 Xinyu Chen
Xinyu Chen Yifan Feng3
Yifan Feng3 Zhixi Fu
Zhixi Fu
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Genet. , 23 July 2024
Sec. Evolutionary and Population Genetics
Volume 15 - 2024 | https://doi.org/10.3389/fgene.2024.1408114
The genus Ainsliaea DC. is one of the major groups within the tribe Pertyeae (Asteraceae). It comprises several important Chinese medicinal species. However, the phylogenetic position has undergone a long process of exploration. The complete chloroplast (cp) genome sequences data has not been employed in species identification and phylogeny of Ainsliaea. In this study, the complete cp genomes of two Ainsliaea species (A. gracilis and A. henryi) were reported, followed by structural, comparative, and phylogenetic analyses within the tribe Peryteae. Both cp genomes displayed a typical quadripartite circular structure, with the LSC and SSC regions separated by the IR regions. The genomes were 152,959 (A. gracilis) and 152,805 (A. henryi) base pairs (bp) long, with a GC content of 37.6%. They were highly conserved, containing 134 genes, including 87 protein-coding genes, 37 tRNA genes, 8 rRNA genes, and 2 pseudogenes (rps19 and ycf1). Moreover, thirteen highly polymorphic regions (e.g., trnK-UUU, trnG-UCC, trnT-GGU, accD-psaI, and rpl22-rps19) were identified, indicating their potential as DNA barcodes. The phylogenetic analysis confirmed the placement of Ainsliaea in the tribe Pertyeae, revealing close relationships with the genera Myripnois and Pertya. In comparison with Ainsliaea, Myripnois was more closely related to Pertya. This study lays a theoretical foundation for future research on species identification, population genetics, resource conservation, and sustainable utilization within Ainsliaea and Pertyeae.
The genus Ainsliaea DC., a member of the tribe Pertyeae (Asteraceae), predominantly flourishes in the tropics and subtropics of East Asia (Mitsui and Setoguchi, 2012; Zhang et al., 2024), with over fifty species of perennial herbs recorded (Freire, 2007; Mitsui et al., 2008; Gao et al., 2011). In China, the species in the genus Ainsliaea demonstrate considerable diversity, with approximately forty distributed, around thirty of them are endemic to the region (Qian, 2000; Wang et al., 2010; Gao et al., 2011; Zhang et al., 2019a; Peng et al., 2020; Zhang et al., 2021). The genus is primarily identified by its basally rosulate and alternate leaves, capitula with few florets, (1-)3(-5), open corollas with deeply irregularly 5-lobed, and pappus of plumose bristles (Freire, 2007; Gao et al., 2011; Zhang et al., 2024). These species thrive in evergreen broad-leaved forests, serving as indicator species for environmental quality. They contribute significantly to ecological maintenance and the conservation of soil and water. Furthermore, Ainsliaea encompasses multiple important medicinal plants utilized as traditional Chinese medicines (Qian, 2000; Peng et al., 2020; Zhang et al., 2021) For instance, Ainsliaea henryi Diels has historically been employed to treat coughs, asthma, and other diseases (Zeng et al., 2016).
However, the process of determining the phylogenetic position of Ainsliaea has undergone a lengthy discovery. Initially, the genus Ainsliaea was treated as a member of the tribe Mutisieae (Mutisioideae) based on incomplete morphological studies (Cabrera, 1977; Hind, 2007; Katinas et al., 2008; Gao et al., 2011). Nevertheless, on the basis of cladistic analyses and molecular systematic studies (Kim et al., 2002; Panero and Funk, 2002; Panero and Funk, 2008; Mitsui et al., 2008; Panero, 2008; Freire, 2012), the five closest genera in the tribe Mutisieae, including Ainsliaea DC., Macroclinidium Maxim. (Japanese endemic), Catamixis T. Thomson (Indian endemic), Myripnois Bunge, and Pertya Sch. Bip. were isolated and collectively constituted a distinct monophyletic taxon, the tribe Pertyeae (Pertyoideae). Furthermore, it was revealed that Myripnois exhibited a closer relationship with Pertya compared to Ainsliaea (Fu et al., 2016). Freire (2017) then proposed the integration of Myripnois into the genus Pertya. However, this proposal has not obtained widespread acceptance. Therefore, further genomic studies should be conducted to strengthen our understanding of the phylogenetic relationships among Ainsliaea, Pertya, and Myripnois.
A crucial organelle in charge of photosynthesis within green plants is the chloroplast (cp) (Jansen and Ruhlman, 2012). Each chloroplast contains a separate genome from the nuclear genome with lengths of 120–160 kilobase pairs (kp) (Wicke et al., 2011; Daniell et al., 2016). In most angiosperms, a typical cp genome contains a quadripartite circular architecture, consisting of two inverted repeat regions (IRs), a large single-copy region (LSC), and a small single-copy region (SSC) (Palmer, 1985; Bendich, 2004; Mower and Vickrey, 2018). The gradual evolution, peculiar genome structure, and highly conserved sequence are distinguishing features of the cp genomes. Consequently, the analysis of complete cp genome is deemed as the optimal approach for phylogenetic research and the creation of molecular markers within Asteraceae (Ma et al., 2020; Abdullah et al., 2021; Jin et al., 2023). Recently, it has become a valuable tool for offering detailed phylogenetic insights into the evolutionary relationships among various taxa in Asteraceae, including Artemisia L. (Jin et al., 2023), Cavea W. W. Sm. & J. Small (Yu et al., 2022), Dolomiaea DC. (Shen et al., 2020), Gerbera L. (Zhang et al., 2019b), Myripnois Bunge (Lin et al., 2019), Nouelia Franch. (Liu et al., 2022), Saussurea DC. (Zhang et al., 2019c; Yun and Kim, 2022), Sinosenecio B. Nord. (Peng et al., 2022), and Taraxacum F. H. Wigg. (Salih et al., 2017). Nonetheless, only a few complete cp genomes of the tribe Pertyeae have been reported in the NCBI database to date (Lin et al., 2019; Wang et al., 2020; Liu et al., 2021). Additionally, the investigation of phylogenetic relationships among plastomes of Ainsliaea and Pertyeae remains unreported.
In this study, the complete cp genomes of two Ainsliaea species (Ainsliaea gracilis Franch. and A. henryi) were obtained and analyzed. The comparative and phylogenetic analyses were subsequently carried out within Pertyeae. The aims of the study were to: (i) illuminate the structure and variation of cp genomes within Ainsliaea and Pertyeae; (ii) establish the phylogenetic status of Ainsliaea and Pertyeae utilizing cp genomes.
The samples of A. gracilis (Voucher specimen, No. DY159) and A. henryi (Voucher specimen, No. DY133) utilized were collected from Guangwu Mountain, Bazhong City, Sichuan Province, China. The voucher specimens were then deposited at the herbarium of Sichuan Normal University (SCNU), Chengdu City, Sichuan Province, China (contact: Dr. Prof. Zhixi Fu, ZnV6eDIwMTdAc2ljbnUuZWR1LmNu). The extraction of total genomic DNA from fresh leaves was performed following the CTAB DNA extraction protocol (Allen et al., 2006). The quantification and evaluation of the total genomic DNA integrity were assessed using the NanoDrop 2000 Spectrophotometer and Qubit 4 Fluorometer (Thermo Fisher Scientific, Wilmington, DE, USA). DNA libraries were constructed using the Illumina Paired-End DNA Library Kit (Illumina Inc., San Diego, CA, USA) and subsequently sequenced on the NovaSeq 6000 platform with 150 bp paired-end reads (NovoGene Inc., Beijing, China). Eventually, the Illumina Genome Analyzer (Hiseq 2000, Illumina, San Diego, CA, USA) was employed to obtain the raw sequence data. The raw data were then subjected to primary and secondary quality control to yield clean data.
Two sets of clean data, comprising the sequence reads of A. gracilis and A. henryi, were mapped to the reference sequence of Ainsliaea latifolia (D. Don) Sch.-Bip. using Bowtie2 v.2.4.5-Linux (Langmead et al., 2019). Following this, SAMtools v.1.15-Linux (Danecek et al., 2021) was employed to selectively retain only the reads mapped to the reference sequence for subsequent assembly. The cp genomes of two Ainsliaea species were assembled by the SPAdes v.3.15.1-Linux with default parameters (Prjibelski et al., 2020). The assembly results were visualized, untangled, inspected, and exported to two complete cp genome sequences using Bandage v.0.9.0-Linux (Wick et al., 2015). Collinearity analysis of the two sequences was performed using MUMmer v4.0.0-Linux (Marcais et al., 2018), with A. latifolia as the reference. Subsequently, the obtained sequences were annotated utilizing the Plastid Genome Annotator (PGA) (Qu et al., 2019), referencing the sequences of Amborella trichopoda Baill. and A. latifolia. The annotation results were examined utilizing Geneious R11 (https://www.geneious.com) and adjusted manually as needed. Eventually, the circular map of cp genomes was generated through OrganellarGenomeDRAW (OGDRAW) (Greiner et al., 2019).
The simple repeat sequence (SSR) was detected with MISA (Beier et al., 2017). The minimum number of repeats for each nucleotide type was set as follows: 10 repeats for mononucleotide (mono-), 5 repeats for dinucleotide (din-), 4 repeats for trinucleotide (tri-), and 3 repeats for tetranucleotide (tetra-), pentanucleotide (penta-), and hexanucleotide (hexan-). The long repeats were analyzed by REPuter (Kurtz et al., 2001). The analysis encompassed forward (F), reverse (R), complement (C), and palindromic (P) repeats. The maximal repeat size was set at 50 bp, the minimal repeat size was set at 30 bp, and the hamming distance was set at 8.
