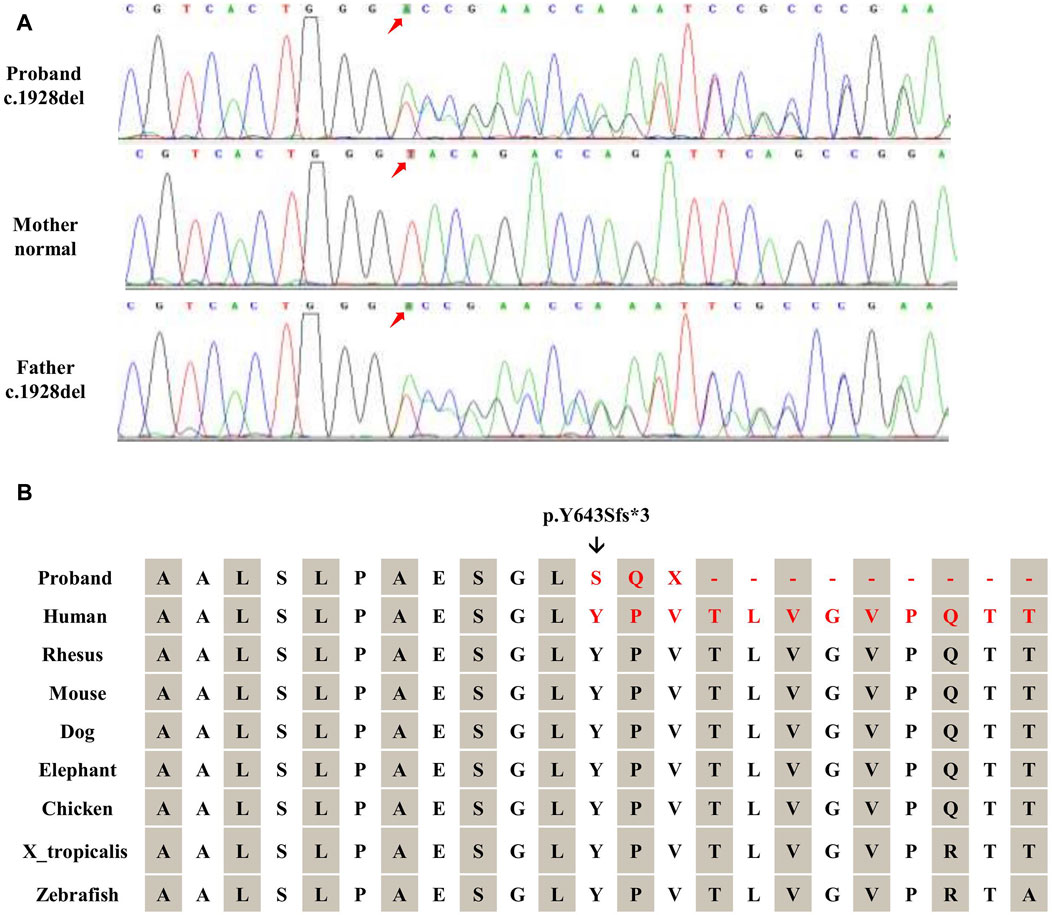
Figure 2. Validation of TRAPPC9 SNVs. (A) Sanger sequencing was utilized to confirm the variant (c.1928del) within the family. (B) The frameshift mutation (c.1928del) in TRAPPC9 leads to the substitution of glycine with a highly conserved arginine, resulting in a frameshift and premature termination (p.Y643Sfs*3).
