 Aysylu Murtazina1*
Aysylu Murtazina1* Dmitrii Subbotin1
Dmitrii Subbotin1 Anna Kuchina1
Anna Kuchina1 Olga Gilvanova2Daniil Degterev2
Olga Gilvanova2Daniil Degterev2 Olga Shchagina1Tatiana Cherevatova1
Olga Shchagina1Tatiana Cherevatova1 Maria Bulakh1
Maria Bulakh1 Darya Sherstyukova1Oksana Ryzhkova1Olga Kurushina3
Darya Sherstyukova1Oksana Ryzhkova1Olga Kurushina3 Mikhail Skoblov1
Mikhail Skoblov1 Artem Borovikov1Sergey Kutsev1
Artem Borovikov1Sergey Kutsev1- 1Research Centre for Medical Genetics, Moscow, Russia
- 2Loginov Moscow Clinical Scientific Center, Moscow, Russia
- 3Department of Neurology, Neurosurgery, Medical Genetics, Volgograd State Medical University, Volgograd, Russia
Recent research has sparked a discussion on the spectrum of diseases linked to the MATR3 gene associated with amyotrophic lateral sclerosis and distal myopathy with vocal cord and pharyngeal weakness (VCPDM). To date, fewer than 50 cases of VCPDM have been reported in the literature. We aim to build upon the work of previous researchers by gathering additional information about VCPDM. In this study, we present six patients from four unrelated families affected by VCPDM. Our observations include patients exhibiting both the typical phenotype associated with MATR3-related distal myopathy and rare symptomatic manifestations of the disease. Notably, two cases presented with an asymmetric scapuloperoneal phenotype, leading in one case to an initial misdiagnosis of facioscapulohumeral muscular dystrophy.
1 Introduction
Distal myopathies are a group of hereditary primary muscle disorders that are characterized by extensive phenotypic and genetic variability. Currently, more than 25 genes associated with this nosology have been identified (Milone and Liewluck, 2019; Savarese et al., 2020). Heterozygous variants in the MATR3 gene cause amyotrophic lateral sclerosis (ALS) type 21 and distal myopathy with vocal cord and pharyngeal weakness (VCPDM) inherited in autosomal dominant manner (Senderek et al., 2009; Johnson et al., 2014).
The MATR3 gene encodes matrin-3, a highly conserved nuclear matrix protein that binds and interacts with nucleic acids (Belgrader et al., 1991; Hibino et al., 2006). It plays a role in RNA transcription and processing, as well as mRNA stabilization (Hibino et al., 2006). Structurally, the protein includes two zinc finger domains that interact with DNA, along with two domains that bind RNA (Belgrader et al., 1991; Hibino et al., 2006; Salton et al., 2011; Johnson et al., 2014).
The association of the 5q31 loсus with VCPDM was initially reported in 1998 (Feit et al., 1998). Almost a decade later, this disease was associated with a pathogenic missense variant, p.S85C, in the MATR3 gene across all patients (Senderek et al., 2009). The central features of the clinical presentation in the reported cases included muscle weakness and atrophy, primarily affecting the distal extremities, with a pronounced impact on the anterior compartment of lower leg muscles. Additionally, patients exhibited dysphonia and dysphagia. Onset of the disease was uniformly observed in adulthood, ranging from 35 to 57 years, and creatine kinase values varied from within the normal range to 8 times higher than normal (Feit et al., 1998; Senderek et al., 2009). Later, the authors reclassified this condition as slowly progressive ALS based on the signs of upper motor neuron involvement in some patients. This occurrence sparked further exploration into the association familial ALS with the MATR3 gene. Subsequently, in cases of clinically definite ALS other causative nucleotide variants in the MATR3 gene were identified (Johnson et al., 2014).
Currently, only one single nucleotide variant in the MATR3 gene NM_018834.6:c.254C>G, p.S85C, has been identified in all reported cases of VCPDM. Despite the identification of a common clinical core, the manifestations of the disease vary significantly among patients, both in terms of the age of onset and the severity of symptoms. The absence of pathognomonic clinical signs, coupled with the high genetic heterogeneity of distal myopathies, complicates the process of a differential diagnosis.
In this study, we present a case series of distal myopathy associated with the heterozygous previously reported p.S85C variant in the MATR3 gene.
2 Subjects and methods
The clinical and genetic characteristics of six patients from four unrelated families were investigated, comprising three male and three female patients. The age of the patients at the time of examination ranged from 36 to 65 years. All patients underwent neurophysiological evaluation, while four patients also underwent lower limb muscle magnetic resonance imaging (MRI) and spirometry testing.
The clinical examination also involved assessing dysphagia through a water swallowing test. Participants were instructed to drink 100 mL of water as quickly as possible without interruption. The duration from onset of swallowing to completing the drinking task was recorded in seconds, and the speed of swallowing (mL/s) was determined by dividing 100 mL by the time taken to drink. A swallowing speed exceeding 10 mL/s was regarded as within the normal range.
A spirometry testing was conducted in the sitting and supine position. Maximum vital capacity and forced vital capacity as percentage of predicted value, as well as the difference in the forced vital capacity in both positions were assessed in three patients. Electrocardiography and cardiac ultrasound were performed on six and three patients, respectively. Neurophysiological examination (Neuro-MVP-micro, Neurosoft, Ivanovo, Russia) included nerve conduction study of peripheral nerves and needle electromyography of proximal and distal limb muscles (four patients), only distal muscles of upper and lower limbs (one patient), proximal and distal lower limb muscles (one patient).
Lower limb muscle MRI (axial T1-weighted and axial T2-weighted images) was conducted for four patients using a 3T magnetic resonance scanner (Siemens MAGNETOM Skyra, Erlangen, Germany). Increased signal intensity on the T1-weighted images was semi quantitatively assessed using the four-point Mercuri scale, which reflects stages of muscle fatty infiltration (Mercuri et al., 2007).
For molecular genetic studies, blood samples were collected from the probands, affected and healthy family members. Genomic DNA was extracted using standard methods. An exome sequencing was performed for all probands. Target enrichment was performed using Illumina TruSeq® ExomeKit (Illumina, San Diego, CA, United States), and custom oligonucleotides (IDT xGen Exome Research Panel v1.0) included coding regions of over 20,000 protein-coding genes. Paired-end sequencing (2 × 150 bp) was carried out on an Illumina NextSeq 500. The sequencing data were processed using Illumina’s Basespace software (Enrichment 3.1.0). Variant filtering was based on their frequency, with variants having a frequency of less than 1% in The Genome Aggregation Database (gnomAD v.2.1.1) and coding region sequence effects such as missense, nonsense, coding indels, and splice sites being considered. The clinical significance of the variants was evaluated using ACMG criteria for variant interpretation (Richards et al., 2015). To validate the pathogenic variant in the proband and affected family members, Sanger sequencing was performed using the Applied Biosystems 3,130 xl Genetic Analyzer (HITACHI, Applied Biosystems Group of The Applera Corporation Japan, Waltham, MA, United States).
One patient underwent a PCR-based approach to detect D4Z4 repeat contraction (Zernov et al., 2021). The genomic DNA extracted from peripheral blood lymphocytes was embedded in agarose plugs. A portion of the plug, which had been pre-treated with the EcoRI restriction enzyme, was used for pulsed-field gel electrophoresis. The gel lines of the samples were then fragmented based on the lengths of the ladder bands. Each gel fragment was completely melted and diluted fivefold with PCR-grade water, and the resulting solutions were directly used for qPCR. For each gel fragment derived from a single patient, two separate qPCR analyses were conducted using specific primers to identify the contracted allele and the presence of a permissive haplotype on this allele. The qPCR data were analyzed using the 2−ΔΔCT method.
Patients and their relatives provided written informed consent for the publication of the data.
3 Case descriptions
The study included six patients (three male, three female) from four families, aged 36–65 years (Table 1). Notably, three unrelated families were ethnic Tatars. Pedigrees are characterized by a significant number of affected family members consistent with autosomal dominant manner of inheritance (Figure 1). The missense variant p.S85C in the MATR3 gene was detected in all of the probands by exome sequencing.
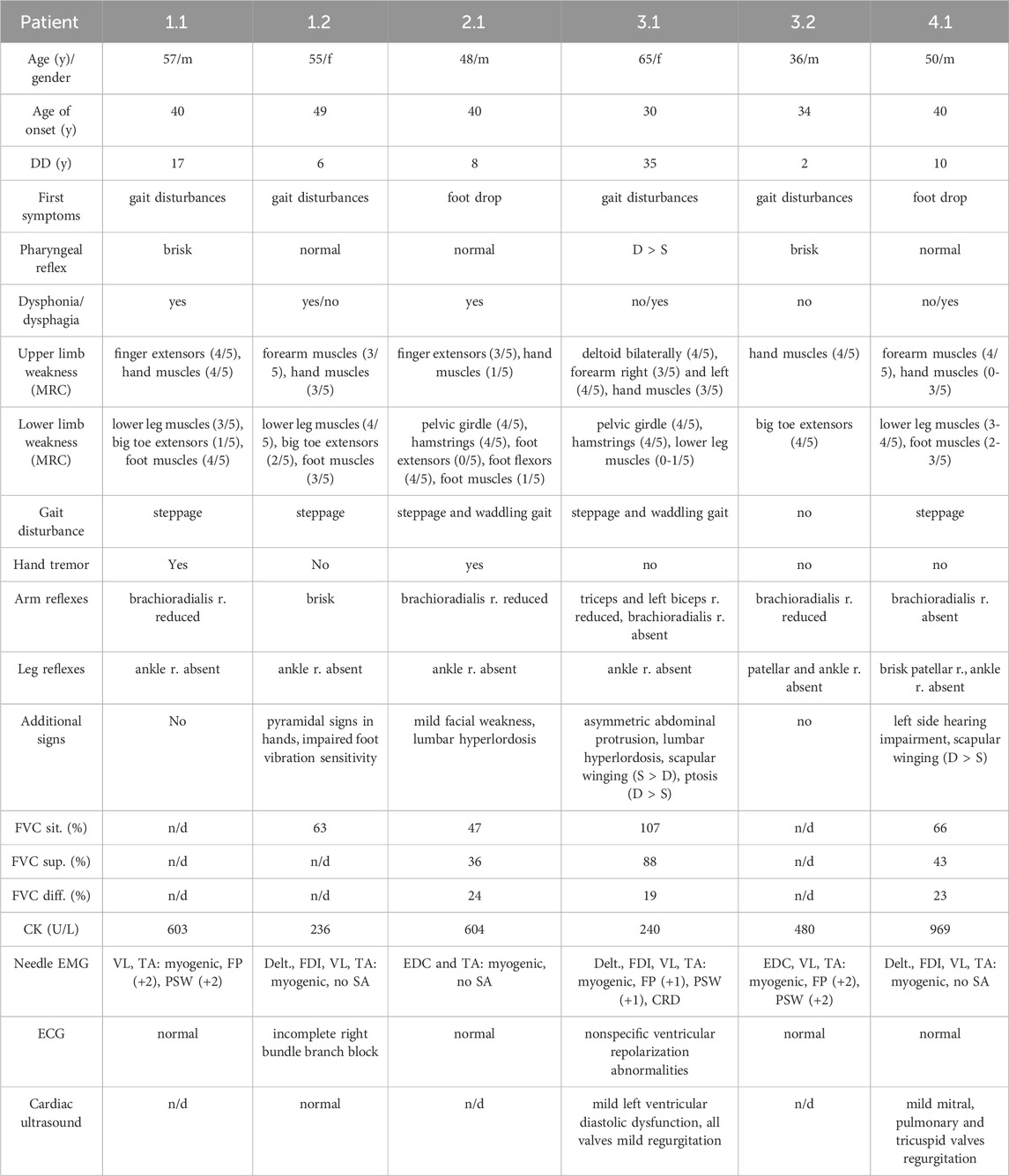
Table 1. Clinical phenotype of patients with VCPDM. VC, vital capacity, percentage of predicted value; FVC sit., forced vital capacity in sitting position; FVC sup., forced vital capacity in supine position; FVC diff., forced vital capacity, the difference in the sitting and supine position; CK, Creatine kinase; EMG, electromyography; VL, vastus lateralis; TA, tibialis anterior; Delt., deltoideus; FDI, first dorsal interosseous; FP, fibrillation potentials; PSW, positive sharp waves; SA, spontaneous activity; CRD, complex repetitive discharges; ECG, electrocardiography; y, years; DD, disease duration; m, male; f, female; n/d, no data; r., reflex.
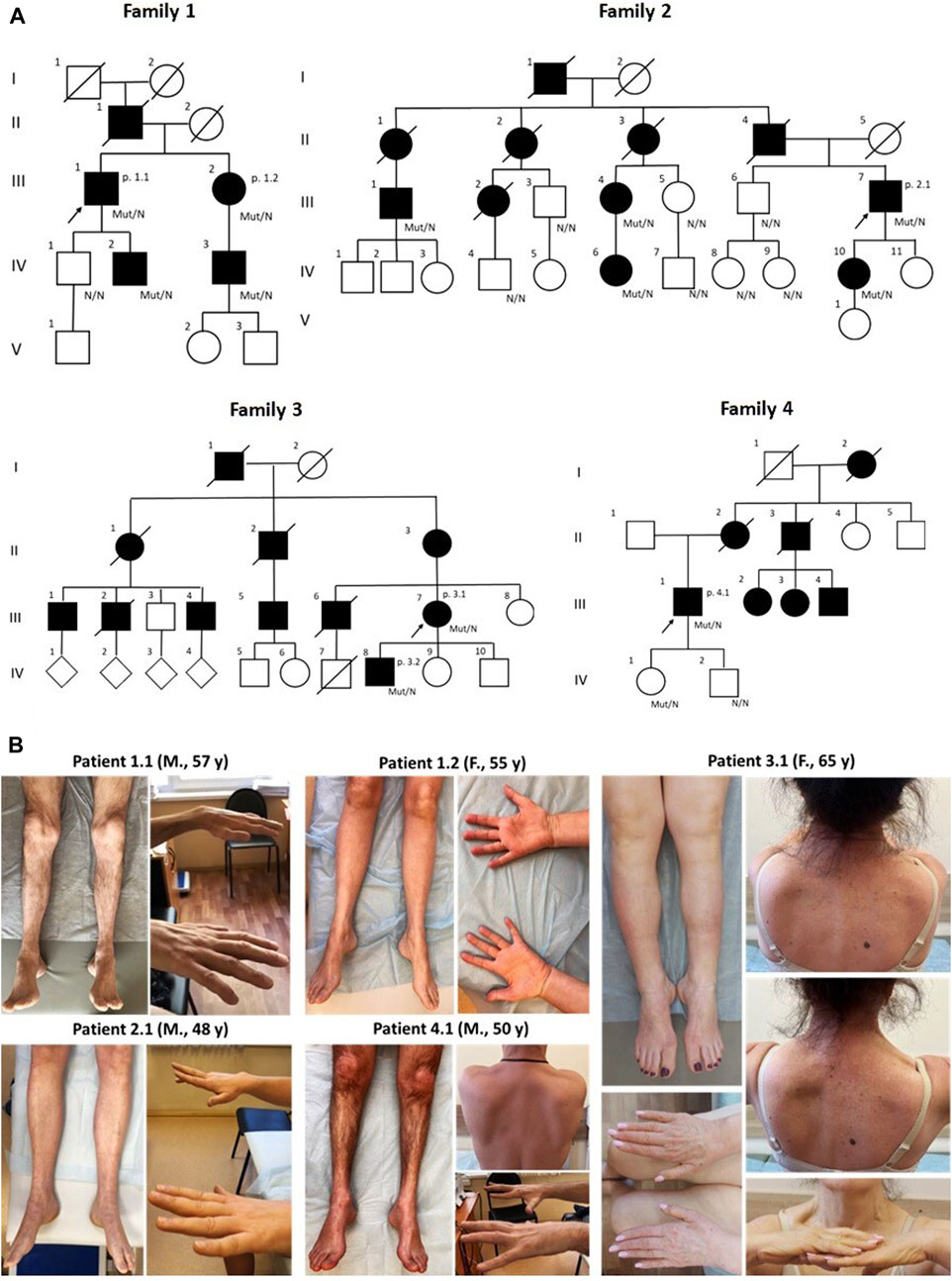
Figure 1. The pedigrees of all patients correspond to the autosomal dominant type of inheritance. Diamond symbols indicate offspring, whose gender is unknown (A) Clinical presentations of five patients with VCPDM (B) The illustrations represent typical distal muscle atrophy observed in all patients. Note scapular winging and a clear asymmetric pattern of muscle involvement in two patients 3.1 and 4.1. M, male; F, female; y, years.
In all patients, the disease manifested between the ages of 30 and 49 years, with initial symptoms of weakness in the distal leg muscles and gait disturbances. None of the patients exhibited evidence of delayed motor development nor reported difficulties in performing physical exercises during childhood or adolescence. Additionally, younger family members below 30 years old, who carried the pathogenic variant, did not display any obvious symptoms of the disease.