The sequences of A. gracilis (No. OQ723680), A. henryi (No. PP175243), A. latifolia (No. MW316662), Myripnois dioica Bunge (No. MK784068), Pertya multiflora Cai F. Zhang & T. G. Gao (No. MW148616), and Pertya phylicoides J. F. Jeffrey. (No. MN935435) were retrieved from GenBank in the NCBI database for comparative cp genome analysis. The contraction and extension of the IR borders within the four major regions (LSC/IRb/SSC/IRa) of the six cp genome sequences were visualized using IRscope (Amiryousefi et al., 2018). The online software mVISTA, with the Shuffle-LAGAN mode (Brudno et al., 2003; Frazer et al., 2004), was employed to compare the plastomes using the sequence of A. gracilis as a reference.
Python scripts were used to extract the coding regions (CDS) and non-coding regions (IGS) from six sequences for subsequent alignment. These sequences included two from Ainsliaea and four from Pertyeae. The sequences were aligned together using the “auto” strategy of MAFFT v.7.475 (Katoh et al., 2019). Nucleotide diversity (Pi) was subsequently calculated using the sliding window of DnaSP v.6.12.03 with window length of 600 bp and step size of 200 bp (Rozas et al., 2017). The Pi values were visualized using an R script.
A total of 25 complete cp genomes from Asteraceae family were selected to construct phylogenetic trees to determine the systematic position of A. gracilis and A. henryi. They represented the major five subfamilies (Asteroideae, Cichorioideae, Gymnarrhenoideae, Pertyoideae, and Carduoideae). Anthriscus cerefolium Hoffm. (Apiaceae) and Kalopanax septemlobus (Thunb.) Koidz. (Araliaceae) were chosen as outgroups. Two data matrices (complete cp genomes and CDS) were selected for phylogenetic analysis. These 27 sequences data were aligned by MAFFT v.7.475 with default parameters (Katoh et al., 2019), followed by manual inspection and adjustment of sequence differences. The maximum likelihood (ML) and Bayesian inference (BI) trees was inferred with RaxML HPC 2 v8.2.12 (Stamatakis, 2014) and MrBayes v3.2.7a (Ronquist et al., 2012) accessible on CIPRES platform (https://www.phylo.org/), employing the GTRGAMMA substitution model (predicted by ModelTest-NG v0.1.7, Darriba et al., 2020), with 1000 bootstrap replicates (Miller et al., 2010). Finally, the ML and BI trees was visualized and optimized in the program FigTree v.1.4.4 (http://tree.bio.ed.ac.uk/software/figtree).
The graphical plastome maps of the newly sequenced Ainsliaea species (A. gracilis and A. henryi) are depicted in Figure 1. A total of 134 genes were identified and annotated in the cp genomes, consisting of 87 protein-coding genes, 37 tRNA genes, 8 rRNA, and 2 pseudogenes (Figure 1). Among these genes, 16 contained one intron (ndhA, ndhB, petB, petD, atpF, rpl2, rps12, rpl16, rps16, rpoC1, trnA-UGC, trnG-UCC, trnI-GAU, trnK-UUU, trnL-UAA, trnV-UAC) and 2 genes possessed two introns (clpP and ycf3). It was found to have 19 double-copy genes, including 5 protein-coding genes (ndhB, rpl2, rpl23, rps7, rps12), 4 rRNA genes (rrn16, rrn23, rrn4.5, rrn5), 8 tRNA genes (trnA-UGC, trnG-UCC, trnI-CAU, trnI-GAU, trnL-CAA, trnN-GUU, trnR-ACG, trnV-GAC), and 2 unknown function genes (ycf2, ycf15) (Table 2). The gene ycf1 also has two copies, but one of them was a pseudogene. In addition, the gene rps12 was a trans-splicing construct. It is noteworthy that the functions of ycf1, ycf2, ycf3, ycf4, and ycf15 are still unknown.
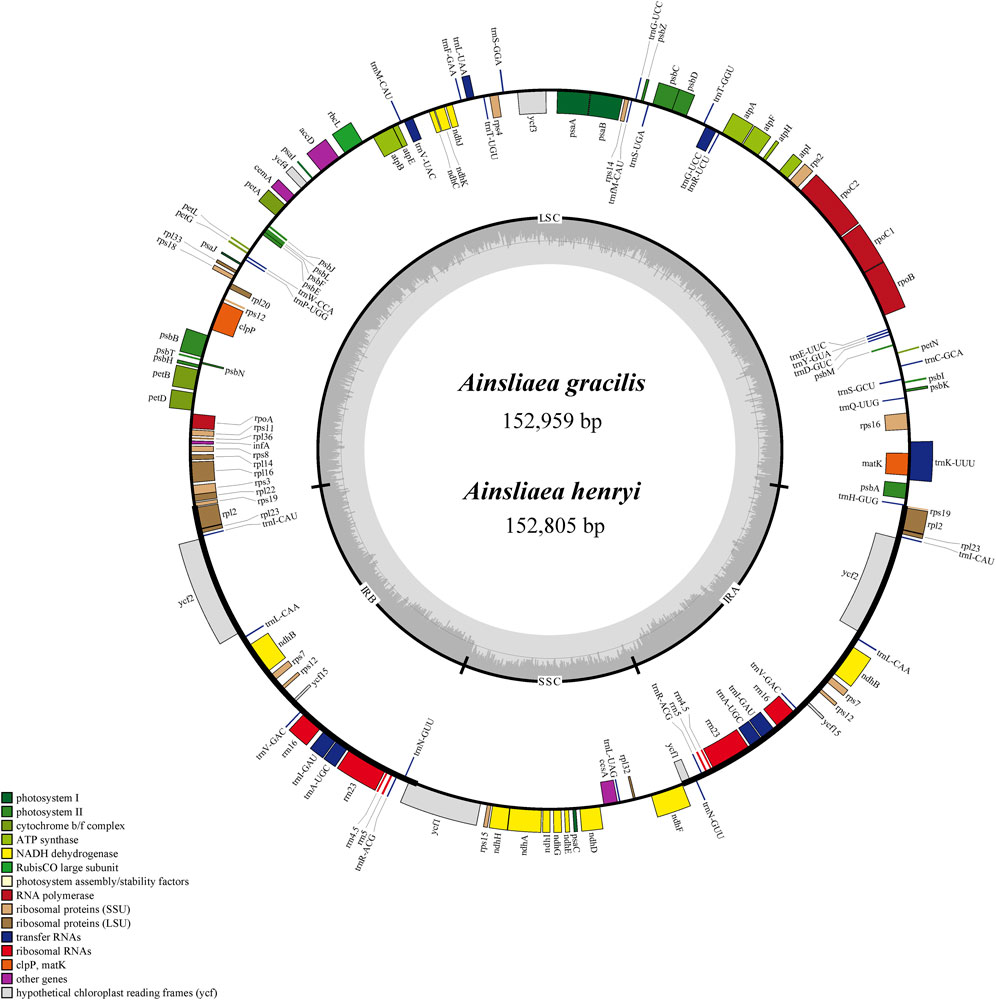
Figure 1. Gene map of the cp genomes of A. gracilis and A. henryi. Genes located on the inner periphery of the large circle are transcribed clockwise, while those on the outer periphery are transcribed counterclockwise. Genes are color-coded based on respective functions. The dashed region denotes the GC composition of the cp genomes.
The gene structure and content of cp genomes of the two Ainsliaea species were highly similar to other four Pertyeae species. The sequence length of the two Ainsliaea and four Pertyeae species ranged from 152,805 bp (A. henryi) to 153,793 bp (M. dioica) (Figure 1; Table 1). These cp genomes exhibited comparable genome structures, comprising 131–135 genes (which include 85–87 protein coding genes, 37 tRNA genes, 8 rRNA genes, and 1-3 pseudogenes, Figure 2 and Table 1). The genes could be categorized into 4 types: photosynthesis-related genes (45), self-replication-related genes (74–75), other genes (6) and genes of unknown function (6–9) (Figure 2). The six genomes contained a pair of IRs (25,049–25,212 bp) separated by the LSC (84,084–85,012 bp) and SSC region (18,378–18,683 bp) (Table 1; Figure 2). The average GC content of the genomes was 37.6%. Compared to the LSC (35.63%–35.83%) and SSC (31.15%–31.31%) regions, the IR regions possessed a higher GC content (43.07%–43.12%)
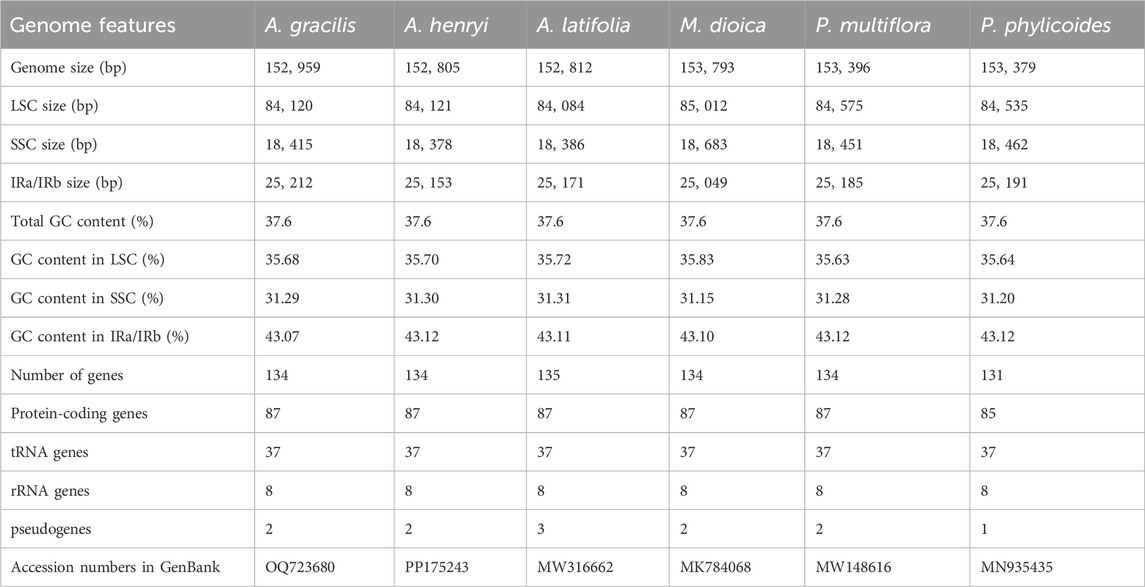
Table 1. Characteristics of complete cp genomes of six Pertyeae species.
