 Larissa Nascimento Antunes1Alex Marcel Moreira Dias1Beatriz Cetalle Schiavo1Beatriz C. A. Mendes2Debora Romeo Bertola1,3
Larissa Nascimento Antunes1Alex Marcel Moreira Dias1Beatriz Cetalle Schiavo1Beatriz C. A. Mendes2Debora Romeo Bertola1,3 Karina Lezirovitz4
Karina Lezirovitz4 Regina Célia Mingroni-Netto1*
Regina Célia Mingroni-Netto1*- 1Centro de Pesquisas sobre o Genoma Humano e Células-Tronco, Departamento de Genética e Biologia Evolutiva, Instituto de Biociências, Universidade de São Paulo, São Paulo, Brazil
- 2Divisão de Educação e Reabilitação dos Distúrbios da Comunicação da Pontifícia Universidade Católica de São Paulo, São Paulo, Brazil
- 3Unidade de Genética do Instituto da Criança, Faculdade de Medicina, Universidade de São Paulo, São Paulo, Brazil
- 4Laboratório de Otorrinolaringologia/LIM32 - Hospital das Clínicas, Faculdade de Medicina, Universidade de São Paulo, São Paulo, Brazil
Introduction: Hearing loss is a frequent sensory impairment type in humans, with about 50% of prelingual cases being attributed to genetic factors. Autosomal recessive hearing loss (ARHL) exhibits great locus heterogeneity and is responsible for 70%–80% of hereditary nonsyndromic cases.
Methods: A total of 90 unrelated Brazilian individuals were selected for having hearing loss of presumably autosomal recessive inheritance, either born from consanguineous marriages or belonging to families with two or more affected individuals in the sibship and most cases were of normal hearing parents. In all cases, common pathogenic variants in GJB2 (c.35delG), GJB6 [del(GJB6-D13S1830) and del(GJB6-D13S1854)] and MT-RNR1 (m.1555A>G) were discarded and most were previously assessed by complete Sanger sequencing of GJB2. Their genetic material was analyzed through next-generation sequencing, targeting 99 hearing loss-related genes and/or whole exome sequencing.
Results: In 32 of the 90 probands (36,7%) causative variants were identified, with autosomal recessive inheritance confirmed in all, except for two cases due to dominant variants (SIX1 and P2RX2). Thirty-nine different causative variants were found in 24 different known hearing loss-associated genes, among which 10 variants are novel, indicating wide genetic heterogeneity in the sample, after exclusion of common pathogenic variants. Despite the genetic heterogeneity, some genes showed greater contribution: GJB2, CDH23, MYO15A, OTOF, and USH2A.
Conclusion: The present results confirmed that next-generation sequencing is an effective tool for identifying causative variants in autosomal recessive hearing loss. To our knowledge, this is the first report of next-generation sequencing being applied to a large cohort of pedigrees with presumable autosomal recessive hearing loss in Brazil and South America.
1 Introduction
Hearing loss is a frequent type of sensory impairment in humans. In developed countries, around 50% of cases are due to genetic alterations (Morton and Nance, 2006), and 70% are nonsyndromic. Estimates of the prevalence of hearing loss in Brazil are scarce. Brazil has older newborn hearing screening programs, when compared to other countries in Latin America, but the law determining that Universal Neonatal Hearing Screening is mandatory for all children was approved only in 2010 (Lezirovitz and Mingroni-Netto, 2022). Even after that, coverage of the screening is incomplete and heterogeneous in different regions of the country, making it difficult to obtain precise estimates of the prevalence of hearing loss among newborns (Dias et al., 2024). The Health Ministry of Brazil presented as 24% the coverage index of newborn hearing screening. Absence of a public policy, national coordination and a national database for newborn screening makes it difficult to estimate how many children are submitted to the hearing screening in the proper time (Neumann et al., 2020).
Hereditary hearing loss displays high heterogeneity in inheritance patterns, locus, alleles, and phenotypes (Shearer et al., 2020). Autosomal recessive hearing loss is responsible for 70%–80% of hereditary nonsyndromic cases (Shearer et al., 2020) and exhibits impressive locus heterogeneity. Causative genetic variants in 83 genes have been related to nonsyndromic autosomal recessive hearing loss (Walls et al., 2024). Syndromic hearing loss represents 30% of genetic cases and is equally genetically heterogeneous.
Pathogenic variants within the DFNB1 locus (13q11-12), which contains the genes GJB2 and GJB6, are estimated to explain near 50% of all cases of autosomal recessive hearing loss in many different countries and regions. (Guilford et al., 1994; Kenneson et al., 2002; Smith and Jones, 2016). The GJB2 (Gap Junction Beta 2) gene (OMIM# 121011) encodes connexin (Cx) 26 (Cx26), a member of the large family of gap junction proteins (Nielsen et al., 2012). Among all known GJB2 pathogenic variants, c.35del:p.(Gly12ValfsTer2), NC_000013.11, OMIM *121011.005) is the most frequent, corresponding to near 75% of all reported alleles with a DFNB1 pathogenic variant in European or European derived populations.
The GJB6 gene encoding connexin 30 (Cx30) is adjacent to the GJB2 gene in the DFNB1 locus. Large deletions encompassing the GJB6 gene or neighbouring regions have been reported either in homozygosis or in trans with a single recessive mutation in the GJB2 gene determining hearing loss. These GJB6-related deletions act as recessive alleles with GJB2 variants (del Castillo et al., 2002; del Castillo et al., 2005).
In Brazil, 10%–15% of nonsyndromic cases were found to be caused by variants in GJB2 and GJB6 related deletions (Batissoco et al., 2009; Batissoco et al., 2022). Due to the high locus heterogeneity, it has been difficult, especially in developing countries, to screen for other genes that are related to autosomal recessive hearing loss. However, some studies from the United States, Belgium, Northern Europe, and East Asia indicated that pathogenic variants in SLC26A4, MYO15A, OTOF, and CDH23 are probably the most common causes of autosomal recessive hearing loss, after the exclusion of variants in the DFNB1 locus (Hilgert et al., 2009; Duman and Tekin, 2012).
