 Sadegh Taheri
Sadegh Taheri Ali Javadmanesh
Ali Javadmanesh Saeed Zerehdaran
Saeed Zerehdaran- Department of Animal Science, Faculty of Agriculture, Ferdowsi University of Mashhad, Mashhad, Iran
Introduction: Identifying genomic regions under selection is the most challenging issue for improving important traits in animals. Few studies have focused on identifying genomic regions under selection in sheep. The aim of this study was to identify selective sweeps and to explore the relationship between these and quantitative trait loci (QTL) in both domestic and wild sheep species using single nucleotide polymorphism markers (SNPs).
Methods: Genomic data were obtained from the NextGen project, which included genotyping 20 domestic and 14 wild sheep using the Illumina Ovine SNP50K BeadChip. The XP-EHH, iHS, and RSB methods were employed to detect signatures of positive selection.
Results: The results of the iHS method indicated 405 and 275 selective sweeps in domestic and wild sheep, respectively. Additionally, RSB and XP-EHH analyses revealed approximately 398 and 479 selective sweeps in domestic and wild sheep, respectively. Some of the genes associated with important QTL traits in domestic sheep include ADGRB3, CADM1, CAPN2, GALNT10, MTR, RELN, and USP25, while in wild sheep, the relevant genes include ACAN, ACO1, GADL1, MGST3, and PRDM16. Selective sweeps identified in domestic sheep were associated with body weight, muscle weight, milk protein percentage, and milk yield. In contrast, selective sweeps found in wild sheep were linked to average daily gain, bone weight, carcass fat percentage, and dressing percentage.
Discussion: These results indicate that selection by humans and the environment have largely progressed in harmony, highlighting the importance of both economic and environmental traits for survival. Additionally, the identification of potential candidate genes associated with economic traits and genomic regions that have experienced selection can be utilized in sheep breeding programs. However, due to the incomplete information regarding the functional annotation of genes in sheep and the limited sample size, further research with a larger sample group is essential to gain a deeper understanding of the candidate genes linked to economic traits in both domestic and wild sheep. Advancing knowledge in this area can significantly enhance the effectiveness of breeding strategies. The quantitative trait loci identified in this study have the potential to be incorporated into breeding plans for both domestic and wild sheep.
1 Introduction
Animal domestication has significantly influenced human life. Due to their high adaptability to various climates and diets, sheep were among the first animals to be domesticated (Zeder, 2008). Evidence suggests that Iran was a world center for farm animal domestication (Zeder, 1999). Archaeological findings indicate that sheep were domesticated approximately 9,000 years ago in what is now northern Iraq and parts of Iran (Kijas et al., 2009; Zeder, 1999). The wild sheep of Iran include three species: Vignei sheep (Ovis vignei), Asian mouflon (O. orientalis), and hybrids with varying chromosome numbers, which are found in the eastern and western regions and the Alborz Mountain range of northern Iran. Iranian wild sheep are recognized as the ancestors of domestic sheep worldwide, which may account for the diversity of domestic sheep breeds in Iran (Hamadalahmad et al., 2020). Domestic sheep are vital to Iran’s agricultural economy and the livelihoods of its people, serving as a source of milk, meat, and other byproducts. To effectively manage the genetic resources of both wild and domestic animals, it is essential to understand the characteristics of different breeds. These include population size and structure, geographical distribution, optimal environmental conditions for production and performance, and levels of genetic diversity within and among breeds (Javadmanesh et al., 2022). Domestication has significantly altered the behavioral and morphological traits of these animals. In addition to domestication, selective breeding for increased production or specific behavioral and morphological traits has led to the emergence of highly divergent modern breeds (Flori et al., 2009). Post-domestication selection for economic and morphological traits can have lasting effects on the genomes of sheep. This selection, combined with natural adaptation to local environments, has resulted in the development of over a thousand distinct breeds of sheep (Kijas et al., 2012). These genetic features, along with the extensive genomic information related to economic traits, offer an opportunity to identify loci that are under selection (Hayes et al., 2008). Selection can lead to specific changes in both selected and neutral sites, with selection signatures—genomic footprints left by selection—used to identify these target areas (Kreitman, 2000). The recent availability of genomic data from domestic animals, coupled with advances in statistical tools, has made it possible to detect these signatures in specific species (Dong et al., 2013). Identifying selection signatures is a key focus for evolutionary geneticists, as it provides insight into the evolutionary processes that shape genomes and translates genomic data into functional information about important regions (Schlötterer, 2003). Several statistical methods are available for the investigation of selection signatures, including the integrated haplotype homozygosity score (iHS) (Voight et al., 2006), cross-population extended haplotype homozygosity (XP_EHH) (Sabeti et al., 2007), and the ratio of extended haplotype homozygosity between populations (RSB) (Tang et al., 2007). Advances in livestock genomics research have led to the recent development of QTL databases linked to the latest reference genomes of various livestock species, such as sheep, cows, pigs, and fish (Hui et al., 2021). These databases provide precise locations of genes and genomic regions associated with known traits in livestock species. As a result, researchers can use these fine-mapped QTL traits to identify selection signatures related to desired traits in livestock species, which could be useful for breeding programs (Zeraatpisheh et al., 2023).
The current study aimed to detect the regions under natural and artificial selection in two wild and domestic Iranian sheep species using iHS, XP-EHH, and RSB methods. The communication between selective sweep and QTL traits was also identified using single nucleotide markers (SNP).
2 Materials and methods
2.1 Sampling and genotyping
The data utilized in this research were retrieved from the NextGen project (http://projects.ensembl.org/nextgen/). The objective of this project is to assess the genetic diversity of some domestic and wild species such as sheep, goat, and cattle by utilizing whole-genome sequencing and genotyping by microarray. For this particular study, samples were collected from 20 Iranian domestic (O. aries) and 14 Iranian wild sheep (O. orientalis). Samples were taken according to the Helsinki Declaration of 1975 (as revised in 2008) concerning animal rights, and this study was approved by the Animal Ethical Committee (3/51431), Ferdowsi University of Mashad, Mashhad, Iran (Taheri et al., 2022). Ear tissue samples were used for DNA extraction using a Macherey Nagel NucleoSpin 96 Tissue kit, following the manufacturer’s instructions, as per Alberto et al. (2018). To integrate the whole-genome sequencing data with the variations in each animal’s entire genome, the NextGen project’s equivalent of the SNP50 BeadChip array was obtained from every individual utilizing PLINK v1.9 software.
2.2 Quality check and PCA analyses
To ensure the quality of the data, the PLINK version 1.9 application (Purcell et al., 2007) was used to perform quality control for the two populations. Individuals with a genotyping call rate of less than 99% were removed. Additionally, SNPs with a genotyping call rate of less than 99%, a minor allele frequency (MAF) of less than 5%, and a significant deviation from Hardy–Weinberg equilibrium (HWE ) (p < 106) were excluded (Taheri et al., 2023). Following this filtering process, the genotype data for both populations were combined, and common SNPs between them were selected.
To investigate the structures of the domestic and wild sheep populations and to identify animals that did not fit into their respective populations, principal component analysis (PCA ) was conducted using a relative genomic matrix. PLINK version 1.9 was employed to perform this analysis.
2.3 Detection of selection sweep and significant SNPs
The three methods used in this study—iHS, RSB, and XP-EHH—are basically the same, based on the concept of extended haplotype homozygosity (EHH), to increase the accuracy in identifying selection signatures. iHS is designed for use on a specific population, while XP-EHH and RSB are for comparing differences in selection between two populations. Three statistics were calculated using the EHH concept developed by Sabeti et al. (2002).
iHS analysis is a statistical method for identifying haplotype-based selection sweeps which involves the calculation of EHH from both ancestral (iHHA) and derived (iHHD) biallelic SNPs. iHS is a within-population value based on the rate of LD decay at a certain variant and is calculated as per Voight et al. (2006):
The “rehh” package (Gautier and Vitalis, 2012) in R was utilized to separately estimate iHS values for SNPs with MAF ≥0.05 within domestic and wild populations. Large positive or negative values of iHS show long haplotypes harboring ancestral or derived alleles, respectively.