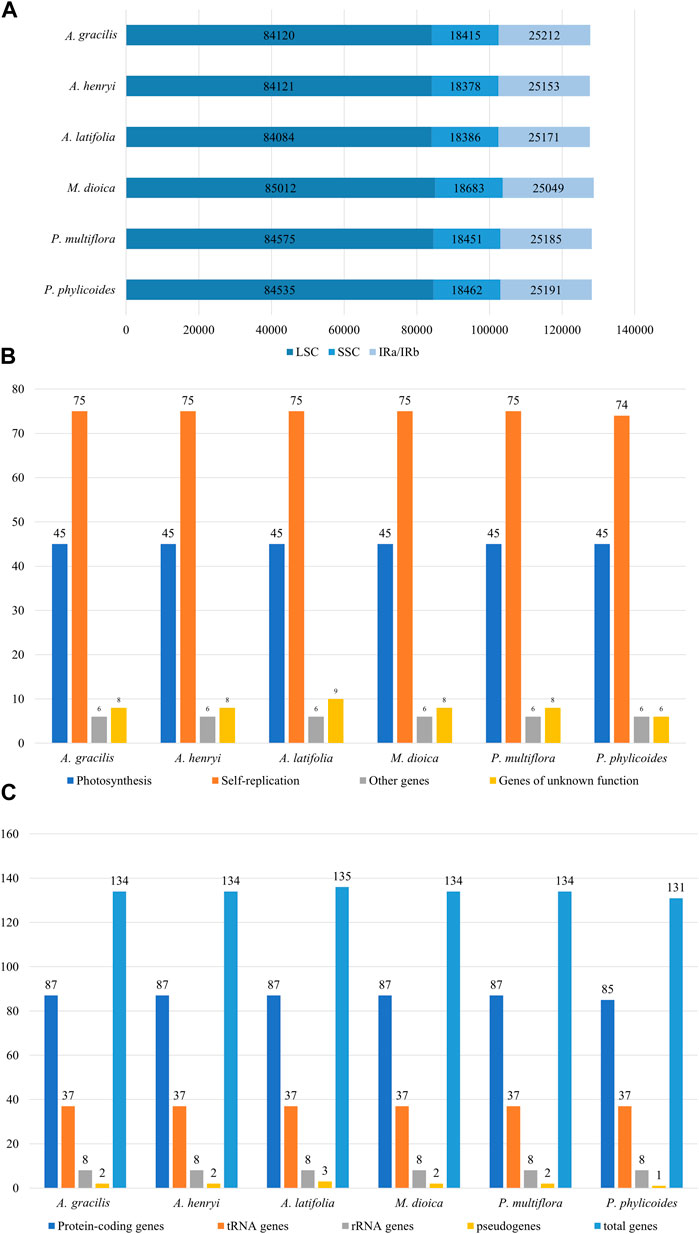
Figure 2. Characteristics of cp genomes in six Pertyeae species. (A) Genomes sizes. (B) Number of genes. (C) Number of genes categorized by functional groups.
In total 57, 58, 56, 48, 63, and 54 SSRs were found in A. gracilis, A. henryi, A. latifolia, M. dioica, P. multiflora, and P. phylicoides, respectively (Figure 3). Among these SSRs, mononucleotide repeats were the most abundant, while pentanucleotides repeats were only detected in A. gracilis and P. multiflora. Further analysis of the long repeats is provided in Figure 4. The results demonstrated that the number of long repeats in the tribe Pertyeae were highly similar. Palindromes (21–31) were the most prevalent, with a majority of them ranging from 40–49 bp in length. This was followed by forward repeats (17–29) and reverse repeats (1–7), while complement repeats were not observed.
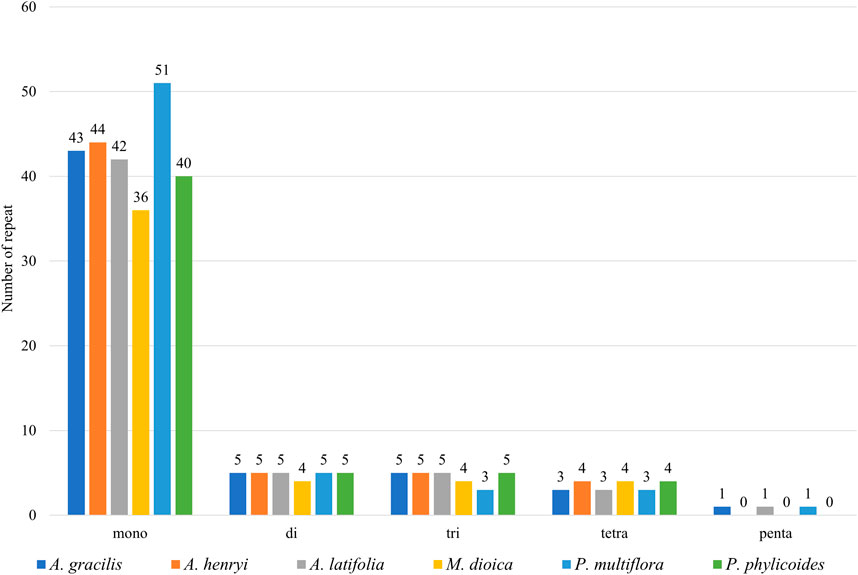
Figure 3. Comparison of the simple sequence repeats (SSRs) across the cp genomes of six Pertyeae species.
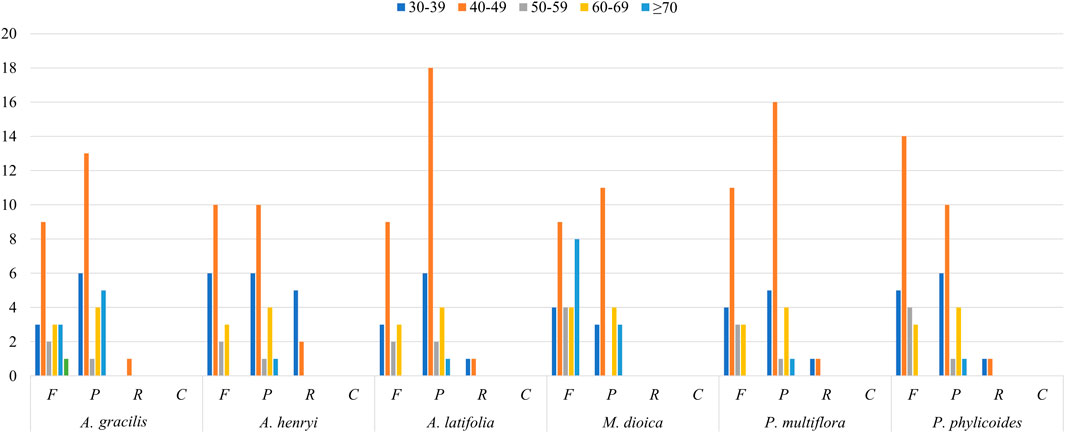
Figure 4. Long repeat sequences in the complete cp genomes of six Pertyeae species categorized as F (forward), P (palindromic), R (reverse), and C (complement).
We compared the IR/SC boundaries of the 6 species within Pertyeae (Figure 5). While the lengths of the IR regions were similar among the six species, variations in the extensions and contractions at the IR boundaries were observed. As shown in Figure 5, notably, A. gracilis exhibited the longest IR region length (25,212 bp) among the six species. Moreover, substantial differences were observed in the range of each region within Pertyeae. In particular, the rps19 gene showed expansions ranging from 60 to 61 bp, extending from the LSC to the IRb region in the five species. In M. dioica, however, this gene was entirely located within the LSC region, 68 bp away from the LSC/IRb boundary. The ndhF gene was found in the SSC regions, both positioned 23–42 bp away from the SSC/IRa boundary, except in M. dioica (101 bp away from the IRb/SSC boundary) and P. phylicoides (37 bp away from the IRb/SSC boundary). The ndhF gene is positioned near the IRa region in the four species and near the IRb region in the other two species. Similarly, the ycf1 gene was positioned at the IRb/SSC border, except for M. dioica and P. phylicoides, where it was located at the SSC/IRa border.

Figure 5. Comparisons of boundaries in the large single copy (LSC), small single copy (SSC), and inverted repeat (IR) regions across cp genomes of six Pertyeae species.
The complete cp genome sequences of six Pertyeae species were compared using mVISTA online software (Figure 6). While some variation was present, the results indicated a large extent of similarity among these cp genomes. It is worth noting that the SC regions were more variable compared to the IR regions. The conserved non-coding sequences (CNSs) were more diverse than the coding sequences.