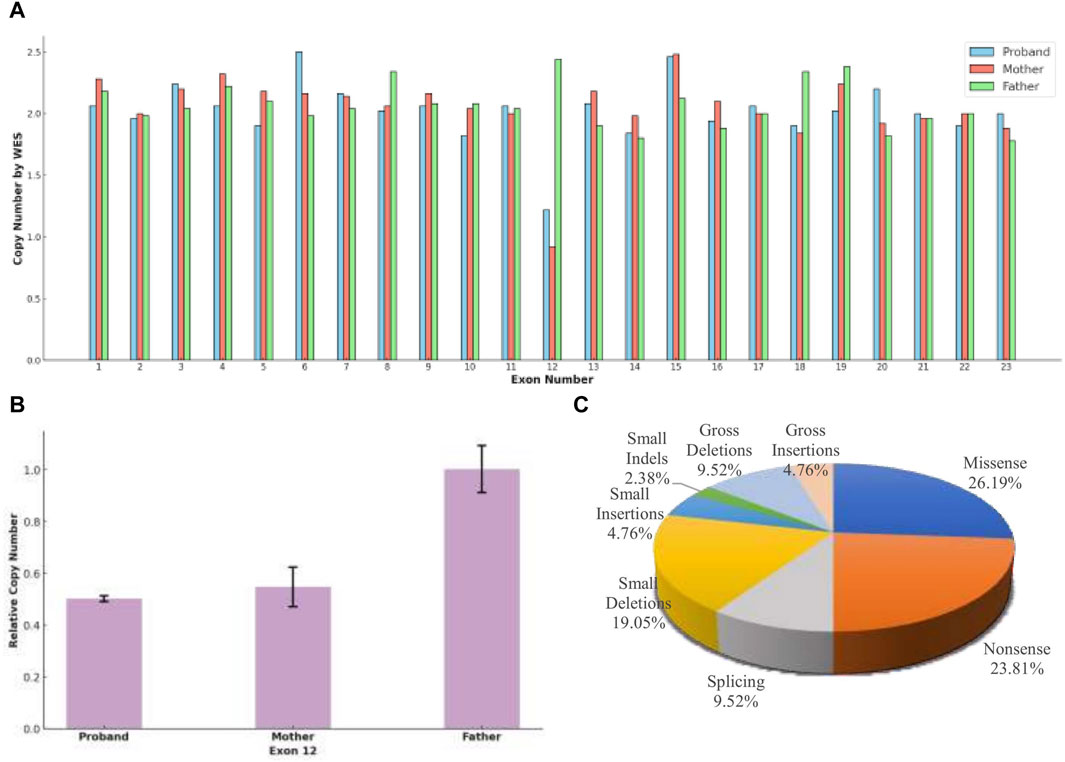
Figure 3. (A) Whole Exome Sequencing (WES) revealed that, except for exon 12, the copy number for all exons in the three subjects was approximately 2, indicating a normal range. Both the proband and the mother exhibited half of the normal copy number for exon 12, while the father’s exon 12 copy number was within the normal range. (B) A heterozygous deletion of TRAPPC9 exon 12 was confirmed using qPCR. The analysis showed that the proband and the unaffected mother had half of the copy number for exon 12 compared to that of the unaffected father. (C) The distribution of disease-causing variation types in TRAPPC9, as cataloged in the Human Gene Mutation Database (HGMD® Professional 2023.4), reflects the variety of genetic alterations associated with the gene.
The proband inherited a frameshift mutation derived from the paternal allele, resulting in a premature stop codon at position 645. This mutation was caused by the substitution of tyrosine with serine at the 643rd amino acid residue encoded by TRAPPC9 exon 13 and the substitution of proline with glutamine at the 644th amino acid residue. Additionally, Figure 2B shows that multiple sequence alignment of the TRAPPC9 gene from different species confirms the evolutionary conservation of amino acid residues at positions 643, 644, and 645 in this gene’s encoded protein. The proband also inherited a heterozygous deletion of exon 12 from their maternal allele, which was detected through WES analysis. qPCR experiments revealed that both the patient and mother had only half of normal copy number for TRAPPC9 exon 12. However, this change was not found in the father (Figures 3A,B).
In summary, the proband’s clinical phenotypes were due to the compound heterozygous variants in the TRAPPC9 gene, which included a maternally inherited heterozygous deletion of exon 12 in TRAPPC9 and a paternally inherited frameshift variant c.1928del in TRAPPC9. According to the ACMG criteria, both compound heterozygous variants were classified as “likely pathogenic”.
4 Discussion
The TRAPPC9 gene, also known as NIBP (NIK-and-IKK2-binding protein) or MRT13 (autosomal recessive mental retardation 13), plays a pivotal role in neurodevelopment (Abbasi et al., 2017; Hu et al., 2023). Animal studies have demonstrated that the defects in TRAPPC9 leads to increased astrocyte proliferation in the hippocampus and corpus callosum of mice, which adversely affects the function of Rab11, a key protein involved in intracellular transport (Barrowman et al., 2010; Ke et al., 2020). TRAPPC9 encodes for a protein believed to be involved in NF-kappa-B signaling (Hu et al., 2005). It acts as a subunit of the Transporter Particle Complex 9, which is a part of the TRAPPII complex. This complex is involved in various cellular processes, including protein secretion, autophagy, cytoplasmic division, and cilia formation (Barrowman et al., 2010). Clinically, variants in the TRAPPC9 gene are closely linked to autosomal recessive intellectual disability (ARID) syndrome. This association is likely due to the gene’s role in regulating critical signaling pathways for brain development, particularly the NF-κB signaling pathway. Genetic studies have linked alterations in TRAPPC9 to changes in brain structure and cognitive function, highlighting its significance in the study of neurodevelopmental biology and genetic disorders that result in intellectual disability (Bodnar et al., 2020). The involvement of this gene in such a wide range of critical developmental processes makes it a significant focus in neurogenetic research.
The TRAPPC9 gene has been reported to have at least 42 variants classified as disease-causing mutations (DM), which encompass 11 missense variants, 10 nonsense variants, four splicing variants, eight small deletions, two small insertions, one small indels, four gross deletions, and two gross insertions in the HGMD database (Figure 3C). Among these variants, there are a total of 28 associated with ID, highlighting the significant role of the gene in cognitive development, and three variants have additionally been linked to DD. Boonsawat’s team reported on a patient with DD who had a TRAPPC9 variant (c.3214C>T, p. Arg1072*), which led to altered neuronal NF-kappa-B signaling and was associated with moderate to severe GDD, mild cortical atrophy, and delayed myelination (Boonsawat et al., 2019). In another study, Masih’s team identified a patient with moderate DD with a TRAPPC9 variant (c. 2458_2459delCT), and Koifman’s research group used chromosome microarray analysis and PCR technology to identify a 141.46 kb homozygous deletion in TRAPPC9, resulting in severe DD and agenesis of the corpus callosum (Koifman et al., 2010; Masih et al., 2022). The single exon deletion of TRAPPC9 exon 12 in our case was initially identified through WES, which differs from the method employed by Koifman et al. WES tends to have high false positive rates in detecting single exon deletion due to uneven capture and less coverage of non-coding regions (Yao et al., 2017; O’Fallon et al., 2022). However, our result was confirmed by qPCR. The detection of this variant in the initial WES analysis may be attributed to the careful selection of control samples, enabling a comparison between control and test samples to identify differences and mitigate systematic errors. This emphasizes the importance of not overlooking copy number variants reported by WES in genetic analysis.
Our findings significantly contribute to the clinical management of the proband. One key insight is the potential link between the absence of TRAPPC9 protein and an imbalance in dopamine D1 and D2 neurons, which might lead to impaired learning and memory (Ke et al., 2020). This suggests that drugs targeting D1 and D2 neurons could potentially alleviate these symptoms. Such a therapeutic approach could be particularly valuable in managing the neurological symptoms associated with TRAPPC9 gene deficiency. Another innovative aspect involves using protein-protein interaction residues to investigate the binding properties of residues in both wild-type and mutant TRAPPC9 proteins (Khattak and Mir, 2014). This approach has the potential to the development of drug therapies tailored to specific TRAPPC9 variants, offering new avenues for therapeutic exploration in affected individuals. In our study, WES identified compound heterozygous variants in the TRAPPC9 gene, including a single exon heterozygous deletion in exon 12 and a frameshift variant in exon 13, pinpointing the etiology of the global developmental of the proband. According to the ACMG criteria, both variants detected in TRAPPC9 were classified as “likely pathogenic” and that parents who carries TRAPPC9 gene variant, but exhibit no developmental delays, suggests a recessive genetic mode for the proband. This observation emphasizes the necessity of prenatal examination for subsequent offspring.
5 Conclusion
First, the observation of proband’s parents, each carrying one likely pathogenic variant of the TRAPPC9 gene, exhibits no clinical symptoms, while the proband with compound heterozygous variants shows GDD, indicates a recessive inheritance pattern. The provision of this information is essential for genetic counseling and indicating that if the parents intend to have additional children, prenatal diagnosis should be offered to them. Additionally, our findings expand the genotypic spectrum of the TRAPPC9 gene. Our findings provided valuable insights for the diagnosis and treatment of related disorders. The comprehensive understanding of the mutation spectrum in the gene can enhance the precision of genetic testing and inform the development of more effective therapeutic strategies for conditions associated with TRAPPC9 variants.
Data availability statement
The variation data reported in this paper have been deposited in the Genome Variation Map (GVM) (https://ngdc.cncb.ac.cn/gvm/) in National Genomies Data Center, Beijing Institute of Genomics, Chinese Academy of Sciences and China National Center for Bioinformation, under accession number GVM000813.
Ethics statement
The studies involving humans were approved by Medical Ethics Committee of West China Second Hospital of Sichuan University. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation was not required from the participants or the participants’ legal guardians/next of kin in accordance with the national legislation and institutional requirements. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author contributions
BY: Visualization, Writing–original draft. JC: Writing–original draft. SY: Formal Analysis, Resources, Writing–review and editing. HW: Data curation, Resources, Writing–review and editing. YX: Methodology, Project administration, Writing–review and editing. SL: Methodology, Project administration, Writing–review and editing.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This study was supported by the National Key Research and Development Program of China (2022YFC2703300 and 2022YFC2703302).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
DD, developmental delay; GDD, global developmental delay; WES, whole exome sequencing; qPCR, quantitative polymerase chain reaction; ACMG, American College of Medical Genetics and Genomics; TRAPPC9, TRAFFICKING PROTEIN PARTICLE COMPLEX, SUBUNIT 9; ID, intellectual disability; NF-κB, nuclear factor κB; ASD, autism spectrum disorder; NGS, next-generation sequencing; OMIM, Online Mendelian Inheritance in Man; SNVs, Single Nucleotide Variants; NIBP, NIK-and-IKK2-binding protein; ARID, autosomal recessive intellectual disability; MRT13, autosomal recessive mental retardation 13.