RSB is also calculated based on EHH. However, unlike iHS, RSB involves comparing the EHH patterns of the common alleles (as “iES”) between the two populations. Tang et al. (2007) defined RSB as:
To estimate RSB values, various iES statistics were estimated using the “rehh” package during the iHS analyses in each of the domestic and wild populations, as described previously. The RSB values were normalized in the R environment. Since the RSB value is directional, positive values indicate selection in the wild population, while negative values indicate selection in the domestic population.
XP-EHH was also utilized to identify any recent selection sweeps within the sheep genome. This method is based on evaluating LD decay across the genome and EHH values. For biallelic SNPs with alleles A and a, EHH was calculated using the following formula (Sabeti et al., 2002):
where nA and na represent haplotype counts with alleles A and a, respectively, ni is the count for the ith haplotype within a sub-population, and hx represents the number of distinct haplotypes in a genomic area up to a distance x from the core locus. All SNPs located within one Mbp interval up- and downstream of a given SNP were taken into account, and EHH was integrated within these intervals for two groups. XP-EHH was estimated according to Sabeti et al. (2007). The highest negative and positive values indicate locations under selection in domestic and wild sheep populations, respectively. The “rehh” software was utilized to estimate XP-EHH, and the top 5% of estimated values were selected for further analysis using both domestic and wild populations (Tao et al., 2020). To increase the power of detecting selection signatures, we selected the overlapping areas between RSB, XP-EHH, and iHS techniques in domestic and wild sheep.
After identifying selection signatures using the three methods, similar locations in both populations were identified separately. The candidate genes related to these common locations were then selected using PLINK V1.9 (Purcell et al., 2007), utilizing a gene list provided from the sheep reference genome (http://ncbi.nlm.nih.gov/genome/?term=sheep).
2.4 Gene ontology and KEGG pathway analyses of genes related to significant SNPs
To identify gene ontology terms and significant metabolic KEGG pathways associated with genes related to significant SNPs, ClueGo version 2.5.6 was used, which is a Cytoscape plug-in that provides biological interpretations of genes (Bindea et al., 2009). Significant genes were then subjected to functional enrichment analysis using the Cytoscape application (Saito et al., 2012) and ClueGO 2.5.6 plug-in (Bindea et al., 2009). Official Gene Symbol was used as the input parameter, and O. aries was selected as the background organism. After Bonferroni correction for multiple testing, p-values <0.05 were considered statistically significant.
2.5 Extraction of QTL traits associated with genes related to significant QTLs
Hu et al. (2022) utilized the AnimalQTL database (www.animalgenome.org/cgi-bin/QTLdb/OA/index) to locate sheep QTL traits previously reported. They then compared the genes identified in regions of high three methods with these QTL traits to determine whether any traits were under selection in Iranian domestic and wild sheep.
3 Results
3.1 Quality control and PCA analyses
After performing quality control on the genomic data, we selected 33,167 and 35,479 SNPs for further analysis in domestic and wild sheep, respectively. To analyze the genetic connection between domestic and wild sheep and characterize their divergence, we conducted principal component analysis (PCA). The results of the PCA showed that domestic and wild sheep were clearly divided as shown in the first and second principal component factors. Furthermore, the results showed that the samples of domestic sheep had lower within-population variation, while the wild sheep had more within-population variation, indicating two possible subpopulations (Figure 1).
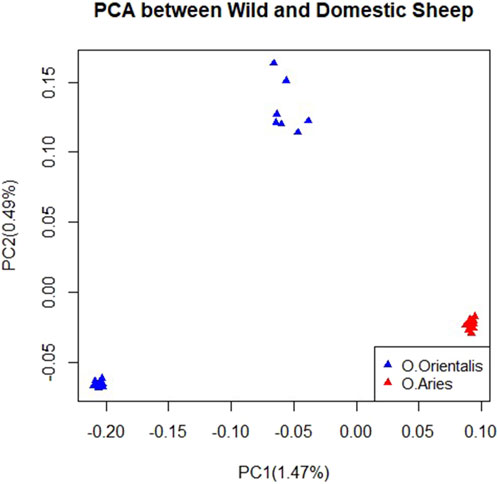
Figure 1. Clustering based on principal component analysis (PCA) using SNP data for domestic and wild sheep.
3.2 Detection of selection sweep, significant SNPs, and their related genes
The study utilized three different statistical tests (iHS, RSB, XP_EHH) to detect any regions of the genome that had potentially undergone recent selection. The iHS test was applied to SNPs across the genome of both domestic and wild sheep populations, and a Manhattan plot was generated to display the distribution of iHS signals across all chromosomes (Figure 2). We adjusted the threshold value to identify the top 5% of SNPs with the highest iHs for each population. The Manhattan plot for the wild sheep population indicated 275 locations with the highest iHS values (iHS>1.91) on chromosomes 13, 18, 6, and 1. The Manhattan plot for the domestic sheep population revealed 405 regions with the highest signals (iHS>1.86) on chromosomes 7, 2, and 3.
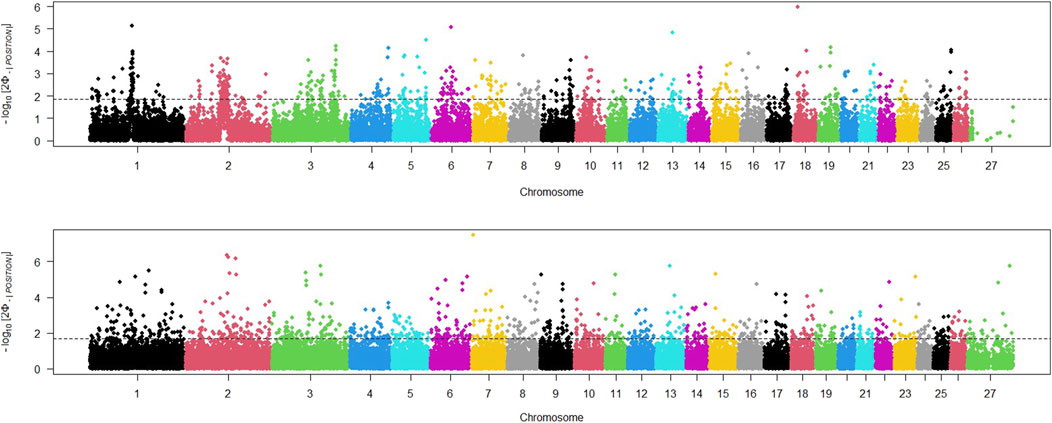
Figure 2. Distribution of iHS values across the genome in wild (above) and domestic (below) sheep; threshold indicates the top 5% SNPs with the largest iHs for each population.
In order to identify selective sweep in the wild and domestic populations, an EHH-based RSB method was also utilized. The distribution of RSB values across sheep autosomal chromosomes is shown in Figure 3. Based on regions with RSB values lower than −1.92, 421 regions were found to be under selection in the domestic sheep population. The lowest RSB values were observed on chromosomes 20, 11, and 6. Similarly, significant positive RSB values (RSB>2.05) were identified in 376 regions in the wild sheep population. The highest RSB values were found on chromosomes 1, 2, and 13.