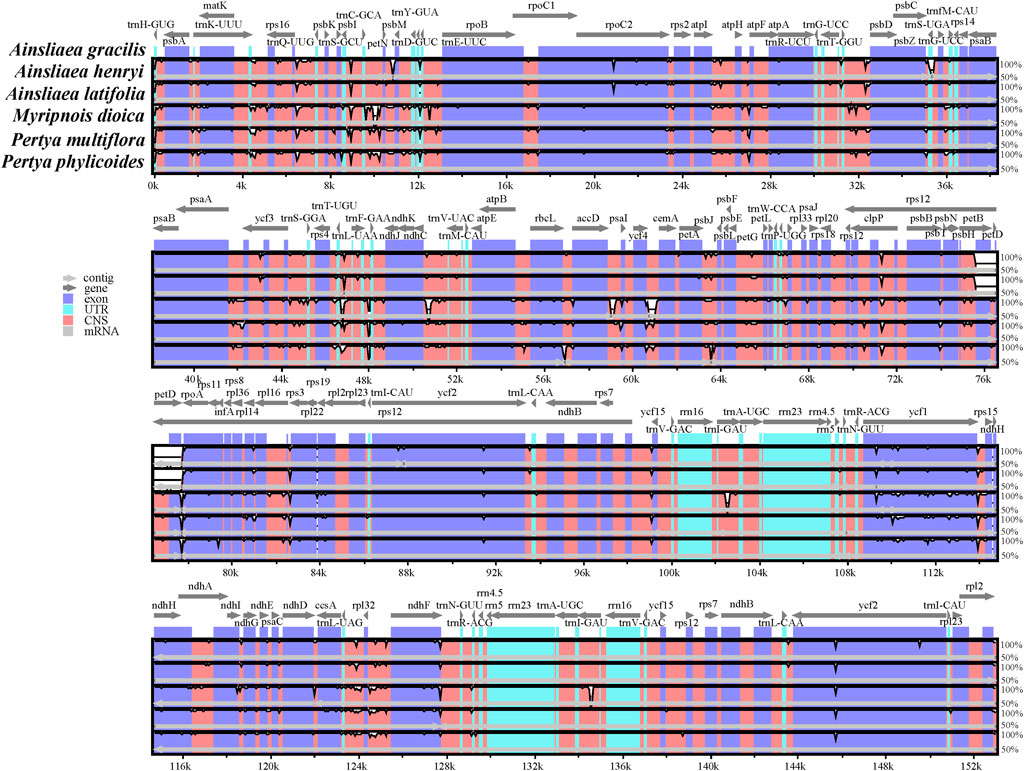
Figure 6. Visualization of genome alignment of cp genomes from six Pertyeae species using A. gracilis as the reference by mVISTA. The horizontal axis represents the coordinate in the cp genomes. The vertical scale depicts sequence similarity of aligned regions, with percent identity ranging from 50%–100%.
Nucleotide diversity (Pi) analysis revealed that a higher proportion of variable sites in non-coding regions on average compared to coding regions (Figure 7). In the coding regions, Pi values ranged from 0 to 0.24545 (trnK-UUU), with an average of 0.00633. In the non-coding regions, Pi values ranged from 0 to 0.04218 (trnT-UGU-trnL-UAA_1), with an average of 0.00804. Notably, thirteen distinct regions showcased high variability, with Pi values exceeding 0.05 (trnK-UUU, trnG-UCC, and trnT-GGU) and 0.02 (trnT-UGU-trnL-UAA_1, rps19-rpl2_2, accD-psaI, rpl22-rps19, rpl2_2-trnH-GUG, ndhC-trnV-UAC_2, trnP-UGG-psaJ, psbB-psbT, ycf4-cemA, and rps18-rpl20). These highly variable regions possess significant potential as informative markers for future phylogenetic analysis at higher taxonomic levels within Pertyeae.
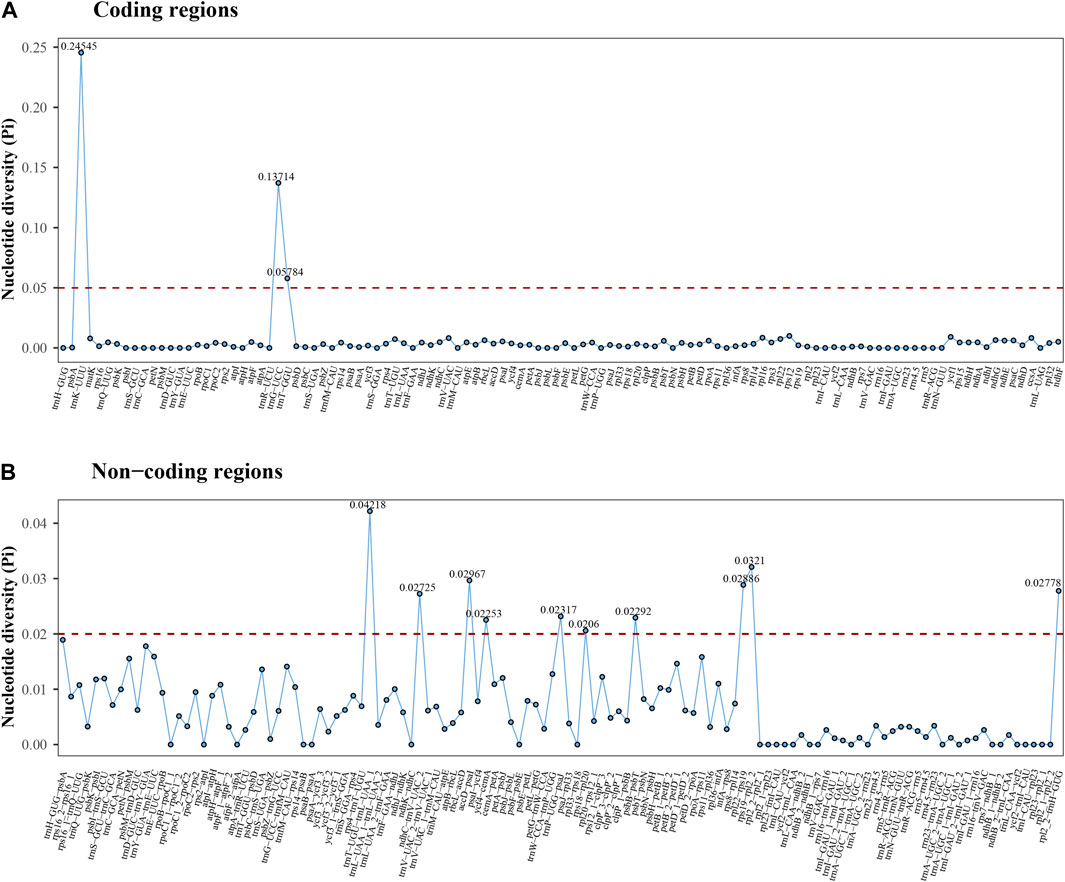
Figure 7. The nucleotide diversity (Pi) values of cp genomes in six Pertyeae species based on (A) coding regions and (B) non-coding regions. X-axis: each coding or non-coding region; Y-axis: nucleotide diversity of each region.
The ML (Figures 8, 9) and BI phylogenetic trees (Figures 10, 11) were constructed using the complete cp sequences and CDS from 27 species, representing five main clades in Asteraceae. In this analysis, A. cerefolium (Apiaceae) and K. septemlobus (Araliaceae) were used as outgroups. The phylogenetic relationships inferred from the ML and BI trees based on complete cp genomes (Figures 8, 10) and CDS (Figures 9, 11) were mostly consistent. The phylogenetic trees revealed that all sampled taxa in Asteraceae formed five significant main subfamilies (Asteroideae, Cichorioideae, Gymnarrhenoideae, Pertyoideae, and Carduoideae), encompassing ten tribes (Astereae, Gnaphalieae, Anthemideae, Senecioneae, Heliantheae, Inuleae, Cichorieae, Gymnarrheneae, Pertyeae, and Cardueae). Among these five subfamilies, Pertyoideae was closely allied with Gymnarrhenoideae and Carduoideae. Furthermore, the two newly sequenced species and four additional species from Pertyeae forme a clade (Pertyeae) with robust support. A. latifolia, A. henryi, and A. gracilis collectively formed a distinct clade, indicating their close evolutionary relationship. Moreover, the clade of Myripnois and Pertya was closely allied with Ainsliaea. Notably, Myripnois was identified as nested within the genus Pertya. Compared to Ainsliaea, Myripnois showed a closer relationship to Pertya. The results supported that the three genera belonged to the tribe Pertyeae and the subfamily Pertyoideae.
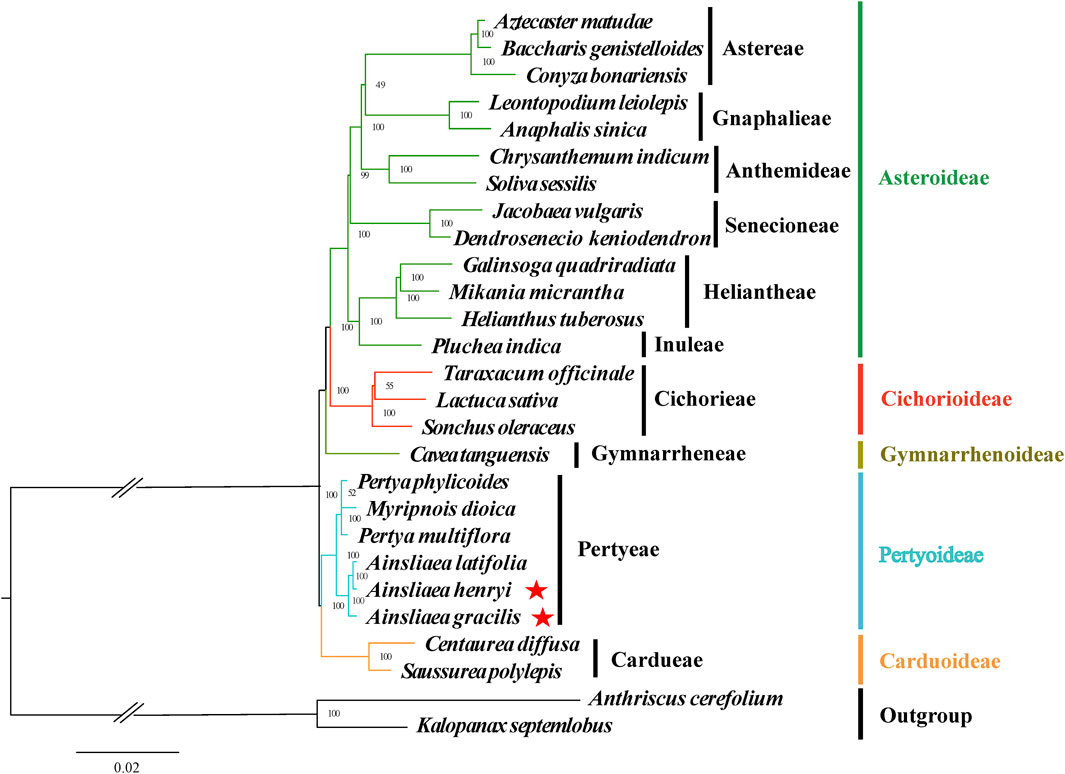
Figure 8. Molecular phylogenetic trees of 25 Asteraceae and 2 outgroups species based on complete cp genomes using maximum likelihood methods. Species are color-coded by subfamily, with branch nodes indicating bootstrap values. The red stars represent the newly sequenced species.