For many years, genetic testing for hearing loss was not a trivial issue. The development of next-generation sequencing (NGS) brought a new perspective to it (Shearer and Smith, 2012). This technique allows the simultaneous sequencing of several genes. Its use in genetically heterogeneous conditions, such as hearing loss, is effective in molecular investigation, and it is recommended for all subjects after screening for variants in the DFNB1 locus, with normal results (Alford et al., 2014; Wright, Fitzpatrick, Firth, 2018). However, it is still not affordable for the majority of the population in developing countries. Thus, its application to big research cohorts may help prove its effectiveness for other neglected populations.
This study aimed to use NGS to contribute to the understanding of the genetic heterogeneity in autosomal recessive hearing loss, in a Brazilian casuistic selected for having hearing loss of presumptive autosomal recessive inheritance.
2 Material and methods
2.1 Subjects
A total of 90 unrelated Brazilian individuals were selected for having hearing loss of presumptive autosomal recessive inheritance. They were referred by the following institutions: DERDIC (Divisão de Educação e Reabilitação de Distúrbios Auditivos da Comunicação da Pontifícia Universidade Católica), Hospital das Clínicas da Faculdade de Medicina da USP e UNIFESP (Universidade Federal de São Paulo), all in the state of São Paulo, and other institutions. They were ascertained between 2000 and 2019 at the Human Genome and Stem Cell Research Center, Instituto de Biociências, Universidade de São Paulo, São Paulo, SP, Brazil. They were selected based on pedigree criteria, being either born from consanguineous couples or belonging to families with two or more affected individuals in the sibship and most cases were of normal-hearing parents. Although most probands presented with non-syndromic hearing loss, individuals with suspected syndromic deafness were not excluded, as long as they followed at least one of the previous criteria. The study was approved by the Ethics Committee, Institute of Biosciences, University of São Paulo (1.133.416–23/06/2015). Written informed consent was obtained from all hearing-impaired individuals, and relatives, or their legal guardians. Information about secondary actionable findings was not offered to patients because samples were collected in research focused on hearing loss, and written informed consent authorized investigation of genetic factors related to hearing loss, exclusively.
In all probands, the molecular cause of hearing loss had been previously investigated by screening of the c.35delG at GJB2 (NM_004004.5, GRC37) (Scott et al., 1998), screening of two GJB6 deletions, del(GJB6-D13S1830) – chr13: 20,797,177–21,105,967:GRCh37 ∼309Kb, and del(GJB6-D13S1854) – chr13: 20,802,726–21,034,755:GRCh37 ∼232 Kb (Del Castillo et al., 2005), and screening of m.1555A>G at MT-RNR1 (NC_012920.1) (Estivil et al., 1998). Most probands also had the coding region of GJB2 previously analyzed through Sanger sequencing. Only individuals without an identified molecular cause for hearing loss after this screening were included. However, four cases identified with hypomorphic variants in GJB2 were included in order to verify if there was another plausible genetic cause for their hearing loss.
2.2 DNA extraction
Blood samples were collected and DNA was extracted using different techniques available at the time of collection of samples: conventional phenol/chloroform extraction, commercial kits, or through the equipment Autopure LS (Gentra Systems, Minneapolis, United States) or QiaSimphony (Qiagen, Hilden, Germany).
2.3 Next-generation sequencing genes panel
The custom panel was designed using the SureDesign tool (https://earray.chem.agilent.com/suredesign/) and the DNA libraries were prepared according to the SureSelect QXT Target Enrichment System Kit for Illumina Multiplexed Sequencing (Agilent, Santa Clara, CA) protocol. Sequencing was performed on the HiSeq 2500 platform (Illumina, San Diego, California, United States). Sequences were aligned with the Burrows-Wheeler Aligner (BWA) (http://bio-bwa.sourceforge.net/). The Picard tool (http://broadinstitute.github.io/picard/) was used to eliminate PCR duplicates, identify indels, and realign reads. Recalibration of base qualities and variant calling were performed using Genome Analysis ToolKit (GATK) (https://github.com/broadinstitute/gatk) and the annotation of variants was performed using Annovar (http://annovar.openbioinformatics.org/en/latest/). The list of 99 genes is presented in Supplementary Material, in Supplementary Table S1.
2.4 Whole exome sequencing
Libraries were captured and constructed through one of the following kits: Nextera Rapid Capture (Illumina, San Diego, California, United States), TruSeq Exome Library Prep Kit (Illumina, San Diego, California, United States), SureSelectQXT Target Enrichment (Agilent Technologies, Santa Clara, California, United States) and xGen Exome Research Panel v1 (Integrated DNA Technologies, Coralville, Iowa, United States). Sequencing was performed on the HiSeq 2500 platform (Illumina, San Diego, California, United States). Bioinformatics procedures were the same as previously described for the next-generation targeted sequencing of 99 genes.
2.5 Variant filtering, interpretation, and classification
Exonic and splicing variants were filtered considering their minor allele frequency under 1% in the following databases: Exome Aggregation Consortium (ExAC, exac.broadinstitute.org), NHLBI Exome Sequencing Project (ESP, http://evs.gs.washington.edu/EVS/), 1000 Genomes Project (www.internationalgenome.org), and the Brazilian ABraOM (http://abraom.ib.usp.br). The pathogenicity of the candidate variants was predicted through REVEL, CADD Phred, and for splicing variants through MAX EntScan, ADA Score, RF Score, and SpliceAI. Pathogenicity information was checked at ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/) and Deafness Variation Database (http://deafnessvariationdatabase.org/). The GRC37 assembly was used as a reference.
The whole exome sequencing analysis focused on variants present in genes previously associated with hearing loss included at “The Hereditary Hearing Loss Homepage” (https://hereditaryhearingloss.org/), Online Mendelian Inheritance in Man (OMIM, https://www.omim.org/phenotypicSeriesTitles/all) or “Deafness Variation Database”.
Candidate variants were classified according to the American College of Medical Genetics and Genomics (ACMG) and Association for Molecular Pathology (AMP) guidelines for genetic hearing loss (Richards et al., 2015; Oza et al., 2018).
2.6 Sanger sequencing
Sanger sequencing was used to confirm the presence of candidate variants and, whenever DNA samples of family members were available, to determine segregation and trans configuration. This information was used in the ACMG classification. Primers to amplify exons containing candidate variants were designed using Primer-BLAST (https://www.ncbi.nlm.nih.gov/tools/primer-blast/). PCR products were purified using Exonuclease I (Affymetrix, Santa Clara, California, United States) and Shrimp Alkaline Phosphatase (Affymetrix, Santa Clara, California, United States). The purified products were prepared using BigDye terminator v3.1 (Thermo Fisher Scientific, Waltham, Massachusetts, United States) and analyzed with the 48-capillary ABI 3730 DNA Analyser (Thermo Fisher Scientific, Waltham, Massachusetts, United States).