References
Abbasi, A. A., Blaesius, K., Hu, H., Latif, Z., Picker-Minh, S., Khan, M. N., et al. (2017). Identification of a novel homozygous TRAPPC9 gene mutation causing non-syndromic intellectual disability, speech disorder, and secondary microcephaly. Am. J. Med. Genet. B 174 (8), 839–845. doi:10.1002/ajmg.b.32602
Alvarez-Mora, M. I., Corominas, J., Gilissen, C., Sanchez, A., Madrigal, I., and Rodriguez-Revenga, L. (2021). Novel compound heterozygous mutation in TRAPPC9 gene: the relevance of whole genome sequencing. Genes-Basel 12 (4), 557. doi:10.3390/genes12040557
Asif, M., Anayat, M., Tariq, F., Noureen, T., Din, G., Becker, C., et al. (2022). Whole-exome sequencing of Pakistani consanguineous families identified pathogenic variants in genes of intellectual disability. Genes-Basel 14 (1), 48. doi:10.3390/genes14010048
Aslanger, A. D., Goncu, B., Duzenli, O. F., Yucesan, E., Sengenc, E., and Yesil, G. (2022). Biallelic loss of TRAPPC9 function links vesicle trafficking pathway to autosomal recessive intellectual disability. J. Hum. Genet. 67 (5), 279–284. doi:10.1038/s10038-021-01007-8
Barrowman, J., Bhandari, D., Reinisch, K., and Ferro-Novick, S. (2010). TRAPP complexes in membrane traffic: convergence through a common Rab. Nat. Rev. Mol. Cell Biol. 11 (11), 759–763. doi:10.1038/nrm2999
Biela, M., Rydzanicz, M., Jankowska, A., Szlagatys-Sidorkiewicz, A., Rozensztrauch, A., Ploski, R., et al. (2022). Further delineation of developmental delay with gastrointestinal, cardiovascular, genitourinary, and skeletal abnormalities caused by ZNF699 gene mutation. Genes-Basel 13 (2), 168. doi:10.3390/genes13020168
Bodnar, B., DeGruttola, A., Zhu, Y., Lin, Y., Zhang, Y., Mo, X., et al. (2020). Emerging role of NIK/IKK2-binding protein (NIBP)/trafficking protein particle complex 9 (TRAPPC9) in nervous system diseases. Transl. Res. 224, 55–70. doi:10.1016/j.trsl.2020.05.001
Bolat, H., Unsel-Bolat, G., Derin, H., Sen, A., and Ceylaner, S. (2022). Distinct autism spectrum disorder phenotype and hand-flapping stereotypes: two siblings with novel homozygous mutation in TRAPPC9 gene and literature review. Mol. Syndromol. 13 (4), 263–269. doi:10.1159/000522041
Boonsawat, P., Joset, P., Steindl, K., Oneda, B., Gogoll, L., Azzarello-Burri, S., et al. (2019). Elucidation of the phenotypic spectrum and genetic landscape in primary and secondary microcephaly. Genet. Med. 21 (9), 2043–2058. doi:10.1038/s41436-019-0464-7
Choo, Y. Y., Agarwal, P., How, C. H., and Yeleswarapu, S. P. (2019). Developmental delay: identification and management at primary care level. Singapore Med. J. 60 (3), 119–123. doi:10.11622/smedj.2019025
Hnoonual, A., Graidist, P., Kritsaneepaiboon, S., and Limprasert, P. (2019). Novel compound heterozygous mutations in the TRAPPC9 gene in two siblings with autism and intellectual disability. Front. Genet. 10, 61. doi:10.3389/fgene.2019.00061
Hu, M., Bodnar, B., Zhang, Y., Xie, F., Li, F., Li, S., et al. (2023). Defective neurite elongation and branching in Nibp/Trappc9 deficient zebrafish and mice. Int. J. Biol. Sci. 19 (10), 3226–3248. doi:10.7150/ijbs.78489
Hu, W. H., Pendergast, J. S., Mo, X. M., Brambilla, R., Bracchi-Ricard, V., Li, F., et al. (2005). NIBP, a novel NIK and IKK(beta)-binding protein that enhances NF-(kappa)B activation. J. Biol. Chem. 280 (32), 29233–29241. doi:10.1074/jbc.M501670200
Ke, Y., Weng, M., Chhetri, G., Usman, M., Li, Y., Yu, Q., et al. (2020). Trappc9 deficiency in mice impairs learning and memory by causing imbalance of dopamine D1 and D2 neurons. Sci. Adv. 6 (47), 6. doi:10.1126/sciadv.abb7781
Khattak, N. A., and Mir, A. (2014). Computational analysis of TRAPPC9: candidate gene for autosomal recessive non-syndromic mental retardation. CNS Neurol. Disord. Drug. Targets 13 (4), 699–711. doi:10.2174/18715273113129990105
Koifman, A., Feigenbaum, A., Bi, W., Shaffer, L. G., Rosenfeld, J., Blaser, S., et al. (2010). A homozygous deletion of 8q24.3 including the NIBP gene associated with severe developmental delay, dysgenesis of the corpus callosum, and dysmorphic facial features. Am J Med Genet A. 152A (5), 1268–1272. doi:10.1002/ajmg.a.33319
Krämer, J., Beer, M., Bode, H., and Winter, B. (2021). Two novel compound heterozygous mutations in the TRAPPC9 gene reveal a connection of non-syndromic intellectual disability and autism spectrum disorder. Front. Genet. 11, 11. doi:10.3389/fgene.2020.00972
Livak, K. J., and Schmittgen, T. D. (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25 (4), 402–408. doi:10.1006/meth.2001.1262
Masih, S., Moirangthem, A., Shambhavi, A., Rai, A., Mandal, K., Saxena, D., et al. (2022). Deciphering the molecular landscape of microcephaly in 87 Indian families by exome sequencing. Eur. J. Med. Genet. 65 (6), 104520. doi:10.1016/j.ejmg.2022.104520
Mbimba, T., Hussein, N. J., Najeed, A., and Safadi, F. F. (2018). TRAPPC9: novel insights into its trafficking and signaling pathways in health and disease (Review). Int. J. Mol. Med. 42 (6), 2991–2997. doi:10.3892/ijmm.2018.3889
O’Fallon, B., Durtschi, J., Kellogg, A., Lewis, T., Close, D., and Best, H. (2022). Algorithmic improvements for discovery of germline copy number variants in next-generation sequencing data. BMC Bioinformatics 23 (1), 285. doi:10.1186/s12859-022-04820-w
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17 (5), 405–424. doi:10.1038/gim.2015.30
Shannon, P., and Anderson, P. R. (2008). Developmental screening in community health care centers and pediatric practices: an evaluation of the Baby Steps Program. Intellect. Dev. Disab. 46 (4), 281–289. doi:10.1352/1934-9556(2008)46[281:DSICHC]2.0.CO;2
Wilton, K. M., Gunderson, L. B., Hasadsri, L., Wood, C. P., and Schimmenti, L. A. (2020). Profound intellectual disability caused by homozygous TRAPPC9 pathogenic variant in a man from Malta. Mol. Genet. Genomic Med. 8 (5), e1211. doi:10.1002/mgg3.1211
Yan, H., Shi, Z., Wu, Y., Xiao, J., Gu, Q., Yang, Y., et al. (2019). Targeted next generation sequencing in 112 Chinese patients with intellectual disability/developmental delay: novel mutations and candidate gene. BMC Med. Genet. 20 (1), 80. doi:10.1186/s12881-019-0794-y
Yao, R., Zhang, C., Yu, T., Li, N., Hu, X., Wang, X., et al. (2017). Evaluation of three read-depth based CNV detection tools using whole-exome sequencing data. Mol. Cytogenet. 10, 30. doi:10.1186/s13039-017-0333-5
Keywords: developmental delay, TRAPPC9, variants, whole exome sequencing, frame shift
Citation: Yu B, Chen J, Yang S, Wang H, Xiao Y and Liu S (2024) Case Report: Whole exome sequencing identifies compound heterozygous variants in the TRAPPC9 gene in a child with developmental delay. Front. Genet. 15:1415194. doi: 10.3389/fgene.2024.1415194
Received: 10 April 2024; Accepted: 29 July 2024;
Published: 09 August 2024.
Edited by:
Katsuhiko Tabuchi, Shinshu University, JapanReviewed by:
Shahid Mahmood Baig, National Institute for Biotechnology and Genetic Engineering, PakistanSemra Gürsoy, Dokuz Eylül University, Türkiye
Copyright © 2024 Yu, Chen, Yang, Wang, Xiao and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yuanyuan Xiao, eXl4aWFvMjAxMEAxNjMuY29t; Shanling Liu, c3Vubnk2MzBAMTI2LmNvbQ==
†These authors have contributed equally to this work