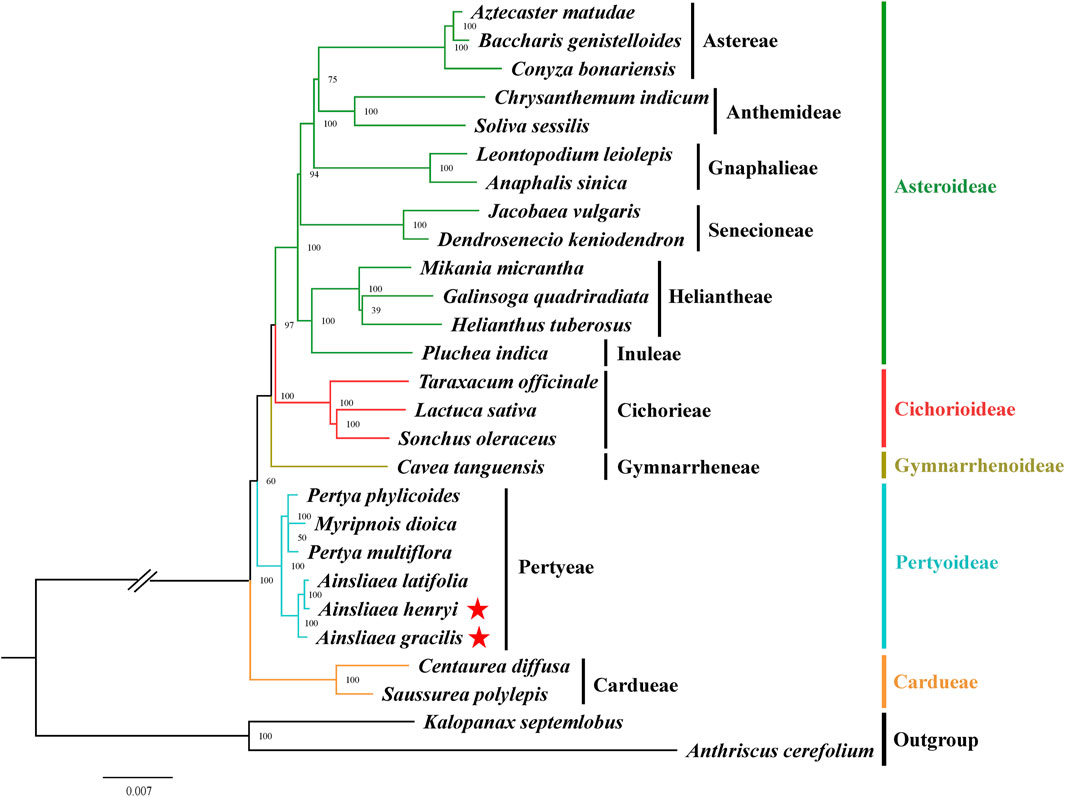
Figure 9. Molecular phylogenetic trees of 25 Asteraceae and 2 outgroups species based on coding DNA sequences using maximum likelihood methods. Species are color-coded by subfamily, with branch nodes indicating bootstrap values. The red stars represent the newly sequenced species.
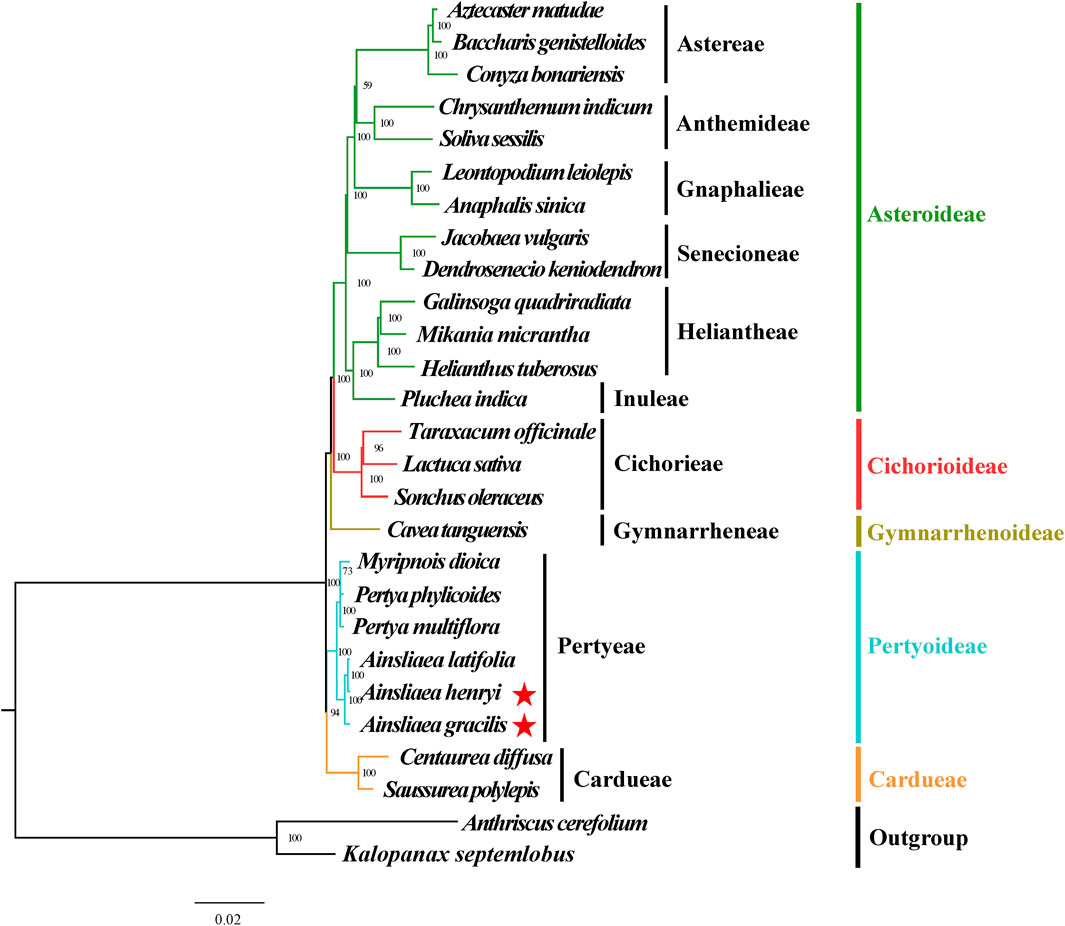
Figure 10. Molecular phylogenetic trees of 25 Asteraceae and 2 outgroups species based on complete cp genomes using Bayesian inference methods. Species are color-coded by subfamily, with branch nodes indicating bootstrap values. The red stars represent the newly sequenced species.
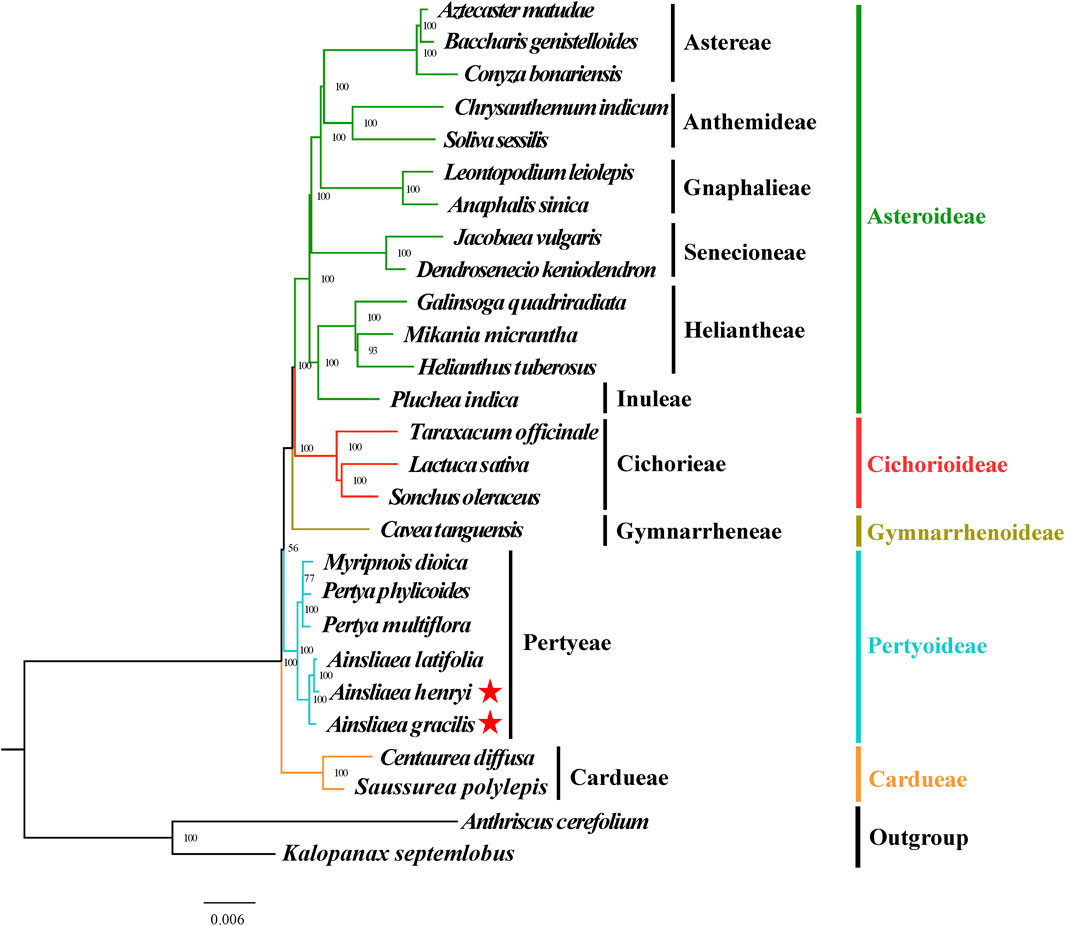
Figure 11. Molecular phylogenetic trees of 25 Asteraceae and 2 outgroups species based on coding DNA sequences using Bayesian inference methods. Species are color-coded by subfamily, with branch nodes indicating bootstrap values. The red stars represent the newly sequenced species.
The Pertyeae is a well-represented and widely distributed tribe with abundant species (around 45 spp. endemic) in China (Gao et al., 2011; Fu et al., 2016). The chloroplast, a semi-autonomous genetic organelle, possesses an independent transcription and transport system (Palmer, 1985). In most terrestrial plants, the chloroplast genomes demonstrate highly conserved structures and organization. The genomes typically exist as circular DNA molecules with a size ranging from 120–170 kb (Wicke et al., 2011).
However, only the plastomes of four Pertyeae species have been sequenced to date (Lin et al., 2019; Wang et al., 2020; Liu et al., 2021). Except for A. latifolia, the cp genomes of M. dioica (Lin et al., 2019), P. phylicoides (Wang et al., 2020), and P. multiflora (Liu et al., 2021) were individually characterized and employed in conducting separate phylogenetic analysis with other Asteraceae species. In addition, only M. dioica was utilized to perform plastome comparative analysis with around 80 species of Asteraceae. Therefore, these four plastomes have not been thoroughly compared. We consequently sequenced and compared the complete chloroplast genomes of A. gracilis and A. henryi with four related species within Pertyeae tribe.
The two cp genomes exhibited lengths of 152,959 (A. gracilis) and 152,805 (A. henryi) bp. The GC content was 37.6% in these cp genomes, as in Dolomiaea denticulate (Y. Ling) C. Shih (Shen et al., 2020) and Sonchus brachyotus DC. (Wang et al., 2021). These genomes each contained 134 genes, congruent with those of Sinosenecio albonervius Y. Liu & Q. E. Yang (Abdullah et al., 2021), which comprised 87 protein-coding genes, 37 tRNA genes, 8 rRNA genes, and 2 pseudogenes (Table 2). Out of 134 genes, 16 contained one intron, while 2 contained two introns. A total of 19 double-copy genes were also detected (Table 2). The detected pseudogenes (rps19 and ycf1) were previously reported in Ligularia Cass. (Chen et al., 2018), Artemisia L. (Liu et al., 2013) and Parasenecio W. W. Sm. & J. Small (Liu et al., 2023). Notably, rps12 was recognized as a trans-splicing gene, previously reported in other species (Choi and Park, 2015). Moreover, the use of GTG, as a start codon for rps19, was reported in other angiosperm cp genomes (Liu and Xue, 2004; Wang et al., 2016).
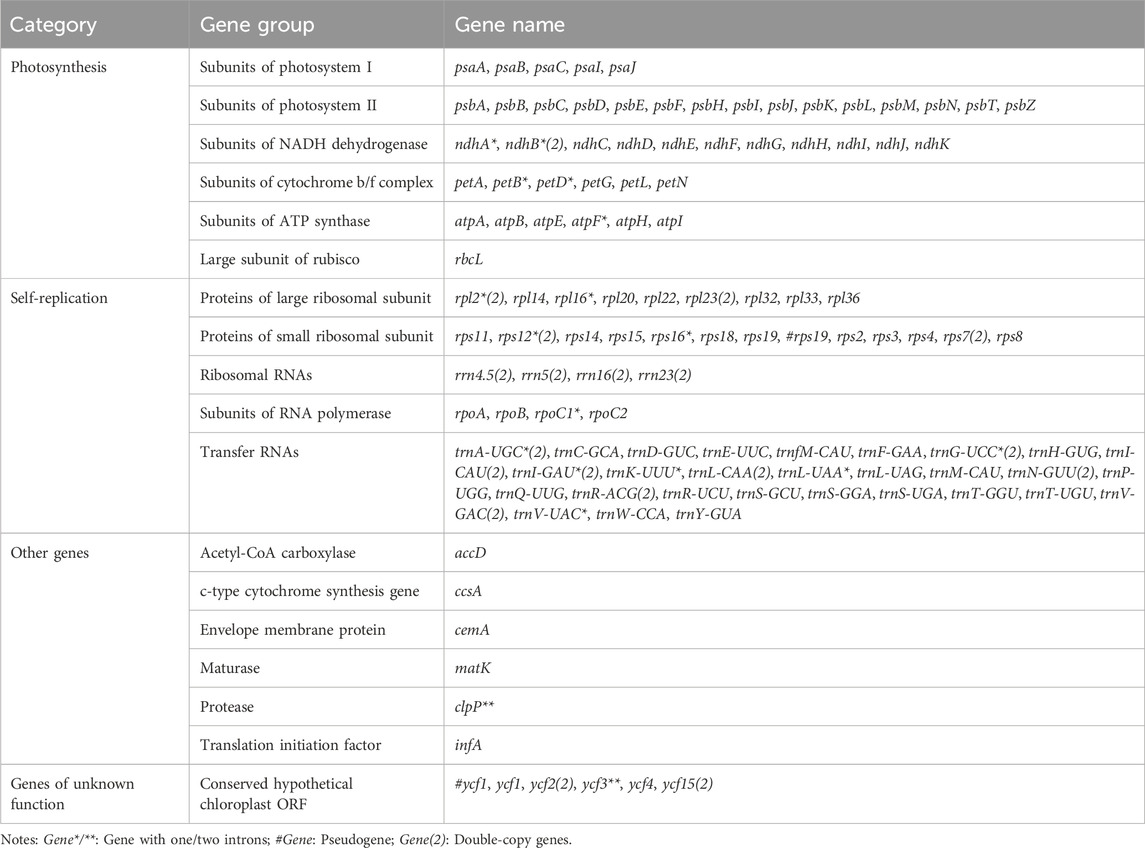
Table 2. Genes present in the cp genome of A. gracilis and A. henryi.
The cp genomes of six Pertyeae species revealed significant conservation in size, gene content, structure, and other characteristics. A typical quadripartite circular structure was present in the genomes, with a distinct separation between the LSC and SSC regions by the IR regions. It was accordant with the cp genomes observed in other Pertyeae species (Lin et al., 2019; Wang et al., 2020; Liu et al., 2021). However, the cp genome size of the genus Ainsliaea (152,805–152,959 bp) is relatively shorter compared to the genera Pertya and Myripnois (153,379–153,793 bp). According to analysis of the cp genome divergence, Ainsliaea appears to possess low levels of sequence divergence and generally conserved plastomes. The intergenic spacers were identified as the most divergent regions, with non-coding regions showing greater divergence than coding regions. Previous multispecies investigations (Hu et al., 2020) had demonstrated that intergenic spacers were highly informative phylogenetic markers.
SSRs, composed of 1-6 nucleotide repeat units, are prevalent across cp genomes. Previous studies have employed them for species identification, genetic diversity, and polymorphism research (Deguilloux et al., 2004; Redwan et al., 2015). A total of 336 SSRs were identified in the cp genomes of Pertyeae. Mononucleotide repeats predominated in sequences. This phenomenon might be caused by the fact that SSRs commonly consist of polyA or polyT repeats (Zhang et al., 2019d). It was congruent with the cp genomes of most Asteraceae species (Choi and Park, 2015; Abdullah et al., 2021; Liu et al., 2023). The discovery of these newly identified SSRs will present valuable resources for future development of genetic markers for Ainsliaea species. Moreover, this study identified thirteen highly polymorphic loci (e.g., trnK-UUU, trnG-UCC, trnT-GGU, accD-psaI, and rpl22-rps19). In the tribe Pertyeae, these variable regions could serve as possible DNA barcodes for species identification and phylogenetic analysis.
The Asteraceae, recognized as one of two largest and most diverse families of blooming plants worldwide (Bremer and Anderberg, 1994; Funk et al., 2005; Anderberg et al., 2007), comprises sixteen subfamilies (Susanna et al., 2020; Zhang et al., 2024). In our study, 5 of the 16 subfamilies were sampled. Most nodes in the phylogenetic trees displayed high support values and were similar. The phylogenetic relationships of five subfamilies were consistent with earlier investigations (Funk et al., 2009; Fu et al., 2016; Panero and Crozier, 2016; Mandel et al., 2017). Previous studies have indicated the phylogenetic position and demographic histories of the tribe Pertyeae utilizing short DNA fragments (e.g., ndhF, rbcL, and matK) or the complete cp genomes data. For instance, Zhang (2024) reconstructed an updated phylogeny of Ainsliaea based on the plastid ndhF and nrDNA (ITS, ETS) sequences. Mitsui and Setoguchi (2012) addressed the demographic histories of adaptively diverged riparian and non-riparian species of Ainsliaea using 10 nuclear DNA loci (e.g., CHS, GTF). Fu et al. (2016) proposed that Pertyeae (as recognized by Panero and Funk, 2002) was sister to the tribes Cardueae and Gymnarrheneae and nested above the subfamily Carduoideae. Moreover, Pertyeae also has been suggested as a sister group to the tribes Cichorieae (Lin et al., 2019) and Cardueae (Wang et al., 2020). However, research on the cp genomes of Ainsliaea has not yet been conducted to date.
The phylogenetic analysis elucidated the taxonomy placement of the genus Ainsliaea within the tribe Pertyeae (Pertyoideae). Compared to A. gracilis, A. henryi and A. latifolia exhibited the closest phylogenetic relationship and clustered together, which is consistent with the findings of Zhang (2024). We also identified that the genus Ainsliaea was closely related to Myripnois and Pertya. This finding was accordant with previous studies (Kim et al., 2002; Fu et al., 2016). Based on previous morphological studies, Myripnois was delineated as a distinct genus (Gao et al., 2011). However, phylogenetic analysis confirmed that Myripnois was nested within Pertya in our study, aligning with the results derived from cladistic analysis (Freire, 2017).
The complete chloroplast genomes of two Ainsliaea species were sequenced, characterized, and analyzed. Phylogenetic analysis confirmed previous studies by placing the genera Ainsliaea, Pertya, and Myripnois within the tribe Pertyeae (Pertyoideae). Additionally, it confirmed that Myripnois was found to be nested within Pertya, indicating a closer relationship to Pertya rather than to Ainsliaea. The phylogenetic results in this study aligned with previous findings and suggested a potential reevaluation and refinement of taxonomic classifications and phylogenetic relationships within the tribe Pertyeae.
The plastomes of two Ainsliaea species demonstrated a typical quadripartite structure, closely resembling those of other Pertyeae species in terms of genomic size, structure, and gene content. Despite overall conservation, the cp genomes of six Pertyeae species presented some degree of variations. Thirteen highly polymorphic regions located at coding regions and non-coding regions (e.g., trnK-UUU, trnT-GGU, accD-psaI, and rpl22-rps19) offered significant potential for developing DNA barcodes. These regions could greatly enhance species identification within tribe Pertyeae. The valuable insights of this study will improve our comprehension of cp genomic data and lay a foundation for phylogenetic relationship of the genus Ainsliaea and tribe Pertyeae.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/genbank/, OQ723680; https://www.ncbi.nlm.nih.gov/genbank/, PP175243; https://www.ncbi.nlm.nih.gov/genbank/, MW316662; https://www.ncbi.nlm.nih.gov/genbank/, MK784068; https://www.ncbi.nlm.nih.gov/genbank/, MW148616; https://www.ncbi.nlm.nih.gov/genbank/, MN935435.
XC: Writing–review and editing, Methodology, Software, Visualization, Writing–original draft. YF: Methodology, Writing–review and editing, Data curation, Formal Analysis, Investigation, Resources. TQ: Data curation, Formal Analysis, Software, Visualization, Writing–review and editing, Investigation, Methodology. HC: Data curation, Writing–review and editing, Software, Visualization. XL: Writing–review and editing, Methodology, Software, Formal Analysis. LP: Writing–review and editing, Funding acquisition, Project administration, Resources, Supervision. MC: Validation, Writing–original draft, Conceptualization, Supervision. ZF: Conceptualization, Writing–original draft, Funding acquisition, Project administration, Supervision, Validation, Writing–review and editing.
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This study was financially supported by the National Natural Science Foundation of China (No. 32000158), the National Science and Technology Fundamental Resources Investigation Program of China (No. 2021XJKK0702), the Foundation of Sustainable Development Research Center of Resources and Environment of Western Sichuan, Sichuan Normal University (No. 2020CXZYHJZX03), the Laboratory equipment research projects, Sichuan Normal University (No. SYJS20220014), and the research project of Xizang Shengyuan Environmental Engineering Co., Ltd., and Sichuan Tianshengyuan Environmental Services Co., Ltd., (No. xzkyxm-2023–001).
The authors would like to thank the editor and reviewers for the constructive criticism of the original manuscript.
Author LP was employed by Sichuan Tianshengyuan Environmental Services Co., Ltd. Author LP was employed by Sichuan Tianshengyuan Information Technology Co., Ltd.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abdullah, , Mehmood, F., Rahim, A., Heidari, P., Ahmed, I., and Poczai, P. (2021). Comparative plastome analysis of Blumea, with implications for genome evolution and phylogeny of Asteroideae. Ecol. Evol. 11, 7810–7826. doi:10.1002/ece3.7614
Allen, G. C., Flores-Vergara, M. A., Krasynanski, S., Kumar, S., and Thompson, W. F. (2006). A modified protocol for rapid DNA isolation from plant tissues using cetyltrimethylammonium bromide. Nat. Protoc. 1, 2320–2325. doi:10.1038/nprot.2006.384
Amiryousefi, A., Hyvonen, J., and Poczai, P. (2018). IRscope: an online program to visualize the junction sites of chloroplast genomes. Bioinformatics 34, 3030–3031. doi:10.1093/bioinformatics/bty220
Anderberg, A. A., Baldwin, B. G., Bayer, R. G., Breitwieser, J., Jeffrey, C., Dillon, M. O., et al. (2007). “Compositae,” in Flowering plants: eudicots: asterales. Editors J. W. Kadereit, and C. Jeffrey (Berlin, Heidelberg: Springer), 61–588. doi:10.1007/978-3-540-31051-8
Beier, S., Thiel, T., Munch, T., Scholz, U., and Mascher, M. (2017). MISA-web: a web server for microsatellite prediction. Bioinformatics 33, 2583–2585. doi:10.1093/bioinformatics/btx198
Bendich, A. J. (2004). Circular chloroplast chromosomes: the grand illusion. Plant Cell 16, 1661–1666. doi:10.1105/tpc.160771
Bremer, K., and Anderberg, A. A. (1994). Asteraceae: cladistics & classification. Portland: Timber Press.
Brudno, M., Malde, S., Poliakov, A., Do, C. B., Couronne, O., Dubchak, I., et al. (2003). Glocal alignment: finding rearrangements during alignment. Bioinformatics 19S1, i54–i62. doi:10.1093/bioinformatics/btg1005
Cabrera, A. (1977). “Mutisieae-systematic review,” in The biology and chemistry of the compositae. Editors V. H. Heywood, J. B. Harborne, and B. L. Turner (London: Academic Press), 1039–1066.
Chen, X., Zhou, J., Cui, Y., Wang, Y., Duan, B., and Yao, H. (2018). Identification of Ligularia herbs using the complete chloroplast genome as a super-barcode. Front. Pharmacol. 9, 695. doi:10.3389/fphar.2018.00695
Choi, K. S., and Park, S. (2015). The complete chloroplast genome sequence of Aster spathulifolius (Asteraceae); genomic features and relationship with Asteraceae. Gene 572, 214–221. doi:10.1016/j.gene.2015.07.020
Danecek, P., Bonfield, J. K., Liddle, J., Marshall, J., Ohan, V., Pollard, M. O., et al. (2021). Twelve years of SAMtools and BCFtools. Gigascience 10, giab008–4. doi:10.1093/gigascience/giab008
Daniell, H., Lin, C. S., Yu, M., and Chang, W. J. (2016). Chloroplast genomes: diversity, evolution, and applications in genetic engineering. Genome Biol. 17, 134. doi:10.1186/s13059-016-1004-2
Darriba, D., Posada, D., Kozlov, A. M., Stamatakis, A., Morel, B., and Flouri, T. (2020). ModelTest-NG: a new and scalable tool for the selection of DNA and protein evolutionary models. Mol. Biol. Evol. 37, 291–294. doi:10.1093/molbev/msz189
Deguilloux, M. F., Pemonge, M. H., and Petit, R. J. (2004). Use of chloroplast microsatellites to differentiate oak populations. Ann. For. Sci. 61, 825–830. doi:10.1051/forest:2004078
Frazer, K. A., Pachter, L., Poliakov, A., Rubin, E. M., and Dubchak, I. (2004). VISTA: computational tools for comparative genomics. Nucleic Acids Res. 32, W273–W279. doi:10.1093/nar/gkh458
Freire, S. E. (2007). Systematic revision and phylogeny of Ainsliaea DC. (Asteraceae, Mutisieae). Ann. Mo. Bot. Gard. 94, 7979–8191. doi:10.3417/0026-6493(2007)94[79:SRAPOA]2.0.CO;2
Freire, S. E. (2012). Systematics of the Japanese Macroclinidium and × macropertya (Asteraceae, Pertyoideae, Pertyeae) and their phylogeny inferred from morphology. Syst. Bot. 37, 554–572. doi:10.1600/036364412x635584
Freire, S. E. (2017). Revision of the asian genus Pertya (Asteraceae, Pertyoideae). Syst. Bot. Monogr. 101, 1–90.
Fu, Z. X., Jiao, B. H., Nie, B., Zhang, G. J., and Gao, T. G. (2016). A comprehensive generic-level phylogeny of the sunflower family: implications for the systematics of Chinese Asteraceae. J. Syst. Evol. 54, 416–437. doi:10.1111/jse.12216
Funk, V. A., Anderberg, A. A., Baldwin, B. G., Bayer, R. J., Bonifacino, J. M., Breitwieser, I., et al. (2009). “Compositae metatrees: the next generation,” in Systematics, evolution, and biogeography of compositae. Editors V. A. Funk, A. Susanna, T. F. Stuessy, and R. J. Bayer (Vienna: IAPT), 747–777.
Funk, V. A., Bayer, R. J., Keeley, S., Chan, R., Watson, L., Gemeinholzer, B., et al. (2005). “Everywhere but Antarctica: using a supertree to understand the diversity and distribution of the Compositae,” in Plant diversity and complexity patterns: local, regional, and global dimensions. Editors I. Friis, and H. Balslev (Copenhagen: Royal Danish Academy of Sciences and Letters), 343–373.
Gao, T. G., Freire, S. E., and Hind, D. (2011). “Ainsliaea, Pertya and Myripnois,” in Flora of China. Editors Z. Y. Wu, P. H. Raven, and D. Y. Hong (Beijing, Saint Louis: Science Press & Missouri Botanical Garden), 15–32.
Greiner, S., Lehwark, P., and Bock, R. (2019). OrganellarGenomeDRAW (OGDRAW) version 1.3.1: expanded toolkit for the graphical visualization of organellar genomes. Nucleic Acids Res. 47, W59–W64. doi:10.1093/nar/gkz238
Hind, D. (2007). “Mutisieae,” in Flowering plants. Eudicots: asterales. Editors J. W. Kadereirt, and C. Jeffrey (Berlin & Heidelberg: Springer), 90–123. doi:10.1007/978-3-540-31051-8
Hu, G., Cheng, L., Huang, W., Cao, Q., Zhou, L., Jia, W., et al. (2020). Chloroplast genomes of seven species of Coryloideae (Betulaceae): structures and comparative analysis. Genome 63, 337–348. doi:10.1139/gen-2019-0153
Jansen, R. K., and Ruhlman, T. A. (2012). “Plastid genomes of seed plants,” in Genomics of chloroplasts and mitochondria. Editors R. Bock, and V. Knoop (Dordrecht: Springer), 103–126. doi:10.1007/978-94-007-2920-9_5
Jin, G., Li, W., Song, F., Yang, L., Wen, Z., and Feng, Y. (2023). Comparative analysis of complete Artemisia subgenus Seriphidium (Asteraceae: Anthemideae) chloroplast genomes: insights into structural divergence and phylogenetic relationships. BMC Plant Biol. 23, 136. doi:10.1186/s12870-023-04113-1
Katinas, L., Pruski, J., Sancho, G., and Tellería, M. C. (2008). The subfamily mutisioideae (Asteraceae). Bot. Rev. 74, 469–716. doi:10.1007/s12229-008-9016-6
Katoh, K., Rozewicki, J., and Yamada, K. D. (2019). MAFFT online service: multiple sequence alignment, interactive sequence choice and visualization. Brief. Bioinform 20, 1160–1166. doi:10.1093/bib/bbx108
Kim, H. G., Loockerman, D. J., and Jansen, R. K. (2002). Systematic implications of ndhF sequence variation in the Mutisieae (Asteraceae). Syst. Bot. 27, 598–609. doi:10.1043/0363-6445-27.3.598
Kurtz, S., Choudhuri, J. V., Ohlebusch, E., Schleiermacher, C., Stoye, J., and Giegerich, R. (2001). REPuter: the manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 29, 4633–4642. doi:10.1093/nar/29.22.4633
Langmead, B., Wilks, C., Antonescu, V., and Charles, R. (2019). Scaling read aligners to hundreds of threads on general-purpose processors. Bioinformatics 35, 421–432. doi:10.1093/bioinformatics/bty648
Lin, N., Zhang, X., Deng, T., Zhang, J., Meng, A., Wang, H., et al. (2019). Plastome sequencing of Myripnois dioica and comparison within Asteraceae. Plant divers. 41, 315–322. doi:10.1016/j.pld.2019.07.003
Liu, Q. P., and Xue, Q. Z. (2004). Codon usage in the chloroplast genome of rice (Oryza sativa L. ssp. japonica). Acta Agron. Sin. 30, 1220–1224.
Liu, X., Han, M., Chen, J., Zhang, X., Chen, Z., Tang, Y., et al. (2022). Characterization of the complete chloroplast genome sequence of Chinese endemic species of Nouelia insignis (Hyalideae, Asteraceae) and its phylogenetic implications. Mitochondrial DNA B Resour. 7, 600–602. doi:10.1080/23802359.2021.1921629
Liu, X., Luo, J., Zhang, M., Wang, Q., Liu, J., Wu, D., et al. (2023). Phylogenomic analysis of two species of Parasenecio and comparative analysis within tribe Senecioneae (Asteraceae). Diversity 15, 563. doi:10.3390/d15040563
Liu, Y., Huo, N., Dong, L., Wang, Y., Zhang, S., Young, H. A., et al. (2013). Complete chloroplast genome sequences of Mongolia medicine Artemisia frigida and phylogenetic relationships with other plants. PLoS One 8, e57533. doi:10.1371/journal.pone.0057533
Liu, Y., Zhang, H., Wu, Q., Huang, W., Wu, T., Guan, M., et al. (2021). Characterization and phylogenetic analysis of the complete chloroplast genome sequence of Pertya multiflora (Asteraceae), a rare and endangered wild plant species in China. Mitochondrial DNA B Resour. 6, 1934–1936. doi:10.1080/23802359.2021.1936671
Ma, Y. P., Zhao, L., Zhang, W. J., Zhang, Y. H., Xing, X., Duan, X. X., et al. (2020). Origins of cultivars of Chrysanthemum—evidence from the chloroplast genome and nuclear LFY gene. J. Syst. Evol. 58, 925–944. doi:10.1111/jse.12682
Mandel, J. R., Barker, M. S., Bayer, R. J., Dikow, R. B., Gao, T. G., Jones, K. E., et al. (2017). The compositae tree of life in the age of phylogenomics. J. Syst. Evol. 55, 405–410. doi:10.1111/jse.12265
Marcais, G., Delcher, A. L., Phillippy, A. M., Coston, R., Salzberg, S. L., and Zimin, A. (2018). MUMmer4: a fast and versatile genome alignment system. PLoS Comput. Biol. 14, e1005944. doi:10.1371/journal.pcbi.1005944
Miller, M. A., Pfeiffer, W., and Schwartz, T. (2010). Creating the CIPRES science gateway for inference of large phylogenetic trees. Proc. 2010 Gatew. Comput. Environ. Workshop (GCE) 14, 1–8. doi:10.1109/GCE.2010.5676129
Mitsui, Y., Chen, S. T., Zhou, Z. K., Peng, C. I., Deng, Y. F., and Setoguchi, H. (2008). Phylogeny and biogeography of the genus Ainsliaea (Asteraceae) in the Sino-Japanese region based on nuclear rDNA and plastid DNA sequence data. Ann. Bot. 101, 111–124. doi:10.1093/aob/mcm267
Mitsui, Y., and Setoguchi, H. (2012). Demographic histories of adaptively diverged riparian and non-riparian species of Ainsliaea (Asteraceae) inferred from coalescent analyses using multiple nuclear loci. BMC Evol. Biol. 12, 254. doi:10.1186/1471-2148-12-254
Mower, J. P., and Vickrey, T. L. (2018). “Chapter nine–structural diversity among plastid genomes of land plants,” in Advances in botanical research Editors S. M. Chaw, and R. K. Jansen (Cambridge: Academic Press), 263–292. doi:10.1016/bs.abr.2017.11.013
Palmer, J. D. (1985). Comparative organization of chloroplast genomes. Annu. Rev. Genet. 19, 325–354. doi:10.1146/annurev.ge.19.120185.001545
Panero, J. L. (2008). Shared molecular signatures support the inclusion of Catamixis in subfamily Pertyoideae (Asteraceae). Phytologia 90, 418–424.
Panero, J. L., and Crozier, B. S. (2016). Macroevolutionary dynamics in the early diversification of Asteraceae. Mol. Phylogenet. Evol. 99, 116–132. doi:10.1016/j.ympev.2016.03.007
Panero, J. L., and Funk, V. A. (2002). Toward a phylogenetic subfamilial classification for the Compositae (Asteraceae). Proc. Biol. Soc. Wash. 115, 905–922.
Panero, J. L., and Funk, V. A. (2008). The value of sampling anomalous taxa in phylogenetic studies: major clades of the Asteraceae revealed. Mol. Phylogenet. Evol. 47, 757–782. doi:10.1016/j.ympev.2008.02.011
Peng, J. Y., Zhang, X. S., Zhang, D. G., Wang, Y., Deng, T., Huang, X. H., et al. (2022). Newly reported chloroplast genome of Sinosenecio albonervius Y. Liu & Q. E. Yang and comparative analyses with other Sinosenecio species. BMC Genomics 23, 639. doi:10.1186/s12864-022-08872-3
Peng, Y., Yang, C., and Luo, Y. (2020). Ainsliaea daheishanensis (Asteraceae): a new species from China. PhytoKeys 138, 233–239. doi:10.3897/phytokeys.138.38566
Prjibelski, A., Antipov, D., Meleshko, D., Lapidus, A., and Korobeynikov, A. (2020). Using SPAdes de novo assembler. Curr. Protoc. Bioinforma. 70, e102. doi:10.1002/cpbi.102
Qu, X. J., Moore, M. J., Li, D. Z., and Yi, T. S. (2019). PGA: a software package for rapid, accurate, and flexible batch annotation of plastomes. Plant Methods 15, 50. doi:10.1186/s13007-019-0435-7
Redwan, R. M., Saidin, A., and Kumar, S. V. (2015). Complete chloroplast genome sequence of MD-2 pineapple and its comparative analysis among nine other plants from the subclass Commelinidae. BMC Plant Biol. 15, 196. doi:10.1186/s12870-015-0587-1
Ronquist, F., Teslenko, M., van der Mark, P., Ayres, D. L., Darling, A., Hohna, S., et al. (2012). MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 61, 539–542. doi:10.1093/sysbio/sys029
Rozas, J., Ferrer-Mata, A., Sanchez-DelBarrio, J. C., Guirao-Rico, S., Librado, P., Ramos-Onsins, S. E., et al. (2017). DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol. Biol. Evol. 34, 3299–3302. doi:10.1093/molbev/msx248
Salih, R. M., Majesky, L., Schwarzacher, T., Gornall, R., and Heslop-Harrison, P. (2017). Complete chloroplast genomes from apomictic Taraxacum (Asteraceae): identity and variation between three microspecies. PLoS One 12, e0168008. doi:10.1371/journal.pone.0168008
Shen, J., Zhang, X., Landis, J. B., Zhang, H., Deng, T., Sun, H., et al. (2020). Plastome evolution in Dolomiaea (Asteraceae, Cardueae) using phylogenomic and comparative analyses. Front. Plant Sci. 11, 376. doi:10.3389/fpls.2020.00376
Stamatakis, A. (2014). RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313. doi:10.1093/bioinformatics/btu033
Susanna, A., Baldwin, B. G., Bayer, R. J., Bonifacino, J. M., Garcia-Jacas, N., Keeley, S. C., et al. (2020). The classification of the compositae: a tribute to vicki ann Funk (1947–2019). Taxon 69, 807–814. doi:10.1002/tax.12235
Wang, B., Zhao, Q., Wang, X. H., and Fu, Z. X. (2020). The complete chloroplast genome of Pertya phylicoides (Asteraceae, Pertyeae): a shurby endemic species from China. Mitochondrial DNA B Resour. 5, 963–964. doi:10.1080/23802359.2020.1722763
Wang, C., Liu, J., Su, Y., Li, M., Xie, X., and Su, J. (2021). Complete chloroplast genome sequence of Sonchus brachyotus helps to elucidate evolutionary relationships with related species of Asteraceae. Biomed. Res. Int. 2021, 9410496. doi:10.1155/2021/9410496
Wang, J., Ye, Y. S., and Ye, H. