 Mengyue Yi
Mengyue Yi Fenglin Zhou*
Fenglin Zhou* Yu Deng
Yu Deng- School of Information Engineering, Jingdezhen Ceramic University, Jingdezhen, China
N4-acetylcysteine (ac4C) is a chemical modification in mRNAs that alters the structure and function of mRNA by adding an acetyl group to the N4 position of cytosine. Researchers have shown that ac4C is closely associated with the occurrence and development of various cancers. Therefore, accurate prediction of ac4C modification sites on human mRNA is crucial for revealing its role in diseases and developing new diagnostic and therapeutic strategies. However, existing deep learning models still have limitations in prediction accuracy and generalization ability, which restrict their effectiveness in handling complex biological sequence data. This paper introduces a deep learning-based model, STM-ac4C, for predicting ac4C modification sites on human mRNA. The model combines the advantages of selective kernel convolution, temporal convolutional networks, and multi-head self-attention mechanisms to effectively extract and integrate multi-level features of RNA sequences, thereby achieving high-precision prediction of ac4C sites. On the independent test dataset, STM-ac4C showed improvements of 1.81%, 3.5%, and 0.37% in accuracy, Matthews correlation coefficient, and area under the curve, respectively, compared to the existing state-of-the-art technologies. Moreover, its performance on additional balanced and imbalanced datasets also confirmed the model’s robustness and generalization ability. Various experimental results indicate that STM-ac4C outperforms existing methods in predictive performance. In summary, STM-ac4C excels in predicting ac4C modification sites on human mRNA, providing a powerful new tool for a deeper understanding of the biological significance of mRNA modifications and cancer treatment. Additionally, the model reveals key sequence features that influence the prediction of ac4C sites through sequence region impact analysis, offering new perspectives for future research. The source code and experimental data are available at https://github.com/ymy12341/STM-ac4C.
1 Introduction
Since the first discovery of RNA chemical modifications 60 years ago, more than 170 types of RNA modifications have been characterized (Zhang Y. et al., 2022). These modifications mainly exist in non-coding RNAs, such as ribosomal RNA (rRNA), transfer RNA (tRNA), and small nuclear RNA (snRNA) (Shi et al., 2020). They are essential for correctly translating the eukaryotic genome and can regulate the same RNA transcript to produce different mRNA and protein products (Schaefer et al., 2017; Cui et al., 2022). Among them, n4-acetylcytidine (ac4C) is a common modification that widely regulates various aspects of RNA metabolism (Karthiya et al., 2020). ac4C mainly occurs in the coding region of mRNA and is catalyzed by the NAT10 enzyme (Jiang et al., 2023; Hu et al., 2024). ac4C modification can affect mRNA’s stability and translation efficiency, and its mechanism is to change the structure and interaction of mRNA and enhance its affinity with ribosome, tRNA, protein, and other RNA (Jin et al., 2020). There have been studies that have found that ac4C plays a role in DNA damage repair, and there have been studies that have found that ac4C is related to the occurrence and development of various diseases, such as cancer, neurodegenerative diseases, viral infections, etc., (Liu et al., 2022; Luo et al., 2023). Therefore, predicting the sites of ac4C is of great significance for understanding its function and designing therapeutic strategies.
In recent years, with the development of high-throughput sequencing technology, Arango et al. used acRIP-seq technology to perform ac4c-specific RNA immunoprecipitation analysis on the transcriptome of human HeLa cells (Arango et al., 2018; 2019). They found more than 4,000 regions containing ac4C. However, acRIP-seq technology cannot accurately locate the position of each ac4C and can only give a rough range. They found that ac4C is widely present in the human transcriptome and is mainly concentrated in the coding regions. They also found that ac4c-modified mRNA is more stable than other mRNA, and its translation level significantly increases after it is acetylated. Because detecting ac4C in mRNA by experimental methods is time-consuming and labor-intensive, computational methods are needed to identify the position of ac4C efficiently and reliably. Currently, some computational methods have been proposed (Zhao et al., 2019; Alam et al., 2020; Wang et al., 2021; Zhang G. et al., 2022; Iqbal et al., 2022; Jia et al., 2023a; 2023c; 2023b; Su et al., 2023; Li et al., 2024).
For example, Zhao et al. developed PACES(Zhao et al., 2019), the first model to use a combination of two random forest (RF) classifiers to predict ac4C sites in human mRNA, using position-specific dinucleotide sequence spectrum (PSDSP) and k-nucleotide frequency (KNF) as features for the two classifiers. Subsequently, Alam et al. developed XG-ac4C (Alam et al., 2020), a model that improved the accuracy of predicting ac4C sites by using the extreme gradient boosting (XGboost) algorithm, combining electron-ion interaction pseudopotentials (EIIP) and electron-ion interaction pseudopotentials of trinucleotide (PseEIIP). Wang et al. designed a model named DeepAc4C(Wang et al., 2021) based on a convolutional neural network (CNN), which used a hybrid feature composed of physicochemical mode and nucleic acid distribution representation and showed better performance in predicting ac4C sites. At the same time, Iqbal et al. proposed a CNN-based deep learning model, DL-ac4C (Iqbal et al., 2022), and compared it with traditional machine learning methods, regression, and Support Vector Machines. Su et al. established a high-quality benchmark dataset and developed a new predictor, iRNA-ac4C (Su et al., 2023). The predictor was based on a gradient boosting decision tree (GBDT), integrating k-mer nucleotide composition, nucleotide chemical property (NCP), and accumulated nucleotide frequency (ANF) as three feature extraction methods, and surpassed the previous models in identifying ac4C sites. Lai and Gao et al. proposed LSA-ac4C (Lai and Gao, 2023), a model that used a two-layer LSTM neural network to learn the dependency relationship of mRNA sequences and used the self-attention mechanism to pay attention to the importance of nucleotides in mRNA sequences. They also trained a transformer-based deep learning model using automated machine learning (AutoML) technology called Auto-ac4C. This model also performed better than the current state-of-the-art (SOTA) models, indicating that it is a reliable baseline and the possibility of further improving the accuracy of ac4C site prediction using deep learning methods. Recently, Jia et al. proposed a deep learning-based prediction model, DLC-ac4C (Jia et al., 2023a). The model uses a variety of features, including three feature encoding schemes, one-dimensional convolutional layers, densely connected convolutional networks (DenseNet), bidirectional long short-term memory networks (Bi-LSTM), channel attention mechanism, and ensemble learning strategy, to capture hidden information features from the sequence perspective. Following DLC-ac4C, Li et al. proposed a deep learning-based multi-module framework, MetaAc4C, which combines pre-trained Bidirectional Encoder Representations from Transformers (BERT) and a Generative Adversarial Network WGAN-GP, where WGAN-GP is used to enhance training data and address data imbalance issues. The core is a Bidirectional Long Short-Term Memory network (BLSTM), which improves model performance through attention mechanisms and residual connections.
Although these models have made some progress, there is still room for improvement in the prediction accuracy of the sites. Therefore, we propose a deep learning-based model named STM-ac4C for identifying N4-acetylcytidine (ac4C) modification sites on human mRNA. The main contributions of this paper are as follows:
1) An innovative hybrid neural network architecture is proposed, which combines the advantages of selective kernel convolution (SKC) (Li et al., 2019), temporal convolutional networks (TCN) (Bai et al., 2018; Raza et al., 2023), and multi-head self-attention mechanisms (MHSA) (Vaswani et al., 2017). This design enables the model to adaptively capture k-mer features of varying lengths, enhances the ability to capture long-term dependencies in mRNA sequences, and achieves adaptive fusion of features, thereby improving the accuracy of predictions and the model’s generalization ability.
2) In terms of feature encoding, this study adopts one-hot encoding (Cheng et al., 2021) to maintain the complete information of the input sequence and avoid information loss. Compared to embedding encoding, one-hot encoding is simple, easy to use, stable, and has no significant impact on model performance while reducing computational resource consumption.
3) The results of ten-fold cross-validation and independent testing show that STM-ac4C outperforms existing models in accuracy, Matthews correlation coefficient, and area under the curve, providing valuable tools and new benchmarks for the related research field.
4) The robustness and generalization ability of the model has been validated on additional balanced and unbalanced datasets, confirming that STM-ac4C maintains stable predictive performance on more challenging datasets.
5) Key sequence features were revealed through sequence region impact analysis, providing a new perspective for understanding the biological significance of mRNA modifications.
In summary, the STM-ac4C model excels in predicting ac4C modification sites on human mRNA, offering a powerful new tool for a deeper understanding of the biological significance of mRNA modifications and cancer treatment.
This paper is divided into the following sections: Section 1 serves as the introduction, presenting the research background, significance, related work, and main contributions. Section 2 elaborates on the research methods, including the selection of datasets, data preprocessing steps, feature encoding techniques, model architecture design details, and performance evaluation criteria. Section 3 displays the experimental results and in-depth analysis, covering the classification performance on benchmark datasets, model comparative analysis, ablation experiments, comparisons with existing technologies, importance assessment of sequence regions, and robustness tests. Section 4 is the conclusion, summarizing the research findings, discussing the study’s limitations, and looking forward to future research directions.
2 Materials and methods
2.1 Dataset and data preprocessing
We used the high-quality dataset constructed by Su et al. to evaluate our model. The dataset was derived from the experimental data of Arango et al. (Arango et al., 2018), which identified 4,250 ac4C peaks using the acRIP-seq method (Zhang X. et al., 2022). Su et al. (Su et al., 2023) extracted sequences of 100 nt up and down as positive samples to create a reliable dataset, starting from the cytosine near each ac4C peak. They also randomly sampled 201 nt sequences centered on cytosines from non-peak regions as negative samples. They used the CD-HIT (Fu et al., 2012) tool with a threshold of 0.8 to remove redundant sequences. To balance the dataset, they selected 2,758 sequences from each positive and negative sample. Finally, they randomly divided the positive and negative data into training and independent test sets at 4:1. The training set contained 2,206 positive and 2,206 negative samples, and the test set contained 552 positive and 552 negative samples. The details of the dataset are shown in Table 1.

Table 1. Statistics of the baseline dataset.
2.2 Preprocessing
2.2.1 Feature encoding method
One-hot encoding (Cheng et al., 2021) is a data preprocessing and feature engineering method that can transform discrete categorical variables into sparse binary codes. This method has wide applications in machine learning, deep learning, bioinformatics, and other fields. The principle of one-hot encoding is given a variable with n categories to generate a new feature vector of length n, where only one element is 1, and the rest are 0. The position of the one indicates the category of the variable. For example, if there are three categories, A, B, and C, their one-hot encodings are
The advantage of one-hot encoding is that each category has an independent binary representation, and these vectors are mutually orthogonal, meaning that their inner product is zero, and there is no size relationship. This can avoid using ordered or continuous values to represent class labels because this would cause problems in calculations such as weight matrices or distance metrics.
We can use one-hot encoding to represent nucleotides in RNA sequences. RNA sequences are composed of four nucleotides, A, U, G, and C, which correspond to different bases. If the type of nucleotide is unknown, use N to represent it. The following formula can be used to convert them into one-hot encoding:
As shown in Figure 1A, this way, an RNA sequence of length 201 bp, can be converted into a 5 x 201 matrix by one-hot encoding, where each column represents a nucleotide, and each row represents a feature.

Figure 1. The schematic diagram of STM-ac4C. (A) Feature encoding. One-hot encoding converts the 201 nt RNA sequence into a 5 × 201 matrix. (B) Model architecture. The encoded feature matrix is sequentially input to three modules: selective kernel convolution (SKC), temporal convolution network (TCN), and multi-head self-attention (MHSA) for feature extraction. Then, the extracted feature matrix is flattened into a one-dimensional vector, and finally, an MLP layer is used for binary classification to predict whether the sequence contains ac4C modification sites.
2.3 Model architecture
In biology, the analytical and predictive capabilities of AI models have been empirically demonstrated across multiple research directions (Ukwuoma et al., 2022b; 2022a; 2023; Fazmiya et al., 2024; Heyat et al., 2024; Sumbul et al., 2024). Based on this, we propose an innovative hybrid neural network model, STM-ac4C, designed to accurately identify N4-acetylcytidine (ac4C) modification sites on human mRNA. As illustrated in Figure 1B, the model skillfully integrates Selective Kernel Convolution (SKC), Temporal Convolutional Network (TCN), and Multi-Head Self-Attention (MHSA) mechanisms. This combination enables STM-ac4C to efficiently capture diverse patterns, structures, and correlations in RNA sequences, which significantly improves prediction accuracy and robustness. SKC overcomes the limitations of traditional fixed kernel sizes by adaptively capturing k-mer features of varying lengths. Compared with LSTM, TCN performs better in modeling long-term dependencies and can effectively handle the temporal and dynamic changes of RNA sequences. The MHSA mechanism, on the other hand, achieves in-depth capture of global and local correlations of RNA sequences by enhancing the model’s characterization capability. At the end of the model, a Multi-Layer Perceptron (MLP) serves as the classifier, transforming the output from the previous layer into the final prediction results using nonlinear activation functions and fully connected layers.
2.3.1 Selective kernel convolution
Selective Kernel Convolution (SKC) (Li et al., 2019) is an innovative convolutional method proposed by Li et al., in 2019. It is designed to simulate the dynamic adaptability of human visual neurons’ receptive fields. SKC intelligently selects the most suitable kernel size by analyzing the multi-scale information of the input features, thereby adjusting the range of the neurons’ receptive fields. This method can be seamlessly integrated into existing convolutional network architectures, significantly enhancing the network’s performance and efficiency while maintaining lightweight parameters and computation.
The core advantage of SKC lies in its subtle improvement over traditional convolutional networks—achieving the effect of multi-size convolutional kernels through grouped and dilated convolutions, thus only slightly increasing the parameters and computational load. The operation process of SKC includes three key steps: split, fuse, and select. In the split phase, the input feature map is convolved with convolutional kernels of different sizes, producing multiple feature sub-maps representing different receptive field sizes. These feature sub-maps are comprehensively integrated during the fusion phase to form a global feature representation containing selection weight information. Finally, based on the calculated selection weights, different-sized feature sub-maps are weighted and aggregated in the selection phase to output the final feature map.
Figure 2 shows the structure of SKC. To further elucidate the mathematical model of SKC, we introduce the following formulas to describe its operation process:
1) Split: For a given input feature map
2) Fuse: Through element-wise addition, all feature sub-maps
3) Select: First, a global descriptor

Figure 2. The structure of selective kernel convolution.
Then, the descriptor
Next, for each channel
Finally, using the attention weights
2.3.2 Temporal convolutional network
Temporal Convolutional Networks (TCN) (Bai et al., 2018; Raza et al., 2023) are network modules for modeling and forecasting sequential data. Their design is inspired by Deep Convolutional Neural Networks (DCNN) (Gu et al., 2018; Abbas et al., 2021), with improvements made to better handle sequential tasks. A significant advantage of TCN is its ability to compute outputs in parallel, significantly enhancing computational efficiency and reducing the computational burden and memory requirements compared to Recurrent Neural Networks (RNN) (Pascanu et al., 2013).
TCN effectively processes time-series data using causal convolutions, ensuring stability and reliability when handling historical information. Causal convolution is a special type only related to the current or previous inputs. It maintains the consistency of the input and output sequence lengths through zero-padding techniques, thereby solving the problem of recursive dependencies. The output of a causal convolution,
Where
To enhance model performance, TCN also employs dilated convolutions and residual connections (He et al., 2016). Dilated convolutions expand the receptive field by increasing the stride of the convolutional kernel, allowing it to capture longer-range dependencies. Residual connections enhance information flow by introducing skip connections, preventing gradient vanishing or explosion issues. The output of a causal dilated convolution,
Here,
A TCN comprises a series of residual blocks containing two layers of dilated causal convolution and ReLU activation function. Weight normalization is applied to the convolutions for stability and efficiency. As depicted in Figure 3, the architecture of a residual block is defined. The TCN processes the input sequence via these residual blocks. The convolutional kernel size, denoted as
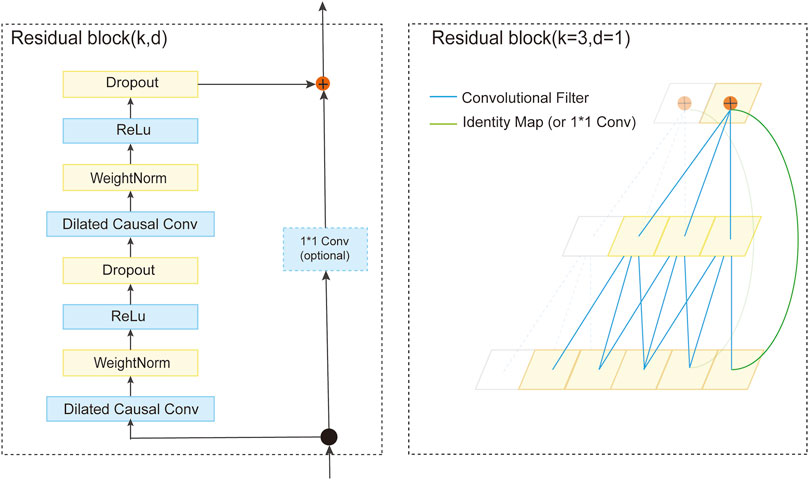
Figure 3. The architecture of the residual block.
2.3.3 Multi-head self-attention
Multi-head self-attention (MHSA) (Vaswani et al., 2017) is an improved version of self-attention, which uses multiple attention heads to process different positions and features of the input sequence, thereby enhancing the model’s feature extraction and interpretability performance on sequential data. Specifically, each attention head multiplies the elements of the input sequence (such as words or pixels) with three matrices, respectively, to obtain three vectors: query, key, and value. Then, according to the similarity between the query and the key, the value vectors are weighted and summed to obtain a sub-representation of the output sequence. Finally, the sub-representations of all attention heads are concatenated to obtain the final representation of the output sequence. This way, the model can flexibly focus on the important parts of the input sequence, thus effectively extracting and utilizing the key information in the sequence. The specific formula of multi-head self-attention is as follows:
Figure 4 shows the structure of MHSA. Assume that the input sequence is
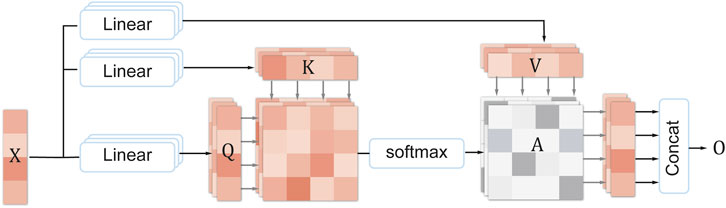
Figure 4. The structure of multi-head self-attention.
Then, the attention weight matrix
Where
2.4 Performance evaluation
In this study, we selected five common evaluation metrics for machine learning classification prediction tasks to evaluate the predictive performance of the model, namely, sensitivity (Sn), specificity (Sp), accuracy (Acc), Matthews correlation coefficient (MCC), and area under the receiver operating characteristic curve (AUC) (Sokolova and Lapalme, 2009). The formulas for these metrics are as follows:
Where TP, FP, TN, and FN represent the number of true positives, false positives, true negatives, and false negatives, respectively. They are the elements of the confusion matrix constructed based on the model’s prediction results and the true labels. AUC is the area under the receiver operating characteristic curve (ROC curve), which is a curve drawn with FP as the horizontal axis and Sn as the vertical axis, reflecting the changes in sensitivity and specificity of the model at different thresholds. Generally speaking, the higher the values of these metrics, the better the model’s predictive performance.
The STM-ac4C was implemented in a Python 3.9.16 environment, utilizing the torch 2.0.0+cu118 framework. The model training employed an Adam optimizer, with a learning rate set to 1e-4, a batch size of 64, and a maximum training epoch set at 300. The loss function chosen was binary cross-entropy. Early stopping was also adopted to prevent overfitting, meaning the training would be halted if the
3 Result and discussion
3.1 Classification performance on the benchmark dataset
In this section, we evaluated the classification performance of the STM-ac4C model on the benchmark dataset. We meticulously recorded the model’s performance in each fold using ten-fold cross-validation. The evaluation metrics include Sensitivity (Sn), Specificity (Sp), Accuracy (Acc), Matthews Correlation Coefficient (MCC), and Area Under the Curve (AUC), with specific data presented in Table 2. The model’s average values and standard deviations for each metric are as follows: Acc is 82.57% ± 1.54%, Sn is 86.49% ± 3.76%, Sp is 78.65% ± 2.36%, MCC is 65.43% ± 2.89%, and AUC is 88.52% ± 1.02%. On the independent test set, the model demonstrated superior performance, with an Acc of 84.78%, Sn of 85.87%, Sp of 83.7%, MCC of 69.58%, and AUC of 90.79%. Both cross-validation and independent testing have demonstrated that the sensitivity (Sn) surpasses the specificity (Sp), signifying that the STM-ac4C model exhibits a pronounced superiority in identifying positive samples. Figure 5 intuitively displays the model’s performance through ROC and PR curves, where the ROC curve reflects the relationship between true positive rate and false positive rate at different thresholds, and the PR curve shows the relationship between precision and recall.

Table 2. Classification results of STM-ac4C on each fold.
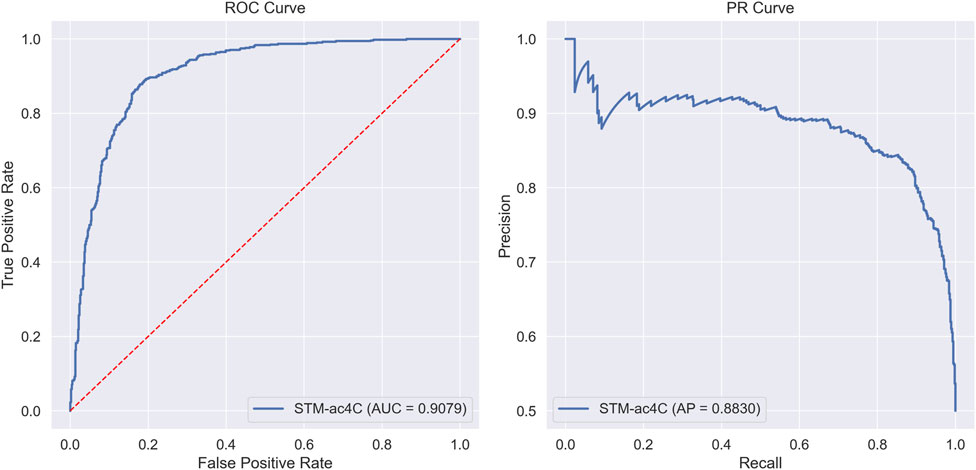
Figure 5. ROC and PR curves of STM-ac4C on the independent test dataset.
3.2 Comparison of using one-hot encoding, NCP encoding and embedding encoding
When encoding sequence features, we aim to preserve as much information as possible from the input sequence while avoiding information loss or redundancy caused by manual feature extraction. To this end, we only considered three encoding schemes: One-hot encoding, NCP encoding (Chen et al., 2016; 2017; Nguyen-Vo et al., 2019), and Embedding encoding (Hasan et al., 2020; Jin et al., 2022b; Lai and Gao, 2023). One-hot encoding is widely used in machine learning, deep learning, bioinformatics, and other fields due to its simplicity, ease of use, stability, and other advantages. NCP encoding converts nucleotide sequences into numerical vectors, which consider nucleotides’ chemical properties and binding characteristics, thereby improving the efficiency and accuracy of sequence analysis. Embedding encoding is a neural network-based method that encodes each categorical element in the sequence into a learnable vector and updates it continuously during the training process, thus learning the semantics and context information of the elements, effectively compressing and abstracting features. To evaluate the impact of the three encoding schemes on model performance, we performed ten-fold cross-validation on the training set, using different encoding methods as model inputs. Figure 6 shows the evaluation metrics under different encoding schemes. From the figure, we can see no significant difference between the results of the three encoding methods. However, embedding encoding increases the model parameters and computational complexity, requiring more time and computational resources during training. In contrast, the one-hot encoding scheme has the highest accuracy (Acc) and Matthews correlation coefficient (MCC): 82.57% and 65.43%. Considering all aspects, we chose one-hot encoding as the final encoding scheme.
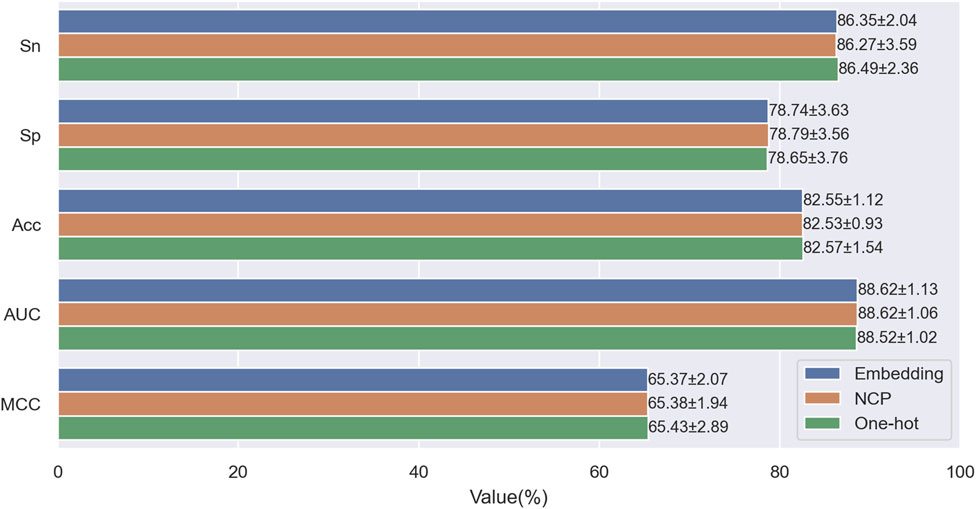
Figure 6. Performance comparison of One-hot encoding with NCP encoding and Embedding encoding on the training dataset.
3.3 Comparison of using TCN, LSTM, GRU and transformer
In STM-ac4C, TCN is mainly used to model the long-term dependencies in sequences. In contrast, some of the current sequence modeling methods are mainly based on variants of RNN, such as long short-term memory networks (LSTM) (Cheng et al., 2021; Zhang G. et al., 2022), gated recurrent units (GRU) (Jin et al., 2022b; Nguyen-Vo et al., 2023; Sultana et al., 2023), or Transformer (Jin et al., 2022a; Tsukiyama et al., 2022; Zeng et al., 2023) network modules based on self-attention mechanisms to build predictors. Although these modules have advantages in processing sequence data, they still have limitations. LSTM and GRU can effectively handle short-term dependencies, but they have difficulty capturing long-term dependencies and are prone to gradient vanishing or exploding problems. Transformer uses self-attention mechanisms and position encoding, which can calculate the dependencies between any positions in the sequence, thus solving the problem of long-term dependencies. However, the over-parameterization of the transformer makes it time-consuming and easy to overfit when dealing with small data sets. To verify the advantages of TCN, we replaced TCN with Transformer and LSTM, GRU, and their bidirectional forms in the model framework of STM-ac4C, built multiple different predictors, and evaluated them on the training set using a ten-fold cross-validation method, and compared their performance on various evaluation indicators. As shown in Figure 7, TCN outperforms other modules on most evaluation indicators, indicating that TCN can better capture the long-term dependencies in the sequence, thus improving prediction accuracy. Therefore, we choose TCN as the core module of the STM-ac4C model to improve the model’s prediction performance.

Figure 7. Performance comparison using TCN, LSTM, GRU, and Transformer on the training dataset.
3.4 Ablation experiments for STM-ac4C
We designed ablation experiments to investigate the contributions of SKC, TCN, and MHSA modules to STM-ac4C. We removed or disabled one or more modules and then performed ten-fold cross-validation on the training set to evaluate the performance of different configurations. Figure 8 and Table 3 show the results of the ablation study, where “√” indicates that the module was used, and “-” indicates that the module was removed or disabled. The table shows that the TCN module has the most significant impact on the model performance, while the SKC and MHSA modules have relatively weaker effects. Using only the TCN module can achieve good performance, with ACC, MCC, and AUC of 81.69%, 63.87%, and 88.01%, respectively. At the same time, each module has some contribution, and removing any module will decrease model performance. When all three modules are used, the model achieves the highest Acc, MCC, and AUC values of 82.57%, 65.57%, and 88.52%. Based on these results, we chose the complete combination of the three modules as the optimal configuration for the STM-ac4C model.
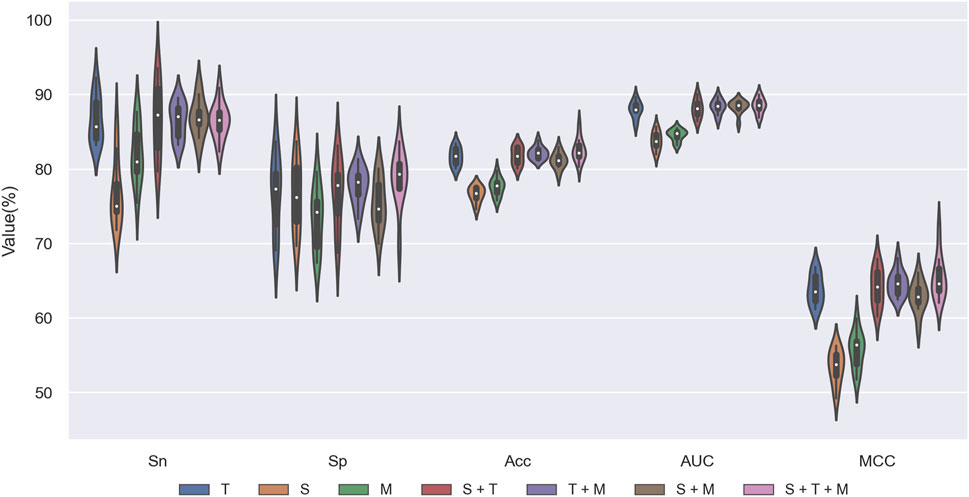
Figure 8. Ablation results of STM-ac4C on the training dataset.
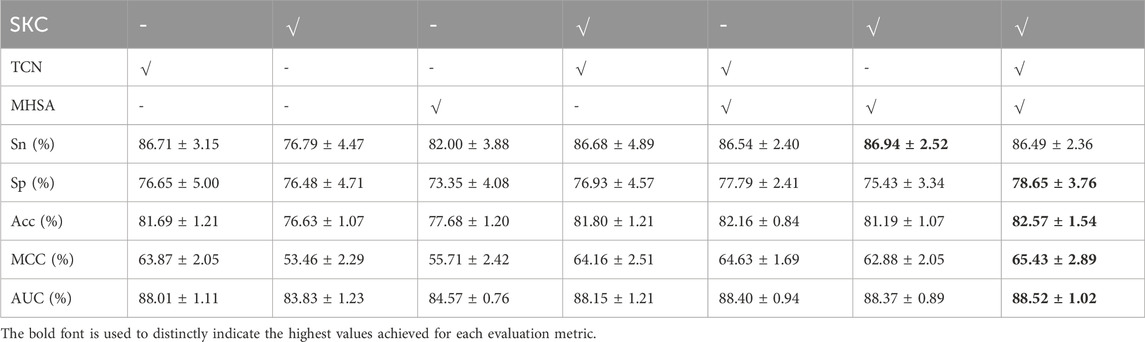
Table 3. Ablation experiments results for STM-ac4C.
3.5 Performance comparison with existing methods
Predicting ac4C sites in human mRNA is a vital bioinformatics problem, and several methods have been proposed and published, such as PACES(Zhao et al., 2019), XG-ac4C (Alam et al., 2020), iRNA-ac4C (Su et al., 2023), DLC-ac4C (Jia et al., 2023a), and LSA-ac4C (Lai and Gao, 2023). However, the performance of these methods is hard to compare directly because they use different datasets and evaluation criteria. In order to evaluate our proposed STM-ac4C method fairly and rigorously, we chose three methods that use the same dataset, iRNA-ac4C, DLC-ac4C, and LSA-ac4C, as comparison objects. We also referred to the results of two methods reported in the LSA-ac4C paper: PACES and XGac4C were retrained using the same dataset, and Auto-ac4C was obtained using the AutoML framework AutoGluon (Erickson et al., 2020; Romero et al., 2022). We compared the performance of these methods on ten-fold cross-validation and independent test sets, and the results are shown in Tables 4 and Table 5. Figure 9 shows the performance comparison on the independent test set. The results indicate that XG-ac4C has very high sensitivity (Sn) in both ten-fold cross-validation and independent test sets, reaching 93.38% and 92.57%, respectively, demonstrating excellent performance in detecting positive samples. However, its specificity (Sp) is relatively low, only 54.76% and 59.78%, which may lead to more false positives when identifying ac4C sequences. In contrast, STM-ac4C surpassed other methods in most evaluation metrics, especially in terms of accuracy (ACC), Matthews correlation coefficient (MCC), and area under the curve (AUC). In ten-fold cross-validation and independent test sets, STM-ac4C’s ACC, MCC, and AUC reached 82.57%, 65.43%, 88.52%, and 84.78%, 69.58%, 90.79%, respectively, all of which are the highest among all models. Particularly in the independent test set, STM-ac4C’s MCC is 3.51% higher than the highest LSA-ac4C. These results suggest that STM-ac4C can effectively balance classification performance and prediction accuracy, effectively identifying ac4C sites in human mRNA.
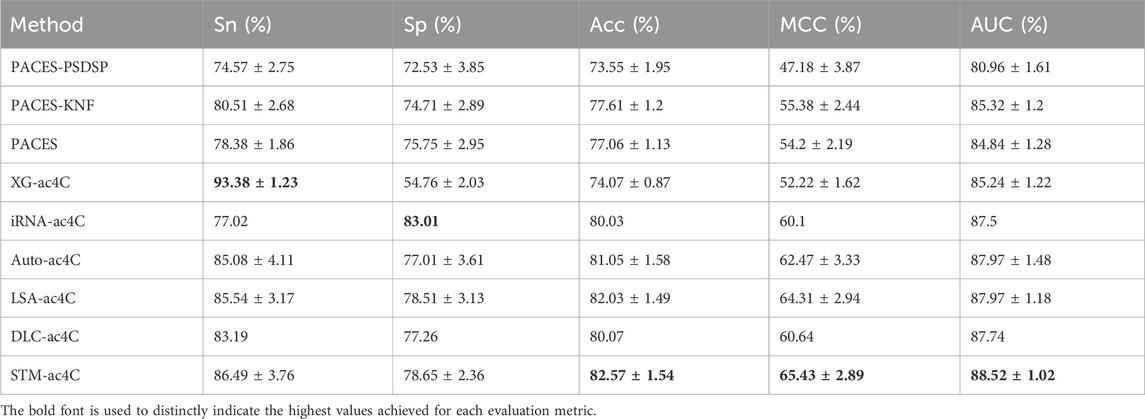
Table 4. The performance over the 10-fold cross-validation.
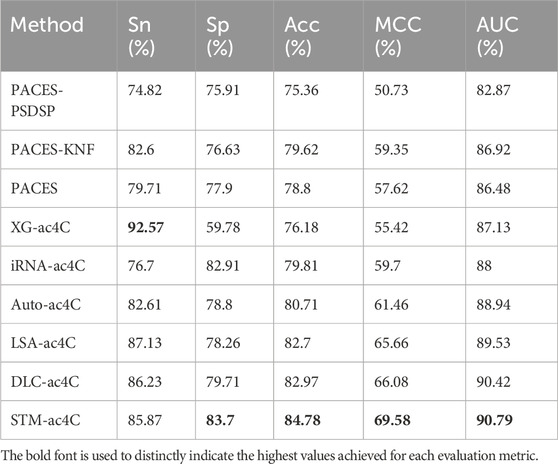
Table 5. The performance over the independent test.
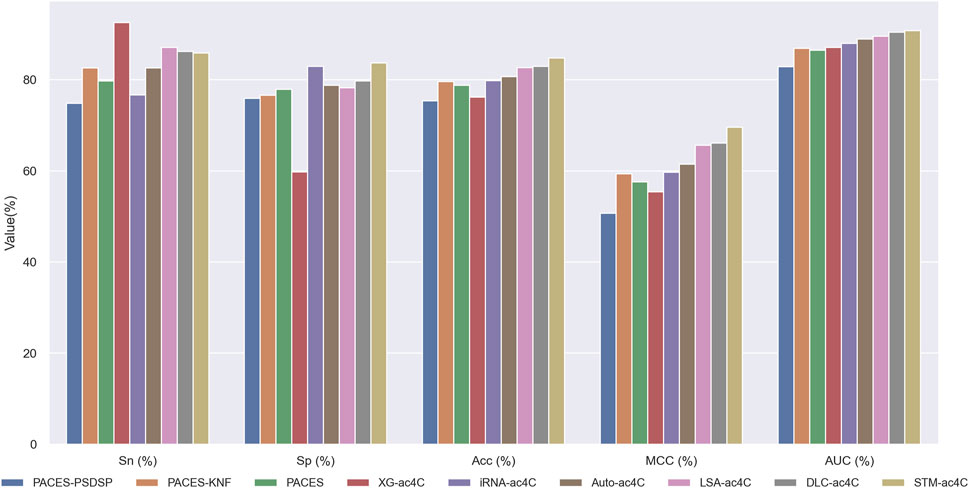
Figure 9. Performance comparison with other predictors on the independent test.
3.6 Sequence region impact analysis
We designed and conducted two experiments to better understand the distribution of critical regions within ac4C sequences and their impact on model prediction performance. These experiments aimed to reveal how different sequence regions specifically affect the model’s predictive capabilities and to identify areas critical for prediction.
In the initial experiment, we evaluated the impact of sequence length on model prediction performance by progressively shortening the sequence length. Specifically, we do this by removing 10 nucleotides from both ends of the sequence each time. To assess the corresponding model’s performance, we performed ten-fold cross-validation on training sets with lengths of 21, 41, 61, 81, 101, 121, 141, 161, 181, and 201 nucleotides (nt). The model’s performance (measured by AUC value) trend with varying sequence lengths is displayed in Figure 10. The results showed that the model’s performance peaked at a sequence length of 201 nucleotides, indicating that this length contained the complete information necessary for accurate prediction. As the sequence length decreased, the model’s performance gradually declined, with an accelerating rate of decline. This may be due to the loss of crucial sequence information, preventing the model from capturing enough features for accurate prediction. Additionally, the accelerated decline in performance may indicate that certain regions within the sequence contribute more significantly to model prediction, possibly containing key biological signals such as specific motifs or structural features crucial for identifying ac4C sites.
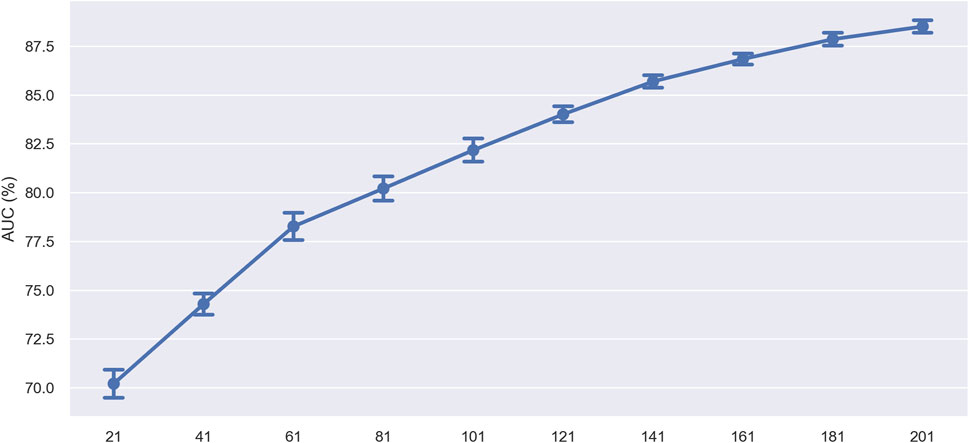
Figure 10. Performance comparison of using different sequence lengths on the training dataset.
In the subsequent experiment, we set a fixed window size of 41 nucleotides (nt). We slid it across the sequence in 10 nt steps to assess the specific impact of different sequence regions on model prediction performance. After each slide, we used only the sequence within the window for ten-fold cross-validation and monitored changes in model performance. Figure 11 presents the experimental results, where the x-axis represents the window’s central position in the sequence, and the y-axis represents the AUC value. The results showed that the model’s performance fluctuated with the movement of the window’s center point, revealing regions within the sequence crucial for predicting ac4C sites. Overall, the model’s performance showed a decreasing trend, suggesting that the sequence region to the left of the ac4C site, i.e., the 5′direction, plays a crucial role in model prediction. The performance increase observed at position 51 of the sequence suggests that this area may contain specific sequence features essential for identifying ac4C sites. These performance increases may reflect key biological signals that the model can recognize, such as specific nucleotide patterns or structural features.
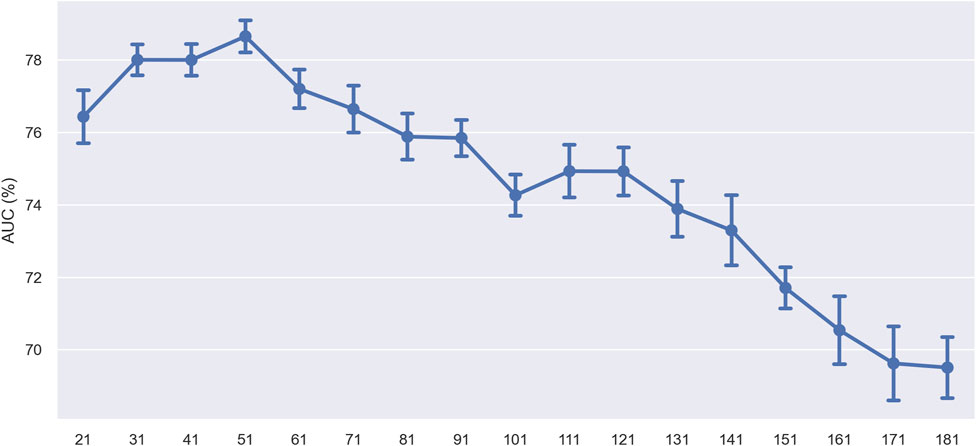
Figure 11. Performance comparison of using different locations of window center points on the training dataset.
Furthermore, the trough phenomenon at the center point may be due to the construction method of the dataset, where both ac4C and non-ac4C sequences are centered around C, resulting in lower information content at that position. To further validate our observations, we analyzed the sequence using kpLogo, and the k-mer Logo (Figure 12) displayed the most significant k-mers at each position, scaled in height according to the p-value (log base 10 transformation), aiding in the identification of enriched motifs in the sequence. The height variation of the k-mers in Figure 12 is consistent with previous experimental results, showing a decreasing trend from left to right in the sequence and revealing significant enrichment of k-mers such as GNNG, GGNGG, and GNG between positions 20 and 60.
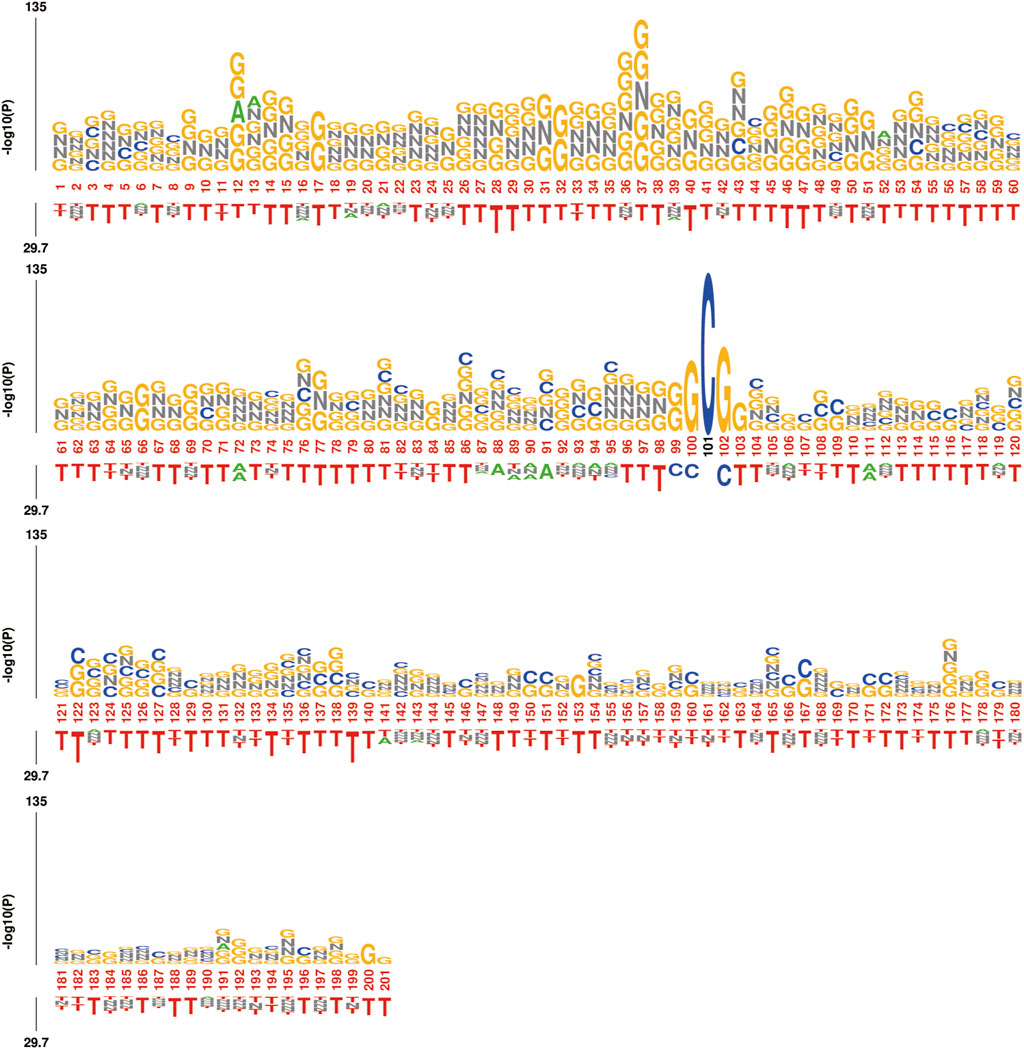
Figure 12. kpLogo: Visualization of k-mer Logo, displaying the most significant k-mers at each position on a 201 nt length sequence. The total height is scaled according to its p-value (log10 transformed) or test statistics, aiding in identifying enriched.
3.7 Robustness and generalizability of STM-ac4C
We collected two datasets from MetaAc4C, also originally derived from ac4C site data of human mRNAs obtained experimentally by Daniel Arango et al. The difference is that these datasets have been processed with CD-HIT, applying a sequence homology threshold of 0.4 to remove redundant sequences. Each sequence is 415 nucleotides (nt) long, with the ac4C modification site located at the center of the sequence. The first dataset is balanced, and the second is unbalanced, sharing the same independent test set. In the training set, the number of negative samples in the unbalanced dataset is five times that of the positive samples. The specific statistics of the two datasets are shown in Table 6. We trained the STM-ac4C on these two datasets and evaluated its performance metrics on the independent test set, with related results detailed in Tables 7 and 8. Other results in the tables are from MetaAc4C. Using a lower CD-HIT threshold can avoid overestimating the prediction model. Even on the balanced dataset processed with a 0.4 homology threshold by CD-HIT, the STM-ac4C model still demonstrated excellent performance, achieving the highest accuracy (Acc), Matthews correlation coefficient (MCC), and area under the curve (AUC) of 82.01, 64.02, and 88.04. On the unbalanced dataset, STM-ac4C achieved the highest MCC (66.64), indicating that STM-ac4C has advantages in dealing with unbalanced data. The above analysis indicates that STM-ac4C is robust and generalizable. However, compared with MetaAc4C, the AUC of STM-ac4C is 0.09% lower, and the sensitivity (Sn) is 2.14% lower, indicating there is still room for improvement of STM-ac4C in recognizing positive samples.

Table 6. Statistical overview of the balanced and unbalanced dataset.
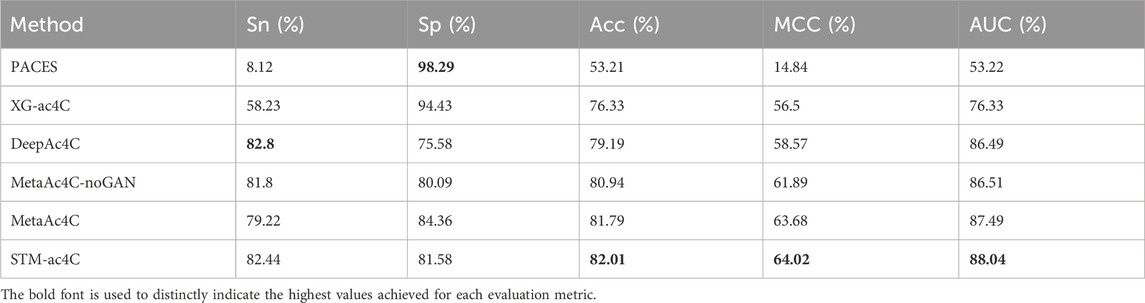
Table 7. Performance evaluation over the balanced dataset.
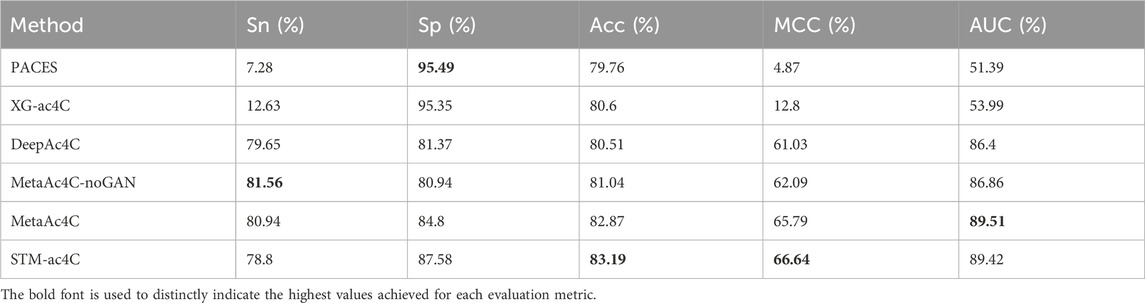
Table 8. Performance evaluation over the unbalanced dataset.
4 Conclusion
This study introduces a novel deep learning model, STM-ac4C, designed to precisely identify N4-acetylcytidine (ac4C) modification sites on human mRNA. The model integrates selective kernel convolution (SKC), temporal convolutional networks (TCN), and multi-head self-attention mechanisms (MHSA), efficiently extracting and integrating multi-level features of RNA sequences for high-precision ac4C site prediction. SKC adapts to capture k-mer features of varying lengths, breaking the constraints of traditional fixed kernel sizes. TCN reveals long-term dependencies within mRNA sequences, enhancing contextual information and effectively learning temporal features. MHSA assigns weights to different features, achieving adaptive integration among them. For feature encoding, the study employs a one-hot encoding strategy to preserve the integrity of input sequences and minimize information loss. After ten-fold cross-validation and independent testing, STM-ac4C performs better in predicting ac4C sites, with key metrics such as accuracy, Matthew’s correlation coefficient, and area under the curve outperforming existing methods. Moreover, the model’s robustness and generalizability are validated on additional balanced and imbalanced datasets. Sequence region influence analysis by the model reveals key sequence features impacting ac4C site prediction, offering new perspectives and directions for future research. Nonetheless, STM-ac4C has certain limitations, such as not considering RNA secondary structures and other factors that may influence ac4C modifications nor being tested across different species or RNA modification types, limiting its generalizability in broader biological applications. Future work will further optimize the model, enhance the accuracy of ac4C site prediction, and extend its application to other species and RNA modification types while incorporating more features and data to explore broader application possibilities.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary material, further inquiries can be directed to the corresponding authors.
Author contributions
MY: Data curation, Methodology, Visualization, Writing–original draft, Writing–review and editing. FZ Funding acquisition, Supervision, Writing–review and editing. YD: Data curation, Visualization, Writing–review and editing.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This work was sponsored in part by the National Science Foundation of China (Nos. 61761023, 62162032, and 31760315), the Natural Science Foundation of Jiangxi Province, China (Nos. 20202BABL202004 and 20202BAB202007), the Scientific Research Plan of the Department of Education of Jiangxi Province, China (GJJ190695 and GJJ2202814). These funders had no role in the study design, data collection and analysis, decision to publish or preparation of manuscript.
Acknowledgments
The authors are grateful for the constructive comments and suggestions made by the reviewers.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Abbas, Z., Tayara, H., and Chong, K. T. (2021). 4mCPred-CNN—prediction of DNA N4-methylcytosine in the mouse genome using a convolutional neural network. Genes. 12, 296. doi:10.3390/genes12020296
Alam, W., Tayara, H., and Chong, K. T. (2020). XG-ac4C: identification of N4-acetylcytidine (ac4C) in mRNA using eXtreme gradient boosting with electron-ion interaction pseudopotentials. Sci. Rep. 10, 20942. doi:10.1038/s41598-020-77824-2
Arango, D., Sturgill, D., Alhusaini, N., Dillman, A. A., Sweet, T. J., Hanson, G., et al. (2018). Acetylation of cytidine in mRNA promotes translation efficiency. Cell. 175, 1872–1886. doi:10.1016/j.cell.2018.10.030
Arango, D., Sturgill, D., and Oberdoerffer, S. (2019). Immunoprecipitation and sequencing of acetylated RNA. Bio Protoc. 9, e3278. doi:10.21769/BioProtoc.3278
Bai, S., Kolter, J. Z., and Koltun, V. (2018). An empirical evaluation of generic convolutional and recurrent networks for sequence modeling. Available at: http://arxiv.org/abs/1803.01271 (Accessed October 23, 2023).
Chen, W., Tang, H., Ye, J., Lin, H., and Chou, K.-C. (2016). iRNA-PseU: identifying RNA pseudouridine sites. Mol. Ther. - Nucleic Acids 5, e332. doi:10.1038/mtna.2016.37
Chen, W., Yang, H., Feng, P., Ding, H., and Lin, H. (2017). iDNA4mC: identifying DNA N4-methylcytosine sites based on nucleotide chemical properties. Bioinformatics 33, 3518–3523. doi:10.1093/bioinformatics/btx479
Cheng, X., Wang, J., Li, Q., and Liu, T. (2021). BiLSTM-5mC: a bidirectional long short-term memory-based approach for predicting 5-methylcytosine sites in genome-wide DNA promoters. Molecules 26, 7414. doi:10.3390/molecules26247414
Cui, L., Ma, R., Cai, J., Guo, C., Chen, Z., Yao, L., et al. (2022). RNA modifications: importance in immune cell biology and related diseases. Signal Transduct. Target Ther. 7, 334. doi:10.1038/s41392-022-01175-9
Erickson, N., Mueller, J., Shirkov, A., Zhang, H., Larroy, P., Li, M., et al. (2020) AutoGluon-tabular: robust and accurate AutoML for structured data. doi:10.48550/arXiv.2003.06505
Fazmiya, M. J. A., Sultana, A., Heyat, M. B. B., Parveen, S., Rahman, K., Akhtar, F., et al. (2024). Efficacy of a vaginal suppository formulation prepared with Acacia arabica (Lam.) Willd. gum and Cinnamomum camphora (L.) J. Presl. in heavy menstrual bleeding analyzed using a machine learning technique. Front. Pharmacol. 15, 1331622. doi:10.3389/fphar.2024.1331622
Fu, L., Niu, B., Zhu, Z., Wu, S., and Li, W. (2012). CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics 28, 3150–3152. doi:10.1093/bioinformatics/bts565
Gu, J., Wang, Z., Kuen, J., Ma, L., Shahroudy, A., Shuai, B., et al. (2018). Recent advances in convolutional neural networks. Pattern Recognit. 77, 354–377. doi:10.1016/j.patcog.2017.10.013
Hasan, Md. M., Manavalan, B., Shoombuatong, W., Khatun, M. S., and Kurata, H. (2020). i4mC-Mouse: improved identification of DNA N4-methylcytosine sites in the mouse genome using multiple encoding schemes. Comput. Struct. Biotechnol. J. 18, 906–912. doi:10.1016/j.csbj.2020.04.001
He, K., Zhang, X., Ren, S., and Sun, J. (2016) Deep residual learning for image recognition, 770–778. doi:10.1109/CVPR.2016.90
Heyat, M. B. B., Akhtar, F., Munir, F., Sultana, A., Muaad, A. Y., Gul, I., et al. (2024). Unravelling the complexities of depression with medical intelligence: exploring the interplay of genetics, hormones, and brain function. Complex Intell. Syst. doi:10.1007/s40747-024-01346-x
Hu, Z., Lu, Y., Cao, J., Lin, L., Chen, X., Zhou, Z., et al. (2024). N -acetyltransferase NAT10 controls cell fates via connecting mRNA cytidine acetylation to chromatin signaling. Sci. Adv. 10, eadh9871. doi:10.1126/sciadv.adh9871
Iqbal, M. S., Abbasi, R., Bin Heyat, M. B., Akhtar, F., Abdelgeliel, A. S., Albogami, S., et al. (2022). Recognition of mRNA N4 acetylcytidine (ac4C) by using non-deep vs. Deep learning. Appl. Sci. 12, 1344. doi:10.3390/app12031344
Jia, J., Cao, X., and Wei, Z. (2023a). DLC-ac4C: a prediction model for N4-acetylcytidine sites in human mRNA based on DenseNet and bidirectional LSTM methods. Curr. Genomics 24, 171–186. doi:10.2174/0113892029270191231013111911
Jia, J., Wei, Z., and Cao, X. (2023b). EMDL-ac4C: identifying N4-acetylcytidine based on ensemble two-branch residual connection DenseNet and attention. Front. Genet. 14, 1232038. doi:10.3389/fgene.2023.1232038
Jia, J., Wei, Z., and Sun, M. (2023c). EMDL_m6Am: identifying N6,2′-O-dimethyladenosine sites based on stacking ensemble deep learning. BMC Bioinforma. 24, 397. doi:10.1186/s12859-023-05543-2
Jiang, X., Cheng, Y., Zhu, Y., Xu, C., Li, Q., Xing, X., et al. (2023). Maternal NAT10 orchestrates oocyte meiotic cell-cycle progression and maturation in mice. Nat. Commun. 14, 3729. doi:10.1038/s41467-023-39256-0
Jin, G., Xu, M., Zou, M., and Duan, S. (2020). The processing, gene regulation, biological functions, and clinical relevance of N4-acetylcytidine on RNA: a systematic review. Mol. Ther. Nucleic Acids 20, 13–24. doi:10.1016/j.omtn.2020.01.037
Jin, J., Yu, Y., Wang, R., Zeng, X., Pang, C., Jiang, Y., et al. (2022a). iDNA-ABF: multi-scale deep biological language learning model for the interpretable prediction of DNA methylations. Genome Biol. 23, 219. doi:10.1186/s13059-022-02780-1
Jin, J., Yu, Y., and Wei, L. (2022b). Mouse4mC-BGRU: deep learning for predicting DNA N4-methylcytosine sites in mouse genome. Methods 204, 258–262. doi:10.1016/j.ymeth.2022.01.009
Karthiya, R., Wasil, S. M., and Khandelia, P. (2020). Emerging role of N4-acetylcytidine modification of RNA in gene regulation and cellular functions. Mol. Biol. Rep. 47, 9189–9199. doi:10.1007/s11033-020-05963-w
Lai, F.-L., and Gao, F. (2023). LSA-ac4C: a hybrid neural network incorporating double-layer LSTM and self-attention mechanism for the prediction of N4-acetylcytidine sites in human mRNA. Int. J. Biol. Macromol. 253, 126837. doi:10.1016/j.ijbiomac.2023.126837
Li, X., Wang, W., Hu, X., and Yang, J. (2019). “Selective kernel networks,” in 2019 IEEE/CVF conference on computer vision and pattern recognition (CVPR), 510–519. doi:10.1109/CVPR.2019.00060
Li, Z., Jin, B., and Fang, J. (2024). MetaAc4C: a multi-module deep learning framework for accurate prediction of N4-acetylcytidine sites based on pre-trained bidirectional encoder representation and generative adversarial networks. Genomics 116, 110749. doi:10.1016/j.ygeno.2023.110749
Liu, S., Zhang, Y., Qiu, L., Zhang, S., Meng, Y., Huang, C., et al. (2022). Uncovering N4-acetylcytidine-related mRNA modification pattern and landscape of stemness and immunity in hepatocellular carcinoma. Front. Cell. Dev. Biol. 10, 861000. doi:10.3389/fcell.2022.861000
Luo, J., Cao, J., Chen, C., and Xie, H. (2023). Emerging role of RNA acetylation modification ac4C in diseases: current advances and future challenges. Biochem. Pharmacol. 213, 115628. doi:10.1016/j.bcp.2023.115628
Nguyen-Vo, T.-H., Nguyen, Q. H., Do, T. T. T., Nguyen, T.-N., Rahardja, S., and Nguyen, B. P. (2019). iPseU-NCP: identifying RNA pseudouridine sites using random forest and NCP-encoded features. BMC Genomics 20, 971. doi:10.1186/s12864-019-6357-y
Nguyen-Vo, T.-H., Trinh, Q. H., Nguyen, L., Nguyen-Hoang, P.-U., Rahardja, S., and Nguyen, B. P. (2023). i4mC-GRU: identifying DNA N4-Methylcytosine sites in mouse genomes using bidirectional gated recurrent unit and sequence-embedded features. Comput. Struct. Biotechnol. J. 21, 3045–3053. doi:10.1016/j.csbj.2023.05.014
Pascanu, R., Mikolov, T., and Bengio, Y. (2013) On the difficulty of training recurrent neural networks. doi:10.48550/arXiv.1211.5063
Raza, A., Alam, W., Khan, S., Tahir, M., and Chong, K. T. (2023). iPro-TCN: prediction of DNA promoters recognition and their strength using temporal convolutional network. IEEE Access 11, 66113–66121. doi:10.1109/ACCESS.2023.3285197
Romero, R. A., A., Deypalan, M. N., Y., Mehrotra, S., Jungao, J. T., Sheils, N. E., Manduchi, E., et al. (2022). Benchmarking AutoML frameworks for disease prediction using medical claims. BioData Min. 15, 15. doi:10.1186/s13040-022-00300-2
Schaefer, M., Kapoor, U., and Jantsch, M. F. (2017). Understanding RNA modifications: the promises and technological bottlenecks of the ‘epitranscriptome. Open Biol. 7, 170077. doi:10.1098/rsob.170077
Shi, H., Chai, P., Jia, R., and Fan, X. (2020). Novel insight into the regulatory roles of diverse RNA modifications: Re-defining the bridge between transcription and translation. Mol. Cancer 19, 78. doi:10.1186/s12943-020-01194-6
Sokolova, M., and Lapalme, G. (2009). A systematic analysis of performance measures for classification tasks. Inf. Process. Manag. 45, 427–437. doi:10.1016/j.ipm.2009.03.002
Su, W., Xie, X.-Q., Liu, X.-W., Gao, D., Ma, C.-Y., Zulfiqar, H., et al. (2023). iRNA-ac4C: a novel computational method for effectively detecting N4-acetylcytidine sites in human mRNA. Int. J. Biol. Macromol. 227, 1174–1181. doi:10.1016/j.ijbiomac.2022.11.299
Sumbul, , Sultana, A., Heyat, M. B. B., Rahman, K., Akhtar, F., Parveen, S., et al. (2024). Efficacy and classification of Sesamum indicum linn seeds with Rosa damascena mill oil in uncomplicated pelvic inflammatory disease using machine learning. Front. Chem. 12, 1361980. doi:10.3389/fchem.2024.1361980
Sultana, A., Mitu, S. J., Pathan, M. N., Uddin, M. N., Uddin, Md. A., and Aryal, S. (2023). 4mC-CGRU: identification of N4-Methylcytosine (4mC) sites using convolution gated recurrent unit in Rosaceae genome. Comput. Biol. Chem. 107, 107974. doi:10.1016/j.compbiolchem.2023.107974
Tsukiyama, S., Hasan, M. M., Deng, H.-W., and Kurata, H. (2022). BERT6mA: prediction of DNA N6-methyladenine site using deep learning-based approaches. Briefings Bioinforma. 23, bbac053. doi:10.1093/bib/bbac053
Ukwuoma, C. C., Cai, D., Heyat, M. B. B., Bamisile, O., Adun, H., Al-Huda, Z., et al. (2023). Deep learning framework for rapid and accurate respiratory COVID-19 prediction using chest X-ray images. J. King Saud Univ. - Comput. Inf. Sci. 35, 101596. doi:10.1016/j.jksuci.2023.101596
Ukwuoma, C. C., Qin, Z., Agbesi, V. K., Cobbinah, B. M., Yussif, S. B., Abubakar, H. S., et al. (2022a). Dual_Pachi: attention-based dual path framework with intermediate second order-pooling for Covid-19 detection from chest X-ray images. Comput. Biol. Med. 151, 106324. doi:10.1016/j.compbiomed.2022.106324
Ukwuoma, C. C., Qin, Z., Agbesi, V. K., Ejiyi, C. J., Bamisile, O., Chikwendu, I. A., et al. (2022b). LCSB-inception: reliable and effective light-chroma separated branches for Covid-19 detection from chest X-ray images. Comput. Biol. Med. 150, 106195. doi:10.1016/j.compbiomed.2022.106195
Vaswani, A., Shazeer, N., Parmar, N., Uszkoreit, J., Jones, L., Gomez, A. N., et al. (2017). Attention is all you need. Available at: https://proceedings.neurips.cc/paper_files/paper/2017/hash/3f5ee243547dee91fbd053c1c4a845aa-Abstract.html (Accessed December 6, 2023).
Wang, C., Ju, Y., Zou, Q., and Lin, C. (2021). DeepAc4C: a convolutional neural network model with hybrid features composed of physicochemical patterns and distributed representation information for identification of N4-acetylcytidine in mRNA. Bioinformatics 38, 52–57. doi:10.1093/bioinformatics/btab611
Zeng, W., Gautam, A., and Huson, D. H. (2023). MuLan-Methyl—multiple transformer-based language models for accurate DNA methylation prediction. GigaScience 12, giad054. doi:10.1093/gigascience/giad054
Zhang, G., Luo, W., Lyu, J., Yu, Z.-G., and Huang, G. (2022a). CNNLSTMac4CPred: a hybrid model for N4-acetylcytidine prediction. Interdiscip. Sci. Comput. Life Sci. 14, 439–451. doi:10.1007/s12539-021-00500-0
Zhang, X., Zeng, J., Wang, J., Yang, Z., Gao, S., Liu, H., et al. (2022b). Revealing the potential markers of N(4)-acetylcytidine through acRIP-seq in triple-negative breast cancer. Genes. 13, 2400. doi:10.3390/genes13122400
Zhang, Y., Lu, L., and Li, X. (2022c). Detection technologies for RNA modifications. Exp. Mol. Med. 54, 1601–1616. doi:10.1038/s12276-022-00821-0
Zhao, W., Zhou, Y., Cui, Q., and Zhou, Y. (2019). PACES: prediction of N4-acetylcytidine (ac4C) modification sites in mRNA. Sci. Rep. 9, 11112. doi:10.1038/s41598-019-47594-7
Glossary

Keywords: N4-acetylcytidine, selective kernel convolution, temporal convolutional network, multi-head self-attention, deep learning
Citation: Yi M, Zhou F and Deng Y (2024) STM-ac4C: a hybrid model for identification of N4-acetylcytidine (ac4C) in human mRNA based on selective kernel convolution, temporal convolutional network, and multi-head self-attention. Front. Genet. 15:1408688. doi: 10.3389/fgene.2024.1408688
Received: 05 April 2024; Accepted: 14 May 2024;
Published: 30 May 2024.
Edited by:
Md Belal Bin Heyat, Westlake University, ChinaReviewed by:
Muhammad Shahid Iqbal, Anhui University, ChinaSwakkhar Shatabda, United International University, Bangladesh
Hilal Tayara, Jeonbuk National University, Republic of Korea
Arshiya Sultana, National Institute of Unani Medicine (NIUM), India
Copyright © 2024 Yi, Zhou and Deng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mengyue Yi, MjIyMDA0MjAxMUBzdHUuamN1LmVkdS5jbg==; Fenglin Zhou, MDA0Mjc4QGpjdS5lZHUuY24=