3 Results
3.1 Cohort characterization
Among the 90 probands, 38% (34/90) were from families with two or more affected individuals in the sibship, 44% (40/90) were born from consanguineous couples, and 18% (16/90) fulfilled both criteria. Nine of the 90 probands had been previously clinically diagnosed with Usher syndrome (Figure 1A).
Forty-six probands had their genetic material analyzed through next-generation targeted sequencing of 99 hearing loss-related genes (Supplementary Material). Due to the discovery of new genes associated with hearing loss that resulted in the need to update repeatedly the customized panel, 63 probands had their genetic material analyzed through whole exome sequencing: 19 probands without identified causative variants identified after the next-generation targeted sequencing of 99 genes panel and 44 additional probands, that had not been previously studied through NGS technologies.
In 32 of the 90 probands (36%) the causative variants were identified: 18 probands after next-generation targeted sequencing of 99 genes, 11 probands after whole exome sequencing and three cases whose causative variants were found after both (Tables 1, 2; Figure 1A). Pedigrees and a summary of the investigation of segregation in families are shown in Supplementary Figure S1. Tables 1, 2 and Figure 1A summarize the identified variants through all the strategies.
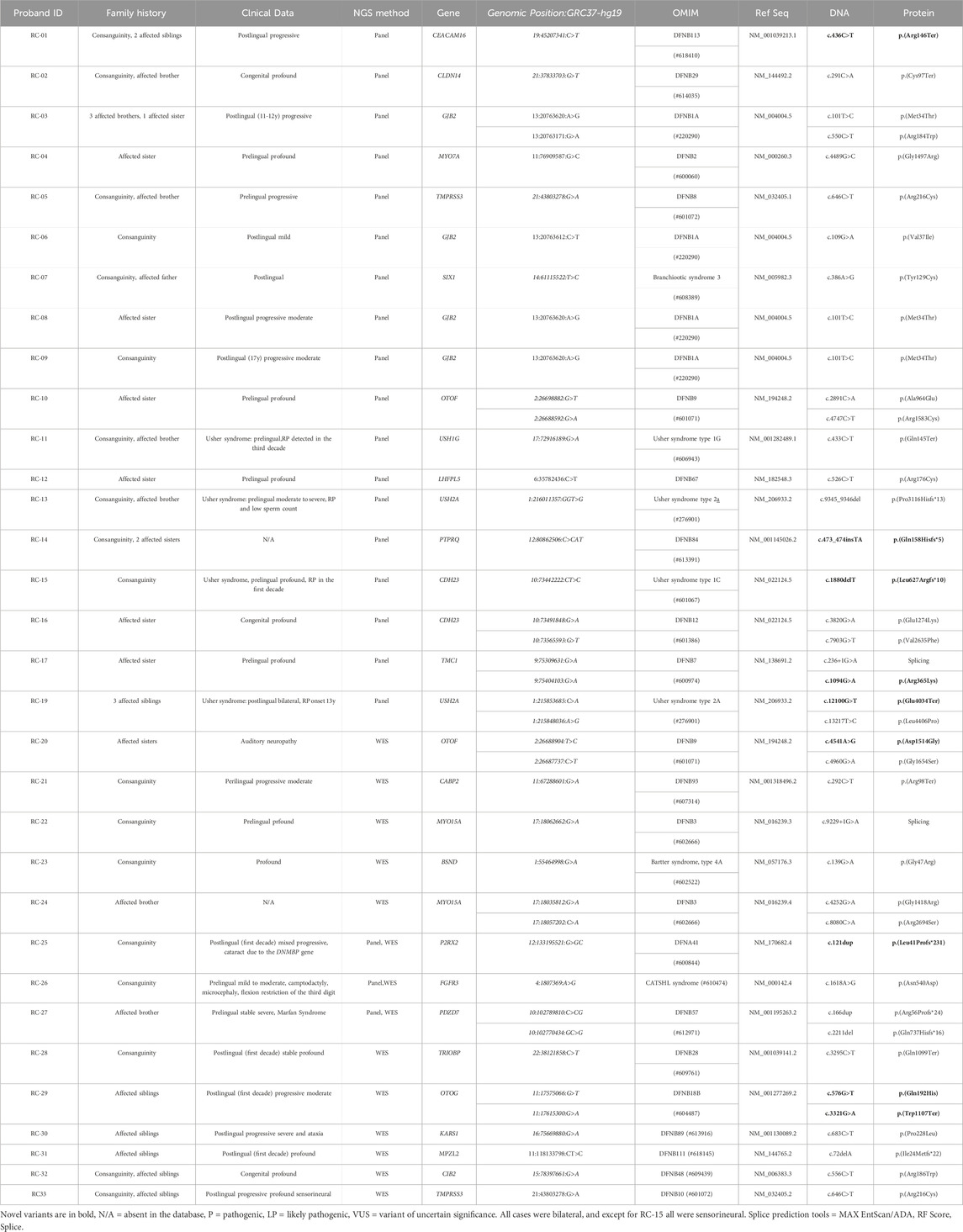
Table 1. Description of the probands phenotype and the causative variants identified.