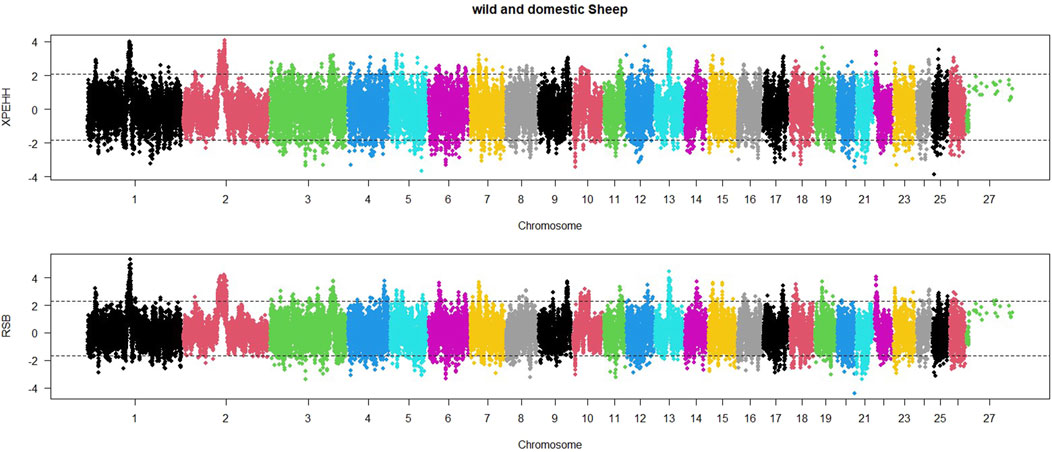
Figure 3. Distribution of XP-EHH and RSB across the genome between domestic (lower than 0) and wild (higher than 0) sheep. Thresholds indicate the top 5% SNPs for each population.
Additionally, a Manhattan plot for XP-EHH values within the sheep genome was generated (Figure 3). Based on regions with XP-EHH values lower than −2.04, 467 regions were identified as under selection in the domestic sheep population. The most negative XP-EHH values were observed on chromosomes 25, 5, and 10. Similarly, according to significant selection signatures for XP-EHH values higher than 1.97, 490 regions were detected in the wild sheep population. The highest XP-EHH values were found on chromosomes 1, 2, and 12.
A Venn diagram for the domestic and wild sheep populations indicated that 34 genes in the domestic sheep and 83 genes in the wild sheep were shared by all three methods (Figure 4).
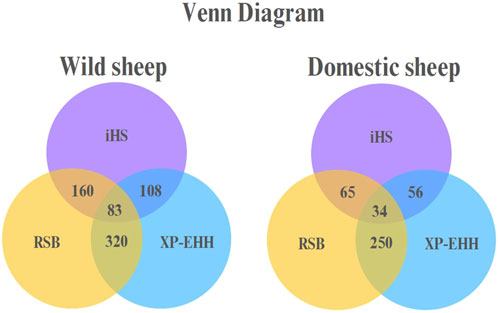
Figure 4. Venn diagram in domestic and wild sheep populations representing overlapping regions between iHS, RSB, and XP-EHH.
3.3 Gene ontology and KEGG pathway analyses of genes related to significant SNPs and associated QTL traits
We discovered that overlapping genes between the iHS, RSB, and XP-EHH methods were related to various processes in domestic (cell adhesion molecules, calcium-dependent cysteine-type endopeptidase activity, and cytoplasm) and wild (metabolic pathways, nucleotide metabolism, and glutamatergic synapse) sheep (Table 1).
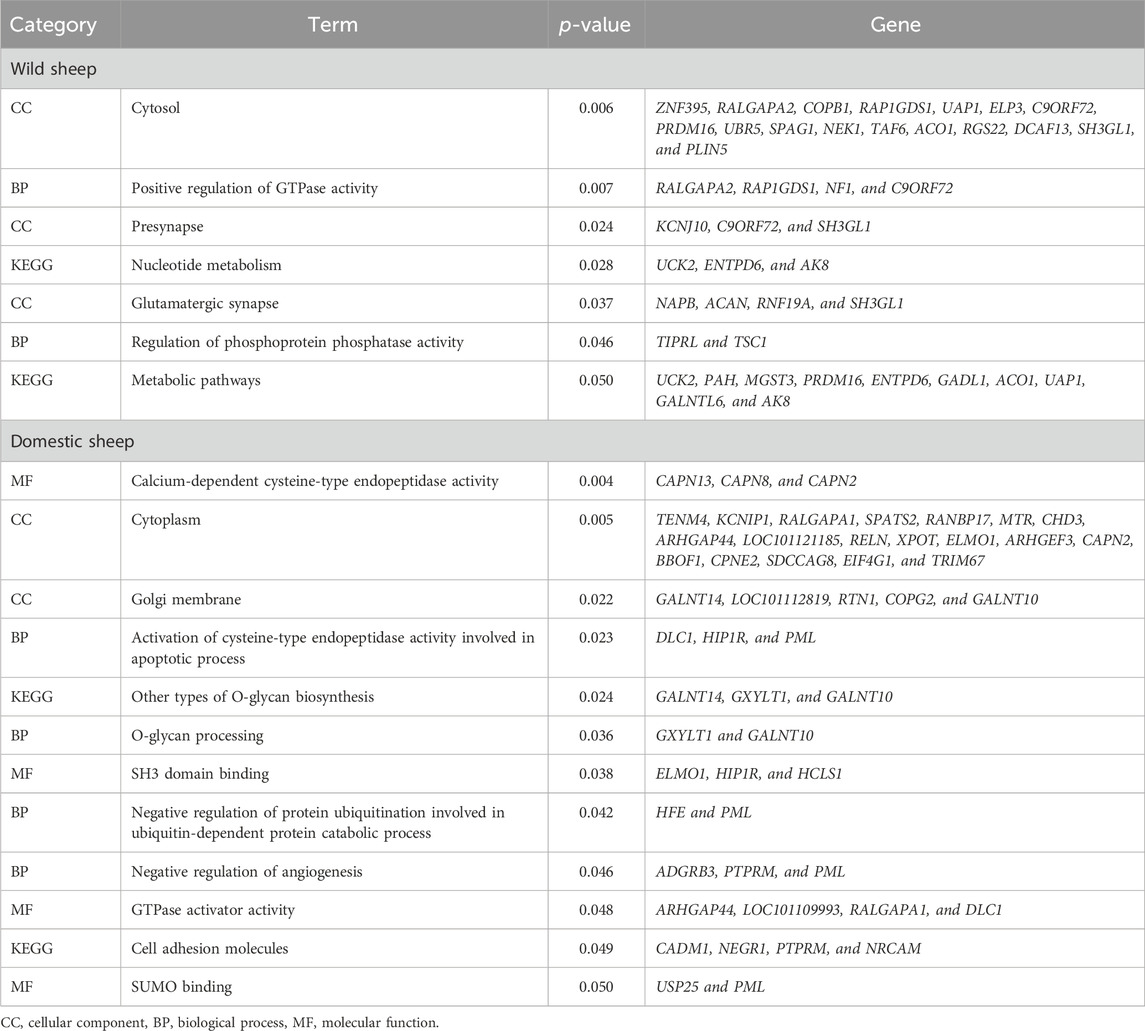
Table 1. GO terms and pathways along with the p-value of genes related to significant SNPs in domestic and wild sheep populations.
The quantitative trait locus report indicated that significant genes identified in the domestic sheep were connected to crucial traits such as body weight, bone density, bone weight, carcass fat percentage, and muscle weight. Additionally, candidate genes identified in the wild sheep were linked to average daily gain, milk fat percentage, milk yield, and muscle weight (Table 2) (more details in Supplementary Tables 1, 2).
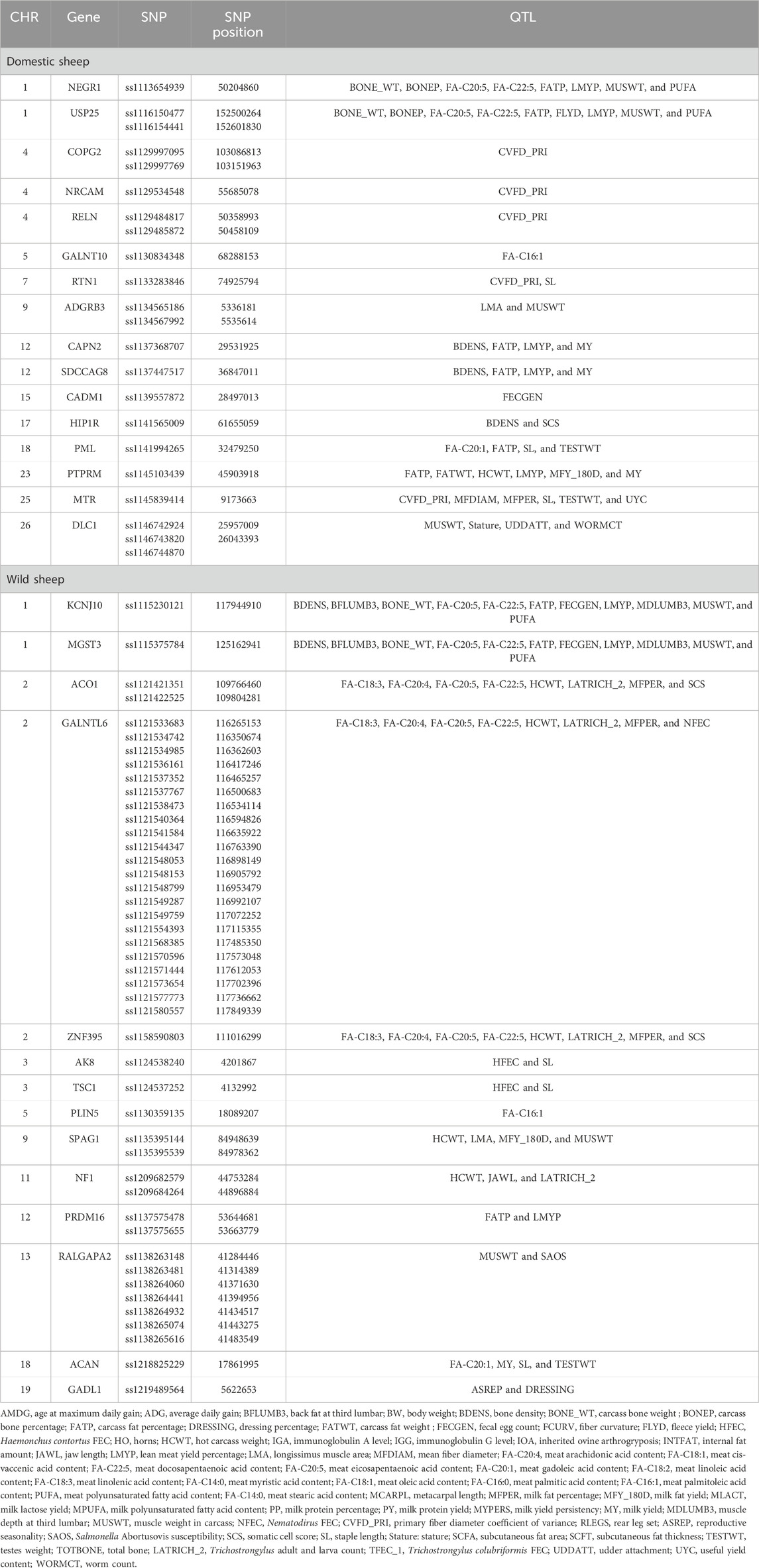
Table 2. QTL traits associated with genes related to significant SNPs along with their genomic locations in domestic and wild populations.
4 Discussion
The results of the PCA indicated a clear distinction between the wild and domestic sheep populations (Figure 1). The use of genotypic relationship-based PCA has been a valuable tool in various studies to understand population structure and genetic connections between individuals (Sabeti et al., 2007). Domestic samples were collected from different regions, while wild samples were collected from the same region (Taheri et al., 2022). Our analysis revealed a more compact structure within the domestic sheep than the wild sheep, possibly due to intense selection pressure favoring economically desirable traits in domestic sheep (Taheri et al., 2022). However, it is important to note that the limited sample size in this study may lead to an underestimation of the true extent of diversity.
This study used three different methods (iHS, RSB, and XP-EHH) to improve the accuracy of identifying selective sweeps. The wild sheep population showed 135 locations, with the highest iHS values on chromosomes 1, 6, 13, and 18. In contrast, the domestic sheep population had 205 regions, with the strongest signals on chromosomes 2, 3, and 7. Using XP-EHH and FST, 93 candidate genomic regions were identified as harboring putative selective sweeps by Manzari et al. (2019) in three Iranian sheep breeds. The identified signatures of selection were related to multiple candidate genes involved e in skeletal system, energy metabolism, growth, reproduction, and immune and nervous system traits. Eydivandi et al. (2021) applied FST, xp-EHH, Rsb, and FLK tests to identify selective sweeps. Their findings revealed 128, 207, 222, and 252 genomic regions, respectively, as candidates for selective sweeps. Additionally, nine overlapping candidate genes linked to disease resistance and climate adaptation were detected by all four tests. Alipanah et al. (2024) found the highest iHS coefficients under natural selection on chromosomes 3 and 2. Additionally, the XP-EHH results revealed that the highest XP-EHH coefficients under natural selection in European wild sheep, compared to Sardinian wild sheep, were observed on chromosome 3, while the reverse was true for Sardinian wild sheep compared to European wild sheep, with significant findings on chromosome 16.
By analyzing shared regions in the domestic population, we identified the genes ADGRB3, CADM1, CAPN2, COPG2, DLC1, GALNT10, HIP1R, MTR, NEGR1, NRCAM, PML, PTPRM, RELN, RTN1, SDCCAG8, and USP25. These are associated with economic traits such as longissimus muscle area, muscle weight, fecal egg count, milk yield, lean meat yield percentage, carcass fat percentage, bone density, udder attachment, meat palmitoleic acid content, somatic cell score, staple length, useful yield content, carcass bone percentage, meat polyunsaturated fatty acid content, bone weight, and hot carcass weight. ADGRB3 has been identified as possibly related to fertilization and litter size by regulating oocyte development in Hu sheep (Tao et al., 2021a). In a GWAS study, two significant SNPs within introns of this gene were found to be associated with a reduced number of parasite eggs in feces (Becker et al., 2022). ADGRB3, which is linked to the positive regulation of synapse assembly, has been identified in prolific sheep (Hernández-Montiel et al., 2022). CADM plays a crucial role in regulating embryo growth, body weight, fat metabolism, and energy balance. It is believed that this gene is involved in determining variations in body size (Xu et al., 2021). CAPN2 is expressed widely in skeletal muscle, and previous research has linked it with gene expression and meat tenderness in various species (Knight et al., 2012). Ratzka et al. (2008) have shown that the CAPN2 protein is essential for the normal growth of the preimplantation murine embryo.
COPG2 was identified in a study of receptor genes related to the progression of Johne’s disease in inbred sheep (Taylor et al., 2008). Additionally, it was found to be more active in the sheep lung and brain on the maternal side prenatally (Duan et al., 2018). Research in cattle revealed that COPG2 is expressed by both copies during fetal tissue development (Khatib, 2005).
Niciura et al. (2022) identified a link between GALNT10 and weight traits. Similarly, Al Kalaldeh et al. (2019) found that GALNT10, which is responsible for producing a sugar molecule known as mucin-type O-glycan, is associated with the ability of sheep to resist gastrointestinal parasites. Jacobs et al. (2020) revealed that HIP1R is crucial for dendrite growth in sheep brain cells. MTR contains instructions for producing an enzyme known as 5-methyltetrahydrofolate-homocysteine methyltransferase, which plays a key role in converting homocysteine into methionine—an essential building block for proteins in the body (Zhang et al., 2012). Research suggests that MTR influences both the quantity and quality of wool produced by sheep (Rong et al., 2015). NEGR1 is a cell adhesion molecule that belongs to the LON family of immunoglobulins, which also includes other molecules such as limbic system-associated membrane protein and neurotrimin (Noh et al., 2019). NEGR1 has been associated with feed efficiency in beef cattle (Seabury et al., 2017) and somatic cell score in sheep (Mohammadi et al., 2022). Specifically, NEGR1 is more prevalent in the cell adhesion molecular pathway, which plays a role in the body’s defense against disease in cattle (Liu et al., 2020). NEGR1 is a protein that can be activated as needed and is involved in the growth, specialization, and death of cells in blood vessels (Wang et al., 2021).