Clinical examination revealed selective involvement of distal limb muscles in all patients. Notably, weakness of the big toe extensors was more pronounced (0-2/5 on the Medical Research Council scale (MRC)) in five patients, with the exception of patient 3.2, while other muscles in the lower leg and feet were moderately affected. Two individuals exhibited proximal leg weakness, reaching up to 4/5 on the MRC scale, leading to a waddling gait. Steppage gait and/or difficulties walking on heels and toes were common signs observed in all patients. Additionally, varying degrees of weakness in hand and forearm muscles were observed in five patients. Special attention was drawn to the selectivity of the hand finger extensor weakness, particularly pronounced in patients 2.1, 3.1, and 4.1 (Figure 1). An interesting finding is the asymmetric atrophy of the left extensor digitorum brevis in patient 3.2. In other patients, although there was some atrophy of the muscles in the feet, the extensor digitorum brevis maintained contour well under tension and was not weak.
In two patients (3.1 and 4.1), we observed weakness of the proximal arm muscles (patient 3.1) and asymmetric scapular winging that in combination with peroneal weakness led us initially to the diagnosis of scapuloperoneal myopathy. One of these individuals, a 65-year-old female patient (3.1), had clear features associated with facioscapulohumeral muscular dystrophy (FSHD). Alongside asymmetric shoulder girdle involvement and scapular winging, she exhibited abdominal protrusion, lumbar hyperlordosis and specific combination of steppage and waddling gait. Due to these clinical features, the patient initially underwent a PCR-based approach to detect D4Z4 repeat contraction, which revealed 12 D4Z4 repeats. Subsequently, whole exome sequencing was conducted to identify causative variants in genes associated with FSHD type 2. However, no variants were detected in these genes, including the SMCHD1 gene.
Ankle reflexes were absent in all patients, while arm reflexes were reduced in four patients. Notably, patient 1.2 exhibited a unique neurological status characterized by brisk tendon reflexes in the hands and the presence of pyramidal signs in the lower limbs at the age of 55, which could be explained by the combination of myopathy and ALS features in one individual. Hand tremor was noticed in patients 1.1 and 2.1.
Bulbar signs were observed in five patients. Dysphagia was detected in four patients, while dysphonia was reported in three cases. In two cases (patients 2.1 and 4.1), dysphagia or dysphonia developed after 8 years from the onset of the disease, and in another case, only after 31 years (patient 3.1). Notably, patient 1.2 had a hoarse voice. None of the cases exhibited atrophy or fasciculation of the tongue muscles. To assess dysphagia, the swallowing test was performed in three patients, revealing a decreased swallowing speed in all examined patients, ranging from 3.6 to 8.3 mL/s (normal reference is more than 10 mL/s).
Two patients (patients 1.2 and 2.1) reported moderate respiratory difficulties in the supine position at night. A spirometry was conducted on three patients (patients 2.1, 3.1 and 4.1), revealing abnormal values in all cases. Vital capacity was lower than 75% of the reference value in two patients (patients 2.1 and 4.1). All three patients exhibited more than a 15% decrease in forced vital capacity while in the supine position, indicating diaphragmatic weakness.
The serum creatine kinase level ranged from slightly elevated to three times higher than normal values in all patients. In all cases, needle electromyography of limb muscles revealed myogenic changes. Pathological spontaneous activity was observed, characterized as mild in one patient, moderate in two patients, and manifested as fibrillation potentials and positive sharp waves. Additionally, complex repetitive discharge was identified in one patient.
According to the electrocardiographic data, patients 1.2 and 3.1 exhibited signs of impaired cardiac conduction (Table 1). Cardiac ultrasound revealed mild changes also in two patients (3.1 and 4.1).
Lower limb muscle MRI was performed on patients 1.1, 3.1, 3.2, and 4.1, each exhibiting varying disease durations (Figure 2). Across all patients, the lower leg muscles showed more extensive involvement compared to the thigh muscles. The pattern and severity of muscle involvement on T1-weighted images correlated with the duration of the disease. Muscle fatty replacement was symmetric in all patients, including patient 3.1, who exhibited clearly asymmetric clinical features. Patients in the mid-stage of the disease (1.1 and 4.1) exhibited a characteristic muscle involvement pattern, with the posterior tibial and lateral gastrocnemius muscles relatively spared. Patient 3.2, in the early disease stage, showed diffuse, slight involvement of lower leg muscles, except for the tibialis anterior and tibialis posterior muscles. Conversely, patient 3.1, in the advanced disease stage, demonstrated total fatty replacement of lower leg muscles. In the thigh, muscles of the posterior compartment, particularly the long head of the biceps femoris and the semimembranosus muscles, were most affected.
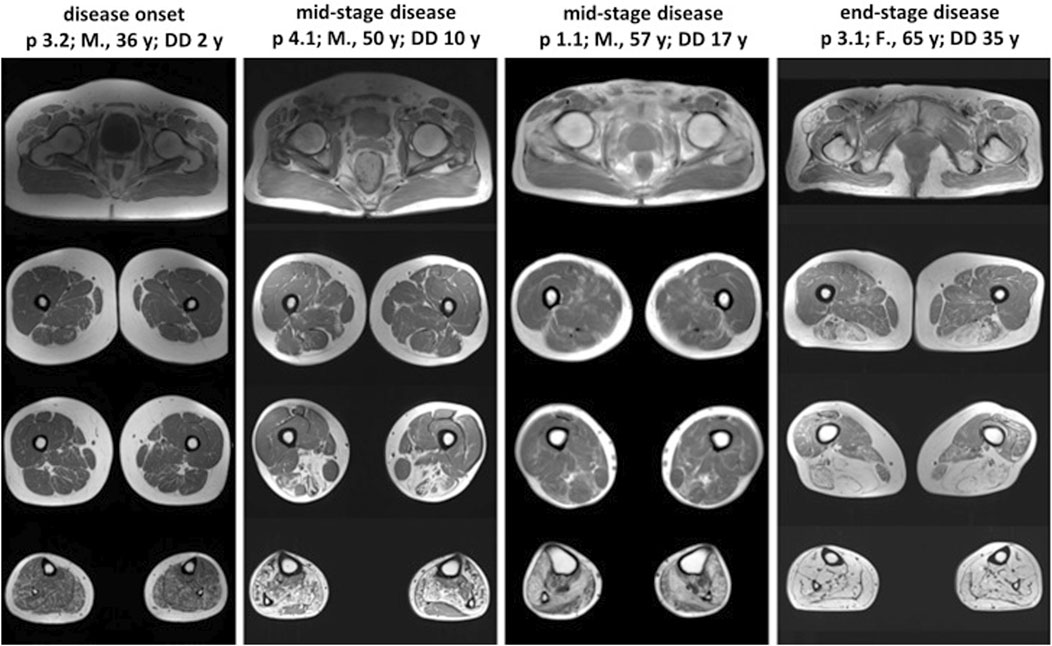
Figure 2. Lower limb muscle MRI in patients with different stages of VCPDM. The characteristic pattern of muscle involvement was observed in patients with the mid-stage of the disease. T1-weighted MRI revealed the typical involvement of lower leg muscles, with relatively spared tibialis posterior and lateral gastrocnemius. The patient with disease onset demonstrated mild diffusely affected posterior compartment of lower leg muscles. In the patient with end-stage disease, complete fatty replacement of lower leg muscles was noted. The most pronounced affected thigh muscles were the long head of the biceps femoris and the semimembranosus with spared gracilis in all patients. M, male; F, female; y, years; p, patient; DD, disease duration.
Due to the lack of specific therapy, all patients were given only general recommendations for physiotherapy, correction of swallowing disorders, and dynamic monitoring of respiratory functions. One patient began to regularly engage in aerobic exercises and noticed an improvement in her condition, with a subjective increase in muscle strength over 1 year of follow-up.
4 Discussion
Here, we present a series of six patients with VCPDM, expanding the worldwide cohort of patients (Feit et al., 1998; Senderek et al., 2009; Müller et al., 2014; Palmio et al., 2016; Barp et al., 2018). In all our patients, the disease is caused by a recurrent pathogenic missense variant p.S85C in the MATR3 gene, as observed in all previously reported cases of this myopathy.
Important functions of MATR3 include interacting with other nuclear matrix proteins to form the internal fibrogranular network and retaining defective RNAs within the nucleus, among others (Iradi et al., 2018). MATR3 is also believed to play a role in gene regulatory functions, including DNA replication and transcription (Cha et al., 2021). In previous studies on cultured cells and Drosophila models, no significant difference was found in the effects specific to the distal myopathy variant p.S85C and the missense variants associated with ALS (Iradi et al., 2018; Ramesh et al., 2020). Given that missense variants have been described for both diseases, it can be assumed that the amino acid substitution at position 85 leads to a gain-of-function event, which is associated with the dysregulation of genes expressed in skeletal muscles.
All cases in our study exhibited a typical disease onset after the age of 30, characterized by weakness of distal leg muscles. The characteristic initial symptoms of VCPDM include the selective involvement of foot extensors, resulting in gait disturbances, foot drop, and frequent falls, observed in 80% of patients as well as in all our cases (Table 2). Hand muscle weakness is also an early sign observed in all our patients, including patient 3.2, at the disease onset stage.

Table 2. The representation of clinical features in previously reported and current study.
Five patients exhibited dysphagia and/or dysphonia, often without actively paying attention to it. One patient reported a long-term hoarse voice that had never been associated with the disease. Therefore, it is crucial to conduct an objective examination of swallowing and respiratory function. In all three patients for whom spirometry was performed, an abnormal forced vital capacity difference between sitting and supine positions was detected. Two patients complained of respiratory difficulties at night.
Some patients with the recurrent variant p.S85C may exhibit additional features of pyramidal involvement (Palmio et al., 2016; Cavalli et al., 2021). In our cohort, we observed one patient who presented with pyramidal signs in her hands at the age of 55 years. However, this patient was diagnosed with distal myopathy, which was confirmed by clear muscle MRI and needle electromyography results. To date, there is no definitive explanation for why some patients present with a combination of ALS and myopathy symptoms, while others manifest only an isolated phenotype (Johnson et al., 2014).
Our study included patients with varying disease durations, enabling us to compare muscle fatty replacement at different stages of the disease. The MRI pattern of muscle involvement in our patients is consistent with previously reported cases (Müller et al., 2014; Palmio et al., 2016; Barp et al., 2018; Mensch et al., 2020). Patients in the mid-stage of the disease exhibited relatively spared tibialis posterior and lateral gastrocnemius muscles at the lower leg level, which are characteristic features of VCPDM (Mensch et al., 2020). It is noteworthy that gracilis was consistently the least affected muscle in all patients, including those in the end-stage of the disease. Despite asymmetric clinical features observed in two individuals (patients 3.1 and 4.1), all patients exhibited symmetric muscle fatty replacement. This observation aligns with findings from previous studies investigating the MRI patterns in patients with VCPDM (Barp et al., 2018; Mensch et al., 2020).
Two out of six patients demonstrated features of asymmetric scapuloperoneal myopathy, which, in one case, necessitated the exclusion of FSHD through targeted genetic molecular analysis. Notably, in this proband, 12 macrosatellite D4Z4 repeats were detected, falling within the lower border of the normal range. Whole exome sequencing did not reveal any causative variants in the genes associated with FSHD type 2. Scapular winging has been rarely described in some patients previously (Müller et al., 2014; Barp et al., 2018). Several other reports have mentioned asymmetry in the early stages of the disease (Feit et al., 1998; Müller et al., 2014; Barp et al., 2018). The clinical overlap between VCPDM and FSHD is an intriguing issue for further investigation. A recent study by Runfola et al. demonstrated that MATR3 serves as a cellular factor regulating the expression and activity of the transcription factor double homeobox 4 (DUX4) (Runfola et al., 2023). It is assumed that DUX4 activates a proapoptotic transcription program, inhibiting myogenic differentiation and leading to FSHD (Mocciaro et al., 2021).
Generally, the differential diagnosis between VCPDM and FSHD is not complicated. VCPDM typically presents with a distal phenotype accompanied by some bulbar disturbances. In contrast, FSHD manifests with facial and shoulder girdle weakness. However, some individuals with VCPDM may develop facial muscle weakness and asymmetric scapular winging, while bulbar symptoms may manifest later or remain inconspicuous. This can seriously complicate the differential diagnosis, particularly considering the existence of a distal phenotype of FSHD without clear facial weakness.
5 Conclusion
The phenotypic variability of VCPDM and the rarity of this pathology are the primary factors contributing to diagnostic challenges. Among the six cases reported here, two exhibited a scapuloperoneal phenotype of MATR3-related distal myopathy, necessitating differential diagnosis with FSHD in one of the cases.
Data availability statement
The datasets for this article are not publicly available due to concerns regarding participant/patient anonymity. Requests to access the datasets should be directed to the corresponding author.
Ethics statement
The studies involving humans were approved by Institutional Review Board of the Research Centre for Medical Genetics. The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author contributions
AM: Conceptualization, Methodology, Writing–original draft, Writing–review and editing. DS: Conceptualization, Visualization, Writing–original draft, Data curation. AK: Investigation, Resources, Visualization, Writing–review and editing. OG: Investigation, Resources, Writing–review and editing. DD: Methodology, Resources, Writing–review and editing. OS: Methodology, Resources, Writing–review and editing. TC: Investigation, Resources, Writing–review and editing. MB: Investigation, Resources, Writing–review and editing. DS: Investigation, Resources, Writing–review and editing. OR: Methodology, Resources, Writing–review and editing. OK: Investigation, Writing–review and editing. MS: Methodology, Resources, Writing–review and editing. AB: Writing–review and editing. SK: Funding acquisition, Supervision, Writing–review and editing.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This research was carried out within the state assignment of the Ministry of Science and Higher Education of the Russian Federation for Research Centre for Medical Genetics.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Barp, A., Malfatti, E., Metay, C., Jobic, V., Carlier, R. Y., and Laforet, P. (2018). The first French case of MATR3-related distal myopathy: clinical, radiological and histopathological characterization. Rev. Neurol. Paris. 174 (10), 752–755. doi:10.1016/j.neurol.2017.08.004
Belgrader, P., Dey, R., and Berezney, R. (1991). Molecular cloning of matrin 3. A 125-kilodalton protein of the nuclear matrix contains an extensive acidic domain. J. Biol. Chem. 266 (15), 9893–9899. doi:10.1016/s0021-9258(18)92902-9
Cavalli, M., Cardani, R., Renna, L. V., Toffetti, M., Villa, L., and Meola, G. (2021). First family of MATR3-related distal myopathy from Italy: the role of muscle biopsy in the diagnosis and characterization of a still poorly understood disease. Front. Neurol. 12, 715386. doi:10.3389/fneur.2021.715386
Cha, H. J., Uyan, Ö., Kai, Y., Liu, T., Zhu, Q., Tothova, Z., et al. (2021). Inner nuclear protein Matrin-3 coordinates cell differentiation by stabilizing chromatin architecture. Nat. Commun. 12 (1), 6241. doi:10.1038/s41467-021-26574-4
Feit, H., Silbergleit, A., Schneider, L. B., Gutierrez, J. A., Fitoussi, R. P., Réyès, C., et al. (1998). Vocal cord and pharyngeal weakness with autosomal dominant distal myopathy: clinical description and gene localization to 5q31. Am. J. Hum. Genet. 63 (6), 1732–1742. doi:10.1086/302166
Hibino, Y., Usui, T., Morita, Y., Hirose, N., Okazaki, M., Sugano, N., et al. (2006). Molecular properties and intracellular localization of rat liver nuclear scaffold protein P130. Biochim. Biophys. Acta 1759 (5), 195–207. doi:10.1016/j.bbaexp.2006.04.010
Iradi, M. C. G., Triplett, J. C., Thomas, J. D., Davila, R., Crown, A. M., Brown, H., et al. (2018). Characterization of gene regulation and protein interaction networks for Matrin 3 encoding mutations linked to amyotrophic lateral sclerosis and myopathy. Sci. Rep. 8 (1), 4049. doi:10.1038/s41598-018-21371-4
Johnson, J. O., Pioro, E. P., Boehringer, A., Chia, R., Feit, H., Renton, A. E., et al. (2014). Mutations in the Matrin 3 gene cause familial amyotrophic lateral sclerosis. Nat. Neurosci. 17 (5), 664–666. doi:10.1038/nn.3688
Mensch, A., Kraya, T., Koester, F., Müller, T., Stoevesandt, D., and Zierz, S. (2020). Whole-body muscle MRI of patients with MATR3-associated distal myopathy reveals a distinct pattern of muscular involvement and highlights the value of whole-body examination. J. Neurol. 267 (8), 2408–2420. doi:10.1007/s00415-020-09862-9
Mercuri, E., Pichiecchio, A., Allsop, J., Messina, S., Pane, M., and Muntoni, F. (2007). Muscle MRI in inherited neuromuscular disorders: past, present, and future. J. Magn. Reson Imaging 25 (2), 433–440. doi:10.1002/jmri.20804
Milone, M., and Liewluck, T. (2019). The unfolding spectrum of inherited distal myopathies. Muscle Nerve 59 (3), 283–294. doi:10.1002/mus.26332
Mocciaro, E., Runfola, V., Ghezzi, P., Pannese, M., and Gabellini, D. (2021). DUX4 role in normal physiology and in FSHD muscular dystrophy. Cells 10 (12), 3322. doi:10.3390/cells10123322
Müller, T. J., Kraya, T., Stoltenburg-Didinger, G., Hanisch, F., Kornhuber, M., Stoevesandt, D., et al. (2014). Phenotype of matrin-3-related distal myopathy in 16 German patients. Ann. Neurol. 76 (5), 669–680. doi:10.1002/ana.24255
Palmio, J., Evilä, A., Bashir, A., Norwood, F., Viitaniemi, K., Vihola, A., et al. (2016). Re-evaluation of the phenotype caused by the common MATR3 p.Ser85Cys mutation in a new family. J. Neurol. Neurosurg. Psychiatry 87 (4), 448–450. doi:10.1136/jnnp-2014-309349
Ramesh, N., Kour, S., Anderson, E. N., Rajasundaram, D., and Pandey, U. B. (2020). RNA-recognition motif in Matrin-3 mediates neurodegeneration through interaction with hnRNPM. Acta Neuropathol. Commun. 8 (1), 138. doi:10.1186/s40478-020-01021-5
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genet. Med. 17 (5), 405–424. doi:10.1038/gim.2015.30
Runfola, V., Giambruno, R., Caronni, C., Pannese, M., Andolfo, A., and Gabellini, D. (2023). MATR3 is an endogenous inhibitor of DUX4 in FSHD muscular dystrophy. Cell Rep. 42 (9), 113120. doi:10.1016/j.celrep.2023.113120
Salton, M., Elkon, R., Borodina, T., Davydov, A., Yaspo, M. L., Halperin, E., et al. (2011). Matrin 3 binds and stabilizes mRNA. PLoS One 6 (8), e23882. doi:10.1371/journal.pone.0023882
Savarese, M., Sarparanta, J., Vihola, A., Jonson, P. H., Johari, M., Rusanen, S., et al. (2020). Panorama of the distal myopathies. Acta Myol. 39 (4), 245–265. doi:10.36185/2532-1900-028
Senderek, J., Garvey, S. M., Krieger, M., Guergueltcheva, V., Urtizberea, A., Roos, A., et al. (2009). Autosomal-dominant distal myopathy associated with a recurrent missense mutation in the gene encoding the nuclear matrix protein, matrin 3. Am. J. Hum. Genet. 84 (4), 511–518. doi:10.1016/j.ajhg.2009.03.006
Keywords: MATR3, distal myopathy, scapuloperoneal phenotype, phenotypic variability, FSHD, ALS
Citation: Murtazina A, Subbotin D, Kuchina A, Gilvanova O, Degterev D, Shchagina O, Cherevatova T, Bulakh M, Sherstyukova D, Ryzhkova O, Kurushina O, Skoblov M, Borovikov A and Kutsev S (2024) Asymmetric scapuloperoneal phenotype of MATR3-related distal myopathy: case series. Front. Genet. 15:1414928. doi: 10.3389/fgene.2024.1414928
Received: 09 April 2024; Accepted: 30 July 2024;
Published: 13 August 2024.
Edited by:
Massimiliano Filosto, University of Brescia, ItalyReviewed by:
Edoardo Malfatti, Hôpitaux Universitaires Henri Mondor, FranceKristl Claeys, University Hospitals Leuven, Belgium
Copyright © 2024 Murtazina, Subbotin, Kuchina, Gilvanova, Degterev, Shchagina, Cherevatova, Bulakh, Sherstyukova, Ryzhkova, Kurushina, Skoblov, Borovikov and Kutsev. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Aysylu Murtazina, YXlzeWx1bXVydGF6aW5hQGdtYWlsLmNvbQ==