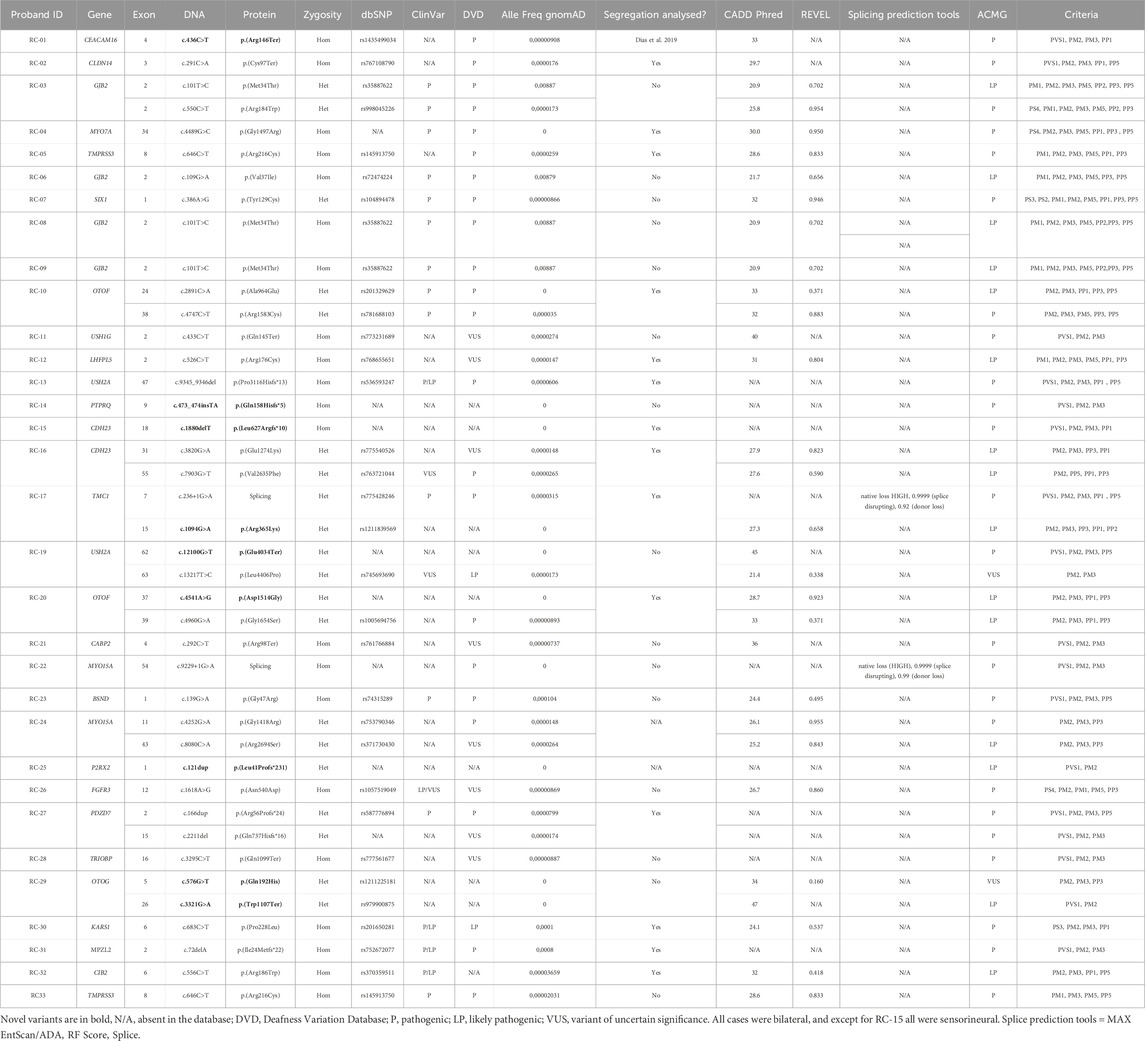
Table 2. Description of further details regarding the causative variants identified
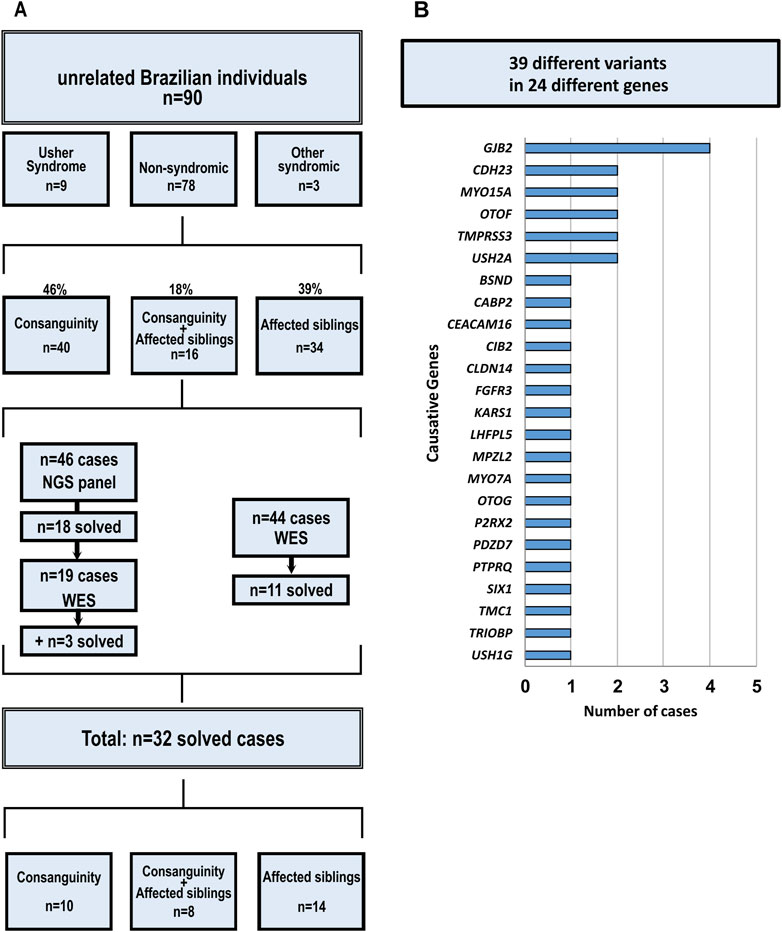
Figure 1. Cohort description and contribution of genes. (A) Cohort Characterization and Strategies; (B) Contribution of each gene to the solved cases.
3.2 Next-generation targeted sequencing of 99 genes
Among the 46 subjects analyzed through the customized panel, 18 cases were solved (39%), and four had previously been clinically diagnosed with Usher syndrome.
The genetic cause of hearing loss in one proband was found to be a variant associated with autosomal dominant syndromic hearing loss (SIX1). In total, 24% (6/25) of the disease-causing variants were novel, being the variant c.436C>T in CEACAM16 first described in Dias et al. (2019).
3.3 Whole exome sequencing
In 14 of the 63 probands (22%) in this group, the causative variants were identified. In three of these probands, no causative variants had been identified previously with the next-generation targeted sequencing of 99 genes: in one of them, the gene harboring the causative variant was not included in the customized panel (FGFR3). In the other two cases (P2RX2 and PDZD7), the variants were not detected after targeted sequencing probably because of technical issues. Causative variants related to syndromic hearing loss were found in two probands (BSND and FGFR3). A causative variant leading to nonsyndromic autosomal dominant hearing loss was found in one proband (P2RX2). In total, 29% (4/14) of the disease-causing variants were novel.
4 Discussion
The detection of causative variants in autosomal recessive hearing loss is challenging due to some factors such as the extreme locus heterogeneity and the high amount of non-recurrent variants. Our study confirms the wide heterogeneity of ARHL, since causative variants were found in 24 different known hearing loss genes and, besides causative variants in GJB2, there was not a single recurrent pathogenic variant causing hearing loss in this sample. Besides, 10 out of the 39 causative variants in known hearing loss genes are novel (Table 1, 2, indicated in bold).
4.1 Characterization of cases with causative variants in genes previously related to syndromic hearing loss
Causative variants in genes described as leading to syndromic hearing loss were found in three probands (RC-26, RC-23, and RC-07) born from consanguineous parents.
Proband RC-26 had a likely pathogenic variant in homozygosis in FGFR3 (NM_000142.4:.c.1618A>G:p.(Asn540Asp). The variant had been described with controversial status, but FGFR3 was already associated with camptodactyly, tall stature, and hearing loss syndrome (CATSHLS). In the first evaluation, at 5 months old, the proband had only camptodactyly in one digit. At the age of 11 years and 2 months, microcephaly was observed. Height was average and flexion restriction of the third digit was observed in both hands. Information about phenotypic variability is limited in CATSHLS syndrome since only four families have been reported (Toydemir et al., 2006; Makrythanasis et al., 2014; Escobar et al., 2016; Cannova et al., 2024). However, the presence of camptodactyly associated with hearing loss and a likely pathogenic variant in FGFR3 pointed to a phenotype related to the CATSHL syndrome spectrum. In three reported cases so far, heterozygous missense variants c.1861C>T:p.(Arg621Cys), and c.1637C>A:p.(Arg621His) were detected and dominant transmission was observed. In a fourth case, c.1637C>A:p.(Thr546Lys) in homozygosis was found in two brothers (Makrythanasis et al., 2014). The catalytic loop of the tyrosine kinase domain of the FGFR3 gene is affected in all cases. The aminoacid residue affected by the variant here described, 540, is very close to the p.(Thr546Lys) variant and interestingly, autosomal recessive inheritance was observed (Makrythanasis et al., 2014), as in the present case. Substitutions of residue 540 were previously associated with hypochondroplasia, but the resulting aminoacid is lysin, p.(Asn540Lys), and not Asp, as in the present case (MIM * 134934). According to UniProt, all variants described above are related to residues located within the protein kinase domain (472–761).
Proband RC-23 presented a pathogenic variant in homozygosis, c.139G>A: p.(Gly47Arg) in BSND, previously associated with Bartter syndrome, type 4A. She was first evaluated at the Genetics Unit at the age of 2 years and 8 months, the second child of consanguineous and healthy parents, presenting failure to thrive and speech delay. She was born at term (37 weeks), with a birth weight of 2,240 g and length of 45 cm, after a pregnancy with no history of polyhydramnios. The mother denies vomiting and the child had an appropriate food intake. No facial dysmorphisms were retrieved. Audiologic evaluation revealed profound hearing loss. In the investigation for her failure to thrive, biochemical analysis revealed metabolic alkalosis (venous blood pH: 7.46, plasma bicarbonate: 30 mmol/L and hypokalemia (2.4mEq/L - NV:3.7–5.0). Other electrolyte abnormalities included hypochloremia (94mEq/L - NV: 97–107), hypocalcemia (1.08 mmol/L - NV:1.12–1.32), as well as increased urine choride/creatinine ratio (390.9mEq/gcreatinine - NV:39–348). She has been referred to the nephrology unit and has been followed with a diagnosis of Bartter syndrome. At the last evaluation, at the age of 8 years and 10 months, she was taking oral potassium supplementation and attending a special school, learning sign language. There was a catch-up on weight gain and growth: W: 22.7 Kg (p.10th-25th centile) and H: 120 cm (p.5th-10th centile). This syndrome exhibits great phenotypic variability (Bartter et al., 1962; Landau et al., 1995). When first ascertained by us, there was no report of kidney problems. The proband, after a recent reassessment, received a clinical diagnosis of Bartter syndrome due to defects in renal reabsorption associated with hearing loss, which is common in Bartter syndrome type 4A.
Proband RC-07 had a heterozygous pathogenic variant c.386A>G: p.(Tyr129Cys) in SIX1 previously associated with Branchiootic syndrome type 3, which exhibits autosomal dominant pattern of inheritance. This syndrome also exhibits great phenotypic variability and hearing loss is the most frequent signal, found in more than 90% of the cases (SMITH, 2014). Clinical revaluation is needed, but it is possible that hearing loss is the only clinical finding in the proband.
This corroborates ACMG recommendations of periodic clinical revaluation since some phenotypic manifestations in syndromic forms appear later in life, after the onset of hearing loss (Alford et al., 2014), and, in some cases, clinical presentation is mild and early recognition of a syndrome may be difficult. These findings favor Whole Exome Sequencing as a first-tier exam in hearing loss, instead of customized panels, and depending on costs or feasibility, exclusion of DFNB1-related hearing loss could be done first, as we have done in our casuistic.
4.2 The relative contribution of different DFNB loci to ARHL
Despite the genetic heterogeneity, some genes showed greater contribution to cases of autosomal recessive deafness in our study: GJB2, CDH23 (10.34%), MYO15A (7%), OTOF (7%), and USH2A (7%) (Figure 1B). It is important to note that the frequency of pathogenic variants in GJB2 cannot be precisely calculated because it would be clearly underestimated, since many families with presumptive ARHL had been screened for variants in GJB2 before this study. In fact, nearly 40 families with DFNB1-related pathogenic variants were known in our diagnostic settings before the sample selection for this study.
We confirmed with our data that the variants classified as “hypomorphic” in GJB2, such as c.101T>C: p.(Met34Thr) and c.109G>A:p.(Val37Ile) were in fact causative of the hearing loss, since no other relevant variants were detected, and the degree of hearing impairment is compatible with the occurrence of these variants.
When comparing data from different studies that investigated variants in known hearing loss genes in large cohorts, especially in probands from pedigrees with presumptive autosomal recessive inheritance, it is interesting to notice that, although there is some variation between different populations, some genes appear as the most frequently altered in more than one study. These genes include MYO15A, MYO7A, SLC26A4, STRC, and USH2A (Bademci et al., 2016; Sloan-Heggen et al., 2016; Yan et al., 2016; Zazo Seco et al., 2017; Morgan et al., 2018). It is important to notice that studies that considered STRC as frequently related to autosomal recessive hearing loss also searched for copy number variations. This analysis did not occur in a systematic way in our sample.
In an investigation performed in the city of Campinas, São Paulo, Brazil, 180 probands had 86 specific variants genotyped in known hearing loss genes through the MassARRAYiPLEX® platform in 17 different genes. This study indicated that GJB2, SLC26A4, MYO15A, OTOF, and CDH23 showed greater contribution in cases of autosomal recessive deafness (Svidnicki et al., 2015) However, this study screened only previously known variants and NGS, as performed in this study, allowed us to identify ten novel variants and solved additional cases.
In the study of Manzoli et al. (2016), conducted in the city of Monte Santo, Bahia, Brazil, 19 probands were selected for having hearing loss of presumptive autosomal recessive inheritance. They had their genetic material analyzed through next-generation targeted sequencing of 180 hearing loss-related genes. The hearing loss genes with causative variants detected in this group were MYO15A (x10) and CLDN14 (x1). The variant c.4198G>A, p.(Val1400Met) in MYO15A was found in eight probands and was probably inherited from a common founder. This study focused on the importance of the p.(Val1400Met) variant in Northeastern Brazil. Other studies also indicated that, apart from GJB2, pathogenic variants in SLC26A4, MYO15A, OTOF, and CDH23 are a frequent cause of hearing loss in European populations (Hilgert, Smith, Van Camp, 2009).
We did not detect variants in the SLC26A4, probably due to a previous study of this cohort (Nonose et al., 2018) that focused on linkage analysis and Sanger sequencing of this gene among candidate pedigrees.
4.3 Considerations about NGS as diagnostic tool in hearing loss
In the sample here presented, 32 of the 90 probands (36%) had the causative variants identified after NGS.
Next-generation sequencing clearly facilitated genetic testing for hearing loss. Targeted sequencing panels allow the identification of causative variants in known hearing loss genes and have been proven effective (Sloan-Heggen et al., 2016). It usually has better coverage and lower costs when compared to whole exome sequencing. However, custom panels need to be periodically updated due to the discovery of new genes, especially in heterogeneous conditions like hearing loss (Atik et al., 2015). In our study, for example, FGFR3 was not included in the custom panel. Therefore, proband RC-26 had a negative result after next-generation targeted sequencing of 99 genes and the causative variant was detected only after whole exome sequencing. It is possible that patients with syndromic hearing loss who present mild clinical features of a syndrome may have been misclassified as “non-syndromic” in some studies.
A limitation of our study was the fact that not all probands were fully screened for copy-number-variants, which is an interesting aspect to investigate, for instance, with arrays or low-pass-whole genome sequencing (Mazzonetto et al., 2024).
Whole exome sequencing also provides the potential for the discovery of novel hearing loss genes, which means that we can focus on unsolved cases to search for candidate variants in genes that were not previously associated with hearing loss.
5 Conclusion
In conclusion, next-generation sequencing is a highly effective tool for the identification of causative variants in hereditary hearing loss, since it is an extremely genetic heterogeneous condition. To our knowledge, ours is the largest report of next-generation sequencing being applied to a cohort with autosomal recessive hearing loss in Brazil and in South America. In our study, the causative variants were identified in 32 of the 90 probands selected because of presumptive autosomal hearing loss (37%) and 28 probands (31%) had causative variants found in genes other than the GJB2. Both, custom panels and whole exome sequencing, exhibit advantages and disadvantages that must be evaluated before their application. We found a greater contribution of CDH23, MYO15A, OTOF, and USH2A in our cohort, besides the expected contribution of GJB2.
Data availability statement
The data presented in the study are deposited in the ClinVar repository (https://www.ncbi.nlm.nih.gov/clinvar/), accession number SUB14687443.
Ethics statement
The studies involving humans were approved by Ethics Committee, Institute of Biosciences, University of São Paulo (1.133.416 - 23/06/2015). The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study. No potentially identifiable images or data are presented in this study.
Author contributions
LA: Conceptualization, Data curation, Formal Analysis, Investigation, Methodology, Validation, Writing–original draft. AD: Data curation, Writing–review and editing, Formal Analysis, Investigation, Methodology, Validation. BS: Data curation, Formal Analysis, Investigation, Methodology, Validation, Writing–original draft, Writing–review and editing. BM: Data curation, Formal Analysis, Investigation, Methodology, Writing–review and editing. DB: Data curation, Formal Analysis, Investigation, Methodology, Writing–review and editing. KL: Data curation, Formal Analysis, Investigation, Supervision, Writing–original draft, Writing–review and editing. RM-N: Conceptualization, Data curation, Formal Analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing–original draft, Writing–review and editing.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This work was supported by Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP—CEPID Human Genome and Stem Cell Research Center 2013/08028–1) and CNPq (406943/2018–4).
Acknowledgments
To all professionals from DERDIC (Divisão de Educação e Reabilitação dos Distúrbios da Comunicação da Pontifícia Universidade Católica de São Paulo) and HCFMUSP for referral of patients and audiologic evaluations. We are indebted to the technical staff of Centro de Pesquisas sobre o Genoma Humano e Células-Tronco for performing NGS lab work and pipelines. We also thank Silvia Souza da Costa and Humberto Marcolino for technical assistance.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2024.1409306/full#supplementary-material
References
Alford, R. A., Arnos, K. S., Fox, M., Lin, J. W., Palmer, C. G., Pandya, A., et al. (2014). American College of Medical Genetics and Genomics guideline for the clinical evaluation and etiologic diagnosis of hearing loss. Genet. Med. 16, 347–355. doi:10.1038/gim.2014.2
Atik, T., Bademci, G., Diaz-Horta, O., Blanton, S. H., and Tekin, M. (2015). Whole-exome sequencing and its impact in hereditary hearing loss. Genet. Res. (Camb) 94, e4–e8. doi:10.1017/S001667231500004X
Bademci, G., Foster Ii, J., Mahdieh, N., Bonyadi, M., Duman, D., Cengiz, F. B., et al. (2016). Comprehensive analysis via exome sequencing uncovers genetic etiology in autosomal recessive nonsyndromic deafness in a large multiethnic cohort. Genet. Med. 18, 364–371. doi:10.1038/gim.2015.89
Bartter, F. C., Pronove, P., Gill, J. R., Mc Cardle, R. C., and Diller, E. (1962). Hyperplasia of the juxtaglomerular complex with hyperaldosteronism and hypokalemic alkalosis: a new syndrome. Am. J. Med. 33, 811–828. doi:10.1016/0002-9343(62)90214-0
Batissoco, A. C., Auricchio, M. T., Kimura, L., Tabith-Junior, A., and Mingroni-Netto, R. C. (2009). A novel missense mutation p.L76P in the GJB2 gene causing nonsyndromic recessive deafness in a Brazilian family. Braz. J. Med. Biol. Res. 42 (2), 168–71. doi:10.1590/s0100-879x2009000200004
Batissoco, A. C., Pedroso-Campos, V., Pardono, E., Sampaio-Silva, J., Sonoda, C. Y., Vieira-Silva, G. A., et al. (2022). Molecular and genetic characterization of a large Brazilian cohort presenting hearing loss. Hum. Genet. 141 (3–4), 519–538. doi:10.1007/s00439-021-02372-2
Cannova, S., Meossi, C., Grilli, F., Milani, D., Alberti, F., Cesaretti, C., et al. (2024). CATSHL syndrome, a new family and phenotypic expansion. Clin. Genet. 105, 313–316. doi:10.1111/cge.14455
del Castillo, F. J., Rodríguez-Ballesteros, M., Alvarez, A., Hutchin, T., Leonardi, E., de Oliveira, C. A., et al. (2005). A novel deletion involving the connexin-30 gene, del(GJB6-d13s1854), found in trans with mutations in the GJB2 gene(connexin-26) in subjects with DFNB1 non-syndromic hearing impairment. J. Hum. Genet. 42, 588–594. doi:10.1136/jmg.2004.028324
del Castillo, I., Villamar, M., Moreno-Pelayo, M. A., del Castillo, F. J., Alvarez, A., Tellería, D., et al. (2002). A deletion involving the connexin 30 gene in nonsyndromic hearing impairment. N. Engl. J. Med. 346, 243–9. doi:10.1056/NEJMoa012052
Dias, A. M. M., Lezirovitz, K., Nicastro, F. S., Mendes, B. C. A., and Mingroni-Netto, R. C. (2019). Further evidence for loss-of-function mutations in the CEACAM16 gene causing nonsyndromic autosomal recessive hearing loss in humans. J. Hum. Genet. 64, 257–260. doi:10.1038/s10038-018-0546-4
Dias, L. R., Tomasi, Y. T., and Boing, A. F. (2024). The newborn screening tests in Brazil: regional and socioeconomic prevalence and inequalities in 2013 and 2019. J. Pediatr. Rio J. 100, 296–304. doi:10.1016/j.jped.2023.11.008
Duman, D., and Tekin, M. (2012). Autosomal recessive nonsyndromic deafness genes: a review. Front. Biosci. (Landmark Ed) 17 (6), 2213–36. doi:10.2741/4046
Escobar, L. F., Tucker, M., and Bamshad, M. (2016). A second family with CATSHL syndrome: confirmatory report of another unique FGFR3 syndrome. Am. J. Med. Genet. A 170, 1908–1911. doi:10.1002/ajmg.a.37676
Estivil, X., Govea, N., Barceló, E., Badenas, C., Romero, E., Moral, L., et al. (1998). Familial progressive sensorineural deafness is mainly due to the mtDNA A1555G mutation and is enhanced by treatment of aminoglycosides. Am. J. Hum. Genet. 62, 27–35. doi:10.1086/301676
Guilford, P., Ben Arab, S., Blanchard, S., Levilliers, J., Weissenbach, J., Belkahia, A., et al. (1994). A non-syndrome form of neurosensory, recessive deafness maps to the pericentromeric region of chromosome 13q. Nat. Genet. 6 (1), 24–8. doi:10.1038/ng0194-24
Hilgert, N., Smith, R. J. H., and Van Camp, G. (2009). Forty-six genes causing nonsyndromic hearing impairment: which ones should be analyzed in DNA diagnostics? Mut. Res. 681, 189–196. doi:10.1016/j.mrrev.2008.08.002
Kenneson, A., Van Naarden Braun, K., and Boyle, C. (2002). GJB2 (connexin 26) variants and nonsyndromic sensorineural hearing loss: a HuGE review Genet. Med. 4, 258–74. doi:10.1097/00125817-200207000-00004
Landau, D., Shalev, H., Ohaly, M., and Carmi, R. (1995). Infantile variant of Bartter syndrome and sensorineural deafness: a new autosomal recessive disorder. Am. J. Med. Genet. 59, 454–459. doi:10.1002/ajmg.1320590411
Lezirovitz, K., and Mingroni-Netto, R. C. (2022). Genetic etiology of non-syndromic hearing loss in Latin America. Hum. Genet. 141, 539–581. doi:10.1007/s00439-021-02354-4
Makrythanasis, P., Temtamy, S., Aglan, M. S., Otaify, G. A., Hamamy, H., and Antonarakis, S. E. (2014). A novel homozygous mutation in FGFR3 causes tall stature, severe lateral tibialdeviation, scoliosis, hearing impairment, camptodactyly, and arachnodactyly. Hum. Mutat. 35, 959–963. doi:10.1002/humu.22597
Manzoli, G. N., Bademci, G., Acosta, A. X., Felix, T. M., Cengiz, F. B., Foster, J., et al. (2016). Targeted resequencing of deafness genes reveals a founder MYO15A variant in northeastern Brazil. Ann. Hum. Genet. 80, 327–331. doi:10.1111/ahg.12177
Mazzonetto, Pc, Villela, D., Da Costa, Ss, Krepischi, A., Milanezi, F., Migliavacca, Mp, et al. (2024). Low-pass whole genome sequencing is a reliable and cost-effective approach for copy number variant analysis in the clinical setting. Ann. Hum. Genet. 88, 113–125. doi:10.1111/ahg.12532
Morgan, A., Lenarduzzi, S., Cappellani, S., Pecile, V., Morgutti, M., Orzan, E., et al. (2018). Genomic studies in a large cohort of hearing impaired Italian patients revealed several newalleles, a rare case of uniparental disomy (UPD) and the importante to search for copy number variations. Front. Genet. 9, 1–16. doi:10.3389/fgene.2018.00681
Morton, C. C., and Nance, W. E. (2006). Newborn hearing screening--a silent revolution. N. Engl. J. Med. 354, 2151–2164. doi:10.1056/NEJMra050700
Neumann, K., Euler, H. A., Chadha, S., and White, K. R. (2020). A survey on the global status of hearing and infant hearing screening. JEHDI 5, 63–84. doi:10.26077/a221-cc28
Nielsen, M. S., Axelsen, L. N., Sorgen, P. L., Verma, V., Delmar, M., and Holstein-Rathlou, N. H. (2012). Gap junctions. Compr Physiol. 2, 1981–2035. doi:10.1002/cphy.c110051
Nonose, R. W., Lezirovitz, K., De Mello Auricchio, M. T. B., Batissoco, A. C., Yamamoto, G. L., and Mingroni-Netto, R. C. (2018). Mutation analysis of SLC26A4 (Pendrin) gene in a Brazilian sample of hearing-impaired subjects. BMC Med. Genet. 19, 73. doi:10.1186/s12881-018-0585-x
Oza, A. M., DiStefano, M. T., Hemphill, S. E., Cushman, B. J., Grant, A. R., and Siegert, R. K.ClinGen Hearing Loss Clinical Domain Working Group (2018). Expert specification of the ACMG/AMP variant interpretation guidelines for genetic hearing loss. Hum. Mutat. 39, 1593–1613. doi:10.1002/humu.23630
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of medical genetics and genomics and the association for molecular Pathology. Genet. Med. 17, 405–424. doi:10.1038/gim.2015.30
Scott, D. A., Kraft, M. L., Carmi, R., Ramesh, A., Elbedour, K., Yairi, Y., et al. (1998). Identification of mutations in the connexin 26 gene that cause autosomal recessive nonsyndromic hearing loss. Hum. Mutat. 11, 387–394. doi:10.1002/(SICI)1098-1004(1998)11:5<387::AID-HUMU6>3.0.CO;2-8
Shearer, A. E., Hildebrand, M. S., and Smith, R. J. H. (2020). Hereditary hearing loss and deafness overview. Maryland, United States: National centre of biotechnology information.
Shearer, A. E., and Smith, R. J. H. (2012). Genetics: advances in genetic testing for deafness. Curr. Opin. Pediatr. 246, 679–686. doi:10.1097/MOP.0b013e3283588f5e
Sloan-Heggen, C. M., Bierer, A. O., Shearer, A. E., Kolbe, D. L., Nishimura, C. J., Frees, K. L., et al. (2016). Comprehensive genetic testing in the clinical evaluation of 1119 patients with hearing loss. Hum. Genet. 135, 441–450. doi:10.1007/s00439-016-1648-8
Smith, R. J. (2014). Branchiootorenal spectrum disorders. Maryland, United States: National centre of biotechnology information.
Smith, R. J. H., and Jones, M. K. N. (2016). “Nonsyndromic hearing loss and deafness, DFNB1.1998 sep 28,” in GeneReviews® internet. Editors M. P. Adam, G. M. Mirzaa, R. A. Pagon, S. E. Wallace, L. J. H. Bean, K. W. Grippet al. (Seattle (WA): University of Washington, Seattle), 1993–2023.
Svidnicki, M. C. C. M., Silva-Costa, S. M., Ramos, P. Z., Santos, N. Z. P., Martins, F. T. A., Castilho, A. M., et al. (2015). Screening of genetic alterations related to non-syndromic hearing loss using MassARRAY iPLEX® technology. BMC Med. Genet. 16, 85–11. doi:10.1186/s12881-015-0232-8
Toydemir, R. M., Brassington, A. E., Baykar-Toydemir, P., Krakowiak, P. A., Jorde, L. B., Whitby, F. G., et al. (2006). A novel mutation in FGFR3 causes camptodactyly, tall stature, and hearing loss (CATSHL) syndrome. Am. J. Hum. Genet. 79, 935–941. doi:10.1086/508433
Walls, W. D., Azaiez, H., and Smith, R. J. H. (2024). Hereditary Hearing Loss Homepage. Available at: https://hereditaryhearingloss.org.
Wright, C. F., Fitzpatrick, D. R., and Firth, H. V. (2018). Paediatric genomics: diagnosing rare disease in children. Nat. Rev. Genet. 19, 253–268. doi:10.1038/nrg.2017.116
Yan, D., Tekin, D., Bademci, G., Foster Ii, J., Cengiz, F. B., Kannan-Sundhari, A., et al. (2016). Spectrum of DNA variants for non-syndromic deafness in a large cohort from multiple continents. Hum. Genet. 135, 953–961. doi:10.1007/s00439-016-1697-z
Keywords: hearing loss, autosomal recessive, next-generation sequencing, hereditary deafness, molecular diagnosis
Citation: Antunes LN, Dias AMM, Schiavo BC, Mendes BCA, Bertola DR, Lezirovitz K and Mingroni-Netto RC (2024) Genetic heterogeneity in autosomal recessive hearing loss: a survey of Brazilian families. Front. Genet. 15:1409306. doi: 10.3389/fgene.2024.1409306
Received: 29 March 2024; Accepted: 12 August 2024;
Published: 21 October 2024.
Edited by:
Miguel Angel Alcántara-Ortigoza, National Institute of Pediatrics , MexicoReviewed by:
Ewelina Bukowska-Olech, Poznan University of Medical Sciences, PolandTerry-Lynn Young, Memorial University of Newfoundland, Canada
Copyright © 2024 Antunes, Dias, Schiavo, Mendes, Bertola, Lezirovitz and Mingroni-Netto. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Regina Célia Mingroni-Netto, cmVuZXR0b0BpYi51c3AuYnI=