NRCAM is linked to cells in the nervous system of sheep and with molecules that aid cell adhesion (Alvarez-Franco et al., 2021). Recent studies have demonstrated that NRCAM, produced by uterine lining cells, can enhance the body’s response to progestin hormones by altering gene behavior (Cheng et al., 2022). In ruminants, research has revealed that a specific gene called PML plays a crucial role in inhibiting tumor growth, indicating a potentially enhanced cancer prevention system in these animals (Wang et al., 2019). Increased expression of PTPRM in small intestinal neuroendocrine tumor cells reduces cell growth and division and induces cell death (Barazeghi et al., 2019). RELN has been observed to be more expressed in susceptible animals (McRae et al., 2014). Comparing Suffolk and Texel sheep infected with Teladorsagia circumcincta, RELN was found to be more expressed in Suffolk sheep 3 days post-infection (Ahmed, 2013). Most notably, the expression of RELN is concentrated in the theca cells of dominant follicles, where it modulates downstream signaling pathways through paracrine interaction with LRP8 in granulosa cells (Fayad et al., 2007), regulating the final stages of follicle growth (Nivet et al., 2013). Studies suggest an association between RELN and protein kinase activity, contributing to progestogenic pathways, while also highlighting a negative impact of MAP due to RELN suppression. RELN, a glycoprotein in the extracellular matrix, is involved in various cellular functions, including the MAPK pathway, which is vital for germinal vesicle breakdown and oocyte maturation (Yang et al., 2018). Aboul-Naga et al. (2022) uncovered crucial genetic variations associated with heat tolerance in the gene RTN1. RTN1 is part of the reticulon family residing in the endoplasmic reticulum (ER), which is implicated in hormone release and membrane mobility in nerve cells and is closely linked to ER stress (Chiurchiu et al., 2014). In genome-wide analyses of goats and sheep, SDCCAG8 was identified as under selection, affecting reproduction and the TGF pathway, which governs the number of offspring in goats and sheep (Tao et al., 2021b). SDCCAG8 is also involved in cellular component organization (Han M. et al., 2022). Zhang et al. (2022) confirmed that USP25 is correlated with the annual reproductive cycle in sheep by investigating various sheep breeds with distinct characteristics.
Identifying common segments in the wild population allowed us to accurately locate the genes ACAN, ACO1, AK8, GADL1, GALNTL6, KCNJ10, MGST3, NF1, PLIN5, PRDM16, RALGAPA2, SPAG1, TSC1, and ZNF395. These genes were associated with several economic traits in the wild population, including muscle weight, milk yield, lean meat percentage, carcass fat percentage, bone density, somatic cell score, staple length, meat polyunsaturated fatty acid content, hot carcass weight, testes weight, milk fat percentage, reproductive seasonality, and jaw length. ACAN encodes the aggrecan protein, which is a type of proteoglycan. Aggrecan is the most abundant proteoglycan found in cartilage, a tough and flexible tissue that forms a significant part of the skeleton during early development. The majority of cartilage eventually transforms into bone through a process called ossification, with the exception of the cartilage that remains to cover and protect the ends of bones, along with that found in the nose, airways, and external ears (Mancioppi et al., 2021). ACAN mutations can cause skeletal disorders such as osteochondrosis and skeletal dysplasia, which affect height (Sabeti-Aghabozorgi et al., 2022).
Manzari et al. (2019) investigated genes related to skeletal and tail growth, noting that genes such as ACAN show signs of selection. GALNTL6 is an important gene for the ability of sheep to resist gastrointestinal parasites (Pratap et al., 2024). MGST3 is associated with muscle tissue in sheep and is important for amino acid metabolism (Thameem et al., 2003). NF1 plays a role in various cellular functions and is linked to sheep reproductive performance. It is crucial for increasing the number of lambs in Texel sheep and for adapting to high-altitude hypoxia (XU et al., 2018). PLIN5 is involved in fat metabolism and insulin regulation (Zhang et al., 2022). PRDM16 plays a role in fat cell formation and embryonic development (Chi and Cohen, 2016). SPAG1 was found to influence fertility in mammals (Faraji et al., 2021). TSC1 is linked to body size variability in sheep breeds (Cao et al., 2015), while ZNF395 is associated with growth traits and the development of fat cells. It may also play a role in obesity and metabolic syndrome. ZNF395 expression levels in Lanzhou fat-tailed sheep were notably higher than in small-tailed Han sheep (Erdenee et al., 2020).
ADAM9, ARHGAP42, and CUTC genes were identified as common genes between domestic and wild sheep species. The ADAM9 gene plays a crucial role in the nervous, cardiovascular, and muscular systems. In the nervous system, ADAM9 is involved in neuron migration, axon growth, and synapse formation (Cho, 2012). In the cardiovascular system, it contributes to the proliferation and migration of vascular endothelial cells (Chou et al., 2020). In muscle tissue, ADAM9 is essential for muscle cell proliferation and differentiation (Ahmed et al., 2017). Furthermore, another study showed significant differences in ADAM9 expression in sheep skin tissues with varying wool fineness (Tian et al., 2017). The ARHGAP42 gene is associated with hair follicle development and wool production traits in sheep (Sun et al., 2021). This gene regulates angiogenesis in cattle (Diao et al., 2019). Additionally, this gene has been linked to hypertension in a study examining the interaction between genomics and diet in adult males (Imaizumi et al., 2017). The CUTC gene was evaluated in the methane yield metagenome and metatranscriptome datasets of sheep (Kelly et al., 2019).
Quantitative trait locus analysis revealed that significant genes identified in domestic sheep populations were primarily linked to economically important traits, including body weight, bone structure, and muscle development. In contrast, candidate genes found in wild sheep populations were associated with structural and immune traits, such as reproductive behaviors and resistance to parasites, Salmonella abortusovis, and mastitis. Additionally, there were candidate genes in wild sheep related to performance traits, including body weight, muscle growth, milk yield, and milk fat. These results clearly show that selection for higher performance in domestic sheep makes them more sensitive to environmental stressors and diseases, while there is a balance between performance and biologically important traits in wild sheep population. Lv et al. (2021) demonstrated that domestic sheep may have acquired beneficial alleles of various immune and sensory genes through natural or managed hybridization with their wild sheep.
5 Conclusion
Our findings suggest that artificial selection by humans, with an emphasis on economically important traits in domestic sheep, has weakened the balance between economic and environmental traits. This balance appears to be crucial for the survival in wild sheep. By identifying potential candidate genes associated with economic and survival traits, along with their genomic regions that have undergone changes due to selection, these insights can be utilized in breeding programs for sheep. However, due to the incomplete information regarding the functional annotation of genes within sheep species and the limited sample size studied, further research with a larger number of samples is necessary to gain a deeper understanding of candidate genes for critical economic traits in both domestic and wild sheep. Advancing knowledge in this area can significantly enhance the design of effective breeding strategies.
Data availability statement
Publicly available datasets were analyzed in this study. These data can be found at: http://projects.ensembl.org/nextgen/.
Ethics statement
The animal study was approved by the Animal Ethics Committee (3/51431), Ferdowsi University of Mashad. The study was conducted in accordance with local legislation and institutional requirements.
Author contributions
ST: data curation, investigation, methodology, software, and writing–original draft. AJ: methodology, supervision, and writing–review and editing. SZ: supervision, validation, writing–original draft, and writing–review and editing.
Funding
The authors declare that financial support was received for the research, authorship, and/or publication of this article. This study was supported by the Ferdowsi University of Mashhad. This study makes use of data generated by the NextGen Consortium. The European Union’s Seventh Framework Programme (FP7/2010-2014) provided funding for the project under grant agreement no 244356 - “NextGen”.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2024.1414717/full#supplementary-material
References
Aboul-Naga, A. M., Alsamman, A. M., El Allali, A., Elshafie, M. H., Abdelsabour, T. H., Hamwieh, A., et al. (2022). The genome-wide analysis identified candidate variants and genes associated with heat stress adaptation in Egyptian sheep breeds. Front. Genet. 13, 898522. doi:10.3389/fgene.2022.898522
Ahmed, A. M. (2013). Immunological and transcriptomic analysis of genetically resistant and susceptible sheep to gastrointestinal nematodes. Dublin: University College Dublin. PhD Thesis.
Ahmed, S. I., Ibrahim, M. E., and Khalil, E. A. (2017). High altitude and pre-eclampsia: adaptation or protection. Med. Hypotheses 104, 128–132.
Alberto, F. J., Boyer, F., Orozco-terWengel, P., Streeter, I., Servin, B., De Villemereuil, P., et al. (2018). Convergent genomic signatures of domestication in sheep and goats. Nat. Commun. 9 (1), 813. doi:10.1038/s41467-018-03206-y
Alipanah, M., Mazloom, S. M., and Gharari, F. (2024). Detection of selective sweep in European wild sheep breeds. 3Biotech 14 (122), 122. doi:10.1007/s13205-024-03964-1
Al Kalaldeh, M., Gibson, J., Lee, S. H., Gondro, C., and Van Der Werf, J. H. (2019). Detection of genomic regions underlying resistance to gastrointestinal parasites in Australian sheep. Genet. Sel. Evol. 51, 37–18. doi:10.1186/s12711-019-0479-1
Alvarez-Franco, A., Rouco, R., Ramirez, R. J., Guerrero-Serna, G., Tiana, M., Cogliati, S., et al. (2021). Transcriptome and proteome mapping in the sheep atria reveal molecular featurets of atrial fibrillation progression. Cardiovasc. Res. 117 (7), 1760–1775. doi:10.1093/cvr/cvaa307
Barazeghi, E., Hellman, P., Westin, G., and Stålberg, P. (2019). PTPRM, a candidate tumor suppressor gene in small intestinal neuroendocrine tumors. Endocr. Connect. 8 (8), 1126–1135. doi:10.1530/EC-19-0279
Becker, G. M., Burke, J. M., Lewis, R. M., Rosen, B. D., Van Tassell, C. P., Notter, D. R., et al. (2022). Variants within genes EDIL3 and ADGRB3 are associated with divergent fecal egg counts in Katahdin sheep at weaning. Front. Genet. 13, 817319. doi:10.3389/fgene.2022.817319
Bindea, G., Mlecnik, B., Hackl, H., Charoentong, P., Tosolini, M., Kirilovsky, A., et al. (2009). ClueGO: a Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics 25 (8), 1091–1093. doi:10.1093/bioinformatics/btp101
Cao, J., Wei, C., Liu, D., Wang, H., Wu, M., Xie, Z., et al. (2015). DNA methylation Landscape of body size variation in sheep. Sci. Rep. 5 (1), 13950. doi:10.1038/srep13950
Cheng, Y., Xie, L., Xu, Z., Hao, M., Yang, B., Shan, W., et al. (2022). NrCAM secreted by endometrial stromal cells enhances the progestin sensitivity of endometrial cancer cells through epigenetic modulation of PRB. Cancer Gene Ther. 29 (10), 1452–1462. doi:10.1038/s41417-022-00467-0
Chi, J., and Cohen, P. (2016). The multifaceted roles of PRDM16: adipose biology and beyond. Trends Endocrinol. Metabolism 27 (1), p11–p23. doi:10.1016/j.tem.2015.11.005
Chiurchiu, V., Maccarrone, M., and Orlacchio, A. (2014). The role of reticulons in neurodegenerative diseases. Neuromol. Med. 16, 3–15. doi:10.1007/s12017-013-8271-9
Cho, C. (2012). Testicular and epididymal ADAMs: expression and function during fertilization. Nat.Rev. Urol. 9 (10), 550–560.
Chou, C. W., Huang, Y. K., Kuo, T. T., Liu, J. P., and Sher, Y. P. (2020). An overview of ADAM9: structure, activation, and regulation in human diseases. Int. J. Mol. Sci. 21 (20), 7790.
Diao, S., Huang, S., Xu, Z., Ye, S., Yuan, X., and Chen, Z. (2019). Genetic diversity of indigenous pigs from South China area revealed by SNP array. Animals 9 (6), 361.
Dong, Y., Xie, M., Jiang, Y. U., Xiao, N., Du, X., Zhang, W., et al. (2013). Sequencing and automated whole-genome optical mapping of the genome of a domestic goat (Capra hircus). Nat. Biotechnol. 31 (2), 135–141. doi:10.1038/nbt.2478
Duan, J., Zhang, M., Flock, K., Seesi, S. A., Mandoiu, I., Jones, A., et al. (2018). Effects of maternal nutrition on the expression of genomic imprinted genes in ovine fetuses. Epigenetics 13 (8), 793–807. doi:10.1080/15592294.2018.1503489
Erdenee, S., Li, J., Kang, Z., Xu, H., Zang, R., Cao, X., et al. (2020). Sheep zinc finger proteins 395 (ZNF395): insertion/deletion variations, associations with growth traits, and mRNA expression. Anim. Biotechnol. 31 (3), 237–244. doi:10.1080/10495398.2019.1585865
Eydivandi, S., Roudbar, M. A., Ardestani, S. S., Momen, M., and Sahana, G. (2021). A selection signatures study among Middle Eastern and European sheep breeds. J. Anim. Breed. Genet. 138 (5), 574–588. doi:10.1111/jbg.12536
Faraji, S., Sharafi, M., Shahverdi, A. H., and Fathi, R. (2021). Sperm associated antigens: vigorous influencers in life. Cell J. 23 (5), 495–502. doi:10.22074/cellj.2021.7377
Fayad, T., Lefebvre, R., Nimpf, J., Silversides, D. W., and Lussier, J. G. (2007). Low-density lipoprotein receptor-related protein 8 (LRP8) is upregulated in granulosa cells of bovine dominant follicle: molecular characterization and spatio-temporal expression studies. Biol. Reprod. 76 (3), 466–475. doi:10.1095/biolreprod.106.057216
Flori, L., Fritz, S., Jaffrézic, F., Boussaha, M., Gut, I., Heath, S., et al. (2009). The genome response to artificial selection: a case study in dairy cattle. PloS. One. 4 (8), e6595. doi:10.1371/journal.pone.0006595
Gautier, M., and Vitalis, R. (2012). rehh: an R package to detect footprints of selection in genome-wide SNP data from haplotype structure. Bioinformatics 28 (8), 1176–1177. doi:10.1093/bioinformatics/bts115
Hamadalahmad, A., Almeziad, M., and Javadmanesh, A. (2020). Genetic similarity comparison between some Iranian and Middle Eastern sheep breeds using mitochondrial control region sequencing. Dyson. Life Sci. 1 (1), 20–24. doi:10.30493/dls.2020.105085
Han, M., Liang, C., Liu, Y., He, X., and Chu, M. (2022). Integrated transcriptome analysis reveals the crucial mRNAs and miRNAs related to Fecundity in the hypothalamus of yunshang black goats during the luteal phase. Animals 12 (23), 3397. doi:10.3390/ani12233397
Hayes, B. J., Lien, S., Nilsen, H., Olsen, H. G., Berg, P., MacEachern, S., et al. (2008). The origin of selection signatures on bovine chromosome 6. Anim. Genet. 39 (2), 105–111. doi:10.1111/j.1365-2052.2007.01683.x
Hernández-Montiel, W., Cob-Calan, N. N., Cahuich-Tzuc, L. E., Rueda, J. A., Quiroz-Valiente, J., Meza-Villalvazo, V., et al. (2022). Runs of homozygosity and gene identification in pelibuey sheep using genomic data. Diversity 14 (7), 522. doi:10.3390/d14070522
Hu, Z. L., Park, C. A., and Reecy, J. M. (2022). Bringing the animal QTLdb and CorrDB into the future: meeting new challenges and providing updated services. Nucleic. acids. Res. 50 (D1), D956–D961. doi:10.1093/nar/gkab1116
Hui, T., Zheng, Y., Yue, C., Wang, Y., Bai, Z., Sun, J., et al. (2021). Screening of cashmere fineness-related genes and their ceRNA network construction in cashmere goats. Sci. Rep. 11 (1), 21977. doi:10.1038/s41598-021-01203-8
Imaizumi, T., Ando, M., Nakatochi, M., Maruyama, S., Yasuda, Y., Honda, H., et al. (2017). Association of interactions between dietary salt consumption and hypertension-susceptibility genetic polymorphisms with blood pressure among Japanese male workers. Clin. Exp. Nephrol. 21, 457–464.
Jacobs, A., Hagin, M., Shugol, M., Shomron, N., Pillar, N., Fañanás, L., et al. (2020). The black sheep of the family-whole-exome sequencing in family of lithium response discordant bipolar monozygotic twins. Eur. Neuropsychopharmacol. 34, 19–27. doi:10.1016/j.euroneuro.2020.03.009
Javadmanesh, A., Ghovvati Rodsari, S., Soltani, M., and Nassiry, M. (2022). Genetic diversity of urial population in Northeast of Iran. Iran. J. Anim. Biosyst. 18 (2), 185–193. doi:10.22067/ijab.2022.74490.1024
Kelly, W. J., Leahy, S. C., Kamke, J., Soni, P., Koike, S., and Mackie, R. (2019). Occurrence and expression of genes encoding methyl-compound production in rumen bacteria. Anim. Microbiome 1, 1–13.
Khatib, H. (2005). The COPG2, DCN, and SDHD genes are biallelically expressed in cattle. Mamm. Genome. 16, 545–552. doi:10.1007/s00335-005-0029-8
Kijas, J. W., Lenstra, J. A., Hayes, B., Boitard, S., Porto Neto, L. R., San Cristobal, M., et al. (2012). Genome-wide analysis of the world's sheep breeds reveals high levels of historic mixture and strong recent selection. PLoS. Biol. 10 (2), e1001258. doi:10.1371/journal.pbio.1001258
Kijas, J. W., Townley, D., Dalrymple, B. P., Heaton, M. P., Maddox, J. F., McGrath, A., et al. (2009). A genome wide survey of SNP variation reveals the genetic structure of sheep breeds. PloS. One. 4 (3), e4668. doi:10.1371/journal.pone.0004668
Knight, M. I., Daetwyler, H. D., Hayes, B. J., Hayden, M. J., Ball, A. J., Pethick, D. W., et al. (2012). Discovery and trait association of single nucleotide polymorphisms from gene regions of influence on meat tenderness and long-chain omega-3 fatty acid content in Australian lamb. Anim. Prod. Sci. 52 (7), 591–600. doi:10.1071/AN11229
Kreitman, M. (2000). Methods to detect selection in populations with applications to the human. Annu. Rev. Genomics Hum. Genet. 1 (1), 539–559. doi:10.1146/annurev.genom.1.1.539
Liu, Y., Xu, L., Yang, L., Li, J., Li, Y., Liu, D., et al. (2020). Discovery of genomic characteristics and selection signatures in Southern Chinese local cattle. Front. Genet. 11, 533052. doi:10.3389/fgene.2020.533052
Lv, F. H., Cao, Y. H., Liu, G. -J., Luo, L. -Y., Lu, R., Liu, M. -J., et al. (2021). Whole-genome resequencing of worldwide wild and domestic sheep elucidates genetic diversity, introgression, a agronomically important loci. Mol. Biol. Evol. 39 (2), msab353. doi:10.1093/molbev/msab353
Mancioppi, V., Prodam, F., Mellone, S., Ricotti, R., Giglione, E., Grasso, N., et al. (2021). Retrospective diagnosis of a novel ACAN pathogenic variant in a family with short stature: a case report and review of the literature. Front. Genet. 12, 708864. doi:10.3389/fgene.2021.708864
Manzari, Z., Mehrabani-Yeganeh, H., Nejati-Javaremi, A., Moradi, M. H., and Gholizadeh, M. (2019). Detecting selection signatures in three Iranian sheep breeds. Anim. Genet. 50 (3), 298–302. doi:10.1111/age.12772
McRae, K. M., McEwan, J. C., Dodds, K. G., and Gemmell, N. J. (2014). Signatures of selection in sheep bred for resistance or susceptibility to gastrointestinal nematodes. Bmc. Genomics 15, 637. doi:10.1186/1471-2164-15-637
Mohammadi, H., Farahani, A. H. K., Moradi, M. H., Mastrangelo, S., Di Gerlando, R., Sardina, M. T., et al. (2022). Weighted single-step genome-wide association study uncovers known and novel candidate genomic regions for milk production traits and somatic cell score in Valle del Belice dairy sheep. Animals 12 (9), 1155. doi:10.3390/ani12091155
Niciura, S. C. M., Benavides, M. V., Okino, C. H., Ibelli, A. M. G., Minho, A. P., Esteves, S. N., et al. (2022). Genome-wide association study for Haemonchus contortus resistance in Morada Nova sheep. Pathogens 11 (8), 939. doi:10.3390/pathogens11080939
Nivet, A. L., Vigneault, C., Blondin, P., and Sirard, M. A. (2013). Changes in granulosa cells' gene expression associated with increased oocyte competence in bovine. Reproduction 145 (6), 555–565. doi:10.1530/REP-13-0032
Noh, K., Lee, H., Choi, T. Y., Joo, Y., Kim, S. J., Kim, H., et al. (2019). Negr1 controls adult hippocampal neurogenesis and affective behaviors. Mol. Psychiatry. 24 (8), 1189–1205. doi:10.1038/s41380-018-0347-3
Pratap, R., Chennuru, S., Krovvidi, S., Chitithoti, J., and Pentala, R. K. (2024). Putative SNPs in Ovar-DRB1 and GALNTL6 genes conferring susceptibility to natural infection of haemonchus contortus in Southern Indian sheep. Acta Parasitol. 69 (1), 583–590. doi:10.1007/s11686-023-00778-8
Purcell, S., Neale, B., Todd-Brown, K., Thomas, L., Ferreira, M. A., Bender, D., et al. (2007). PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 81 (3), 559–575. doi:10.1086/519795
Ratzka, A., Kalus, I., Moser, M., Dierks, T., Mundlos, S., and Vortkamp, A. (2008). Redundant function of the heparan sulfate 6-O-endosulfatases Sulf1 and Sulf2 during skeletal development. Dev. Dyn. 237 (2), 339–353. doi:10.1002/dvdy.21423
Rong, E. G., Yang, H., Zhang, Z. W., Wang, Z. P., Yan, X. H., Li, H., et al. (2015). Association of methionine synthase gene polymorphisms with wool production and quality traits in Chinese Merino population. J. Anim. Sci. 93 (10), 4601–4609. doi:10.2527/jas.2015-8963
Sabeti, P. C., Reich, D. E., Higgins, J. M., Levine, H. Z., Richter, D. J., Schaffner, S. F., et al. (2002). Detecting recent positive selection in the human genome from haplotype structure. Nature 419 (6909), 832–837. doi:10.1038/nature01140
Sabeti, P. C., Varilly, P., Fry, B., Lohmueller, J., Hostetter, E., Cotsapas, C., et al. (2007). Genome-wide detection and characterization of positive selection in human populations. Nature 449 (7164), 913–918. doi:10.1038/nature06250
Sabeti-Aghabozorgi, A., Ahangari, N., Mobini, M., Vakili, R., Doosti, M., Vakili, S., et al. (2022). A novel heterozygous ACAN variant in an Iranian family with short stature: a case report. Int. J. Pediatr. 10 (9), 16729–16737. doi:10.22038/ijp.2022.63946.4859
Saito, R., Smoot, M. E., Ono, K., Ruscheinski, J., Wang, P. L., Lotia, S., et al. (2012). A travel guide to Cytoscape plugins. Nat. Methods. 9 (11), 1069–1076. doi:10.1038/nmeth.2212
Schlötterer, C. (2003). Hitchhiking mapping–functional genomics from the population genetics perspective. Trends. Genet. 19 (1), 32–38. doi:10.1016/S0168-9525(02)00012-4
Seabury, C. M., Oldeschulte, D. L., Saatchi, M., Beever, J. E., Decker, J. E., Halley, Y. A., et al. (2017). Genome-wide association study for feed efficiency and growth traits in US beef cattle. Bmc. Genomics. 18, 386. doi:10.1186/s12864-017-3754-y
Sun, W., Ma, S., and Ma, Y. (2021). On hair follicle development and wool production traits in sheep: a review. Intl. J. Agric. Biol. 25, 450–454. doi:10.17957/IJAB/15.1687
Taheri, S., Saedi, N., Zerehdaran, S., and Javadmanesh, A. (2023). Identification of selection signatures in Capra hircus and Capra aegagrus in Iran. Anim. Sci. J. 94 (1), e13864. doi:10.1111/asj.13864
Taheri, S., Zerehdaran, S., and Javadmanesh, A. (2022). Genetic diversity in some domestic and wild sheep and goats in Iran. Small. Rum. Res. 209, 106641. doi:10.1016/j.smallrumres.2022.106641
Tang, K., Thornton, K. R., and Stoneking, M. (2007). A new approach for using genome scans to detect recent positive selection in the human genome. PLoS. Biol. 5 (7), e171. doi:10.1371/journal.pbio.0050171
Tao, L., He, X., Jiang, Y., Liu, Y., Ouyang, Y., Shen, Y., et al. (2021a). Genome-wide analyses reveal genetic convergence of prolificacy between goats and sheep. Genes 12 (4), 480. doi:10.3390/genes12040480
Tao, L., He, X., Wang, F., Zhong, Y., Pan, L., Wang, X., et al. (2020). Luzhong mutton sheep: inbreeding and selection signatures. J. Anim. Sci. Technol. 62 (6), 777–789. doi:10.5187/jast.2020.62.6.777
Tao, L., He, X., Wang, X., Di, R., and Chu, M. (2021b). Litter size of sheep (Ovis aries): inbreeding depression and homozygous regions. Genes 12 (1), 109. doi:10.3390/genes12010109
Taylor, D. L., Zhong, L., Begg, D. J., de Silva, K., and Whittington, R. J. (2008). Toll-like receptor genes are differentially expressed at the sites of infection during the progression of Johne's disease in outbred sheep. Vet. Immunol. Immunopathol. 124 (1-2), 132–151. doi:10.1016/j.vetimm.2008.02.021
Thameem, F., Yong, X., Permana, P. A., Wolford, J. K., Bogardus, C., and Prochazka, M. (2003). Evaluation of the microsomal glutathione S-transferase 3 (MGST3) locus on 1q23 as a Type 2 diabetes susceptibility gene in Pima Indians. Hum. Genet. 113 (4), 353–358. doi:10.1007/s00439-003-0980-y
Tian, Y. Z., Usman, T., Tian, K. C., Di, J., Huang, X. X., and Xu, X. M. (2017). Comparative study of 13 candidate genes applying multi-reference normalization to detect the expression of different fineness in skin tissues of wool sheep. Genet. Mol. Res. 16 (10.4238).
Voight, B. F., Kudaravalli, S., Wen, X., and Pritchard, J. K. (2006). A map of recent positive selection in the human genome. PLoS. Biol. 4 (3), e72. doi:10.1371/journal.pbio.0040072
Wang, B., Guo, H., Yu, H., Chen, Y., Xu, H., and Zhao, G. (2021). The role of the transcription factor EGR1 in cancer. Front. Oncol. 11, 642547. doi:10.3389/fonc.2021.642547
Wang, Y., Zhang, C., Wang, N., Li, Z., Heller, R., Liu, R., et al. (2019). Genetic basis of ruminant headgear and rapid antler regeneration. Science 364 (6446), eaav6335. doi:10.1126/science.aav6335
Xu, S. S., Gao, L., and Lyu, F. (2021). Whole-genome selective scans detect genes associated with important phenotypic traits in sheep (Ovis aries). Front. Genet. 12, 738879. doi:10.3389/fgene.2021.738879
Xu, S. S., Gao, L., Ren, Y. L., Wang, F., Eypórsdóttir, E., Hallsson, J. H., et al. (2018). Genome-wide association analyses highlight the potential for different genetic mechanisms for litter size among sheep breeds. Front. Genet. 9, 340573. doi:10.3389/fgene.2018.00118
Yang, F., Wang, M., Zhang, B., Xiang, W., Zhang, K., Chu, M., et al. (2018). Identification of new progestogen-associated networks in mammalian ovulation using bioinformatics. Bmc. Syst. Biol. 12, 36–12. doi:10.1186/s12918-018-0577-7
Zeder, M. A. (1999). Animal domestication in the Zagros: a review of past and current research. Paléorient 25, 11–25. doi:10.3406/paleo.1999.4684
Zeder, M. A. (2008). Domestication and early agriculture in the Mediterranean Basin: origins, diffusion, and impact. Proc. Nation. Acad. Sci. 105 (33), 11597–11604. doi:10.1073/pnas.0801317105
Zeraatpisheh, Y., Zerehdaran, S., and Javadmanesh, A. (2023). Investigation of metabolic pathways related to the QTLs of parasite resistance trait in sheep genome using gene network and gene ontology. Vet. Res. Biolo. Prod. 36 (1), 83–90. In Persian. doi:10.22092/vj.2022.357660.1941
Zhang, C. L., Liu, C., Zhang, J., Zheng, L., Chang, Q., Cui, Z., et al. (2022). Analysis on the desert adaptability of indigenous sheep in the southern edge of Taklimakan Desert. Sci. Rep. 12 (1), 12264. doi:10.1038/s41598-022-15986-x
Zhang, Z., Tian, C., Zhou, S., Wang, W., Guo, Y., Xia, J., et al. (2012). Mechanism-based design, synthesis and biological studies of N⁵-substituted tetrahydrofolate analogs as inhibitors of cobalamin-dependent methionine synthase and potential anticancer agents. Eur. J. Med. Chem. 58, 228–236. doi:10.1016/j.ejmech.2012.09.027
Keywords: domestic and wild sheep, iHS, RSB, selective sweep, QTL, XP-EHH
Citation: Taheri S, Javadmanesh A and Zerehdaran S (2024) Identification of selective sweep and associated QTL traits in Iranian Ovis aries and Ovis orientalis populations. Front. Genet. 15:1414717. doi: 10.3389/fgene.2024.1414717
Received: 09 April 2024; Accepted: 19 November 2024;
Published: 19 December 2024.
Edited by:
Fei Hao, Northumbria University, United KingdomReviewed by:
Yingpeng Han, Northeast Agricultural University, ChinaKalpana Singh, Guru Angad Dev Veterinary and Animal Sciences University, India
Copyright © 2024 Taheri, Javadmanesh and Zerehdaran. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Saeed Zerehdaran, emVyZWhkYXJhbkB1bS5hYy5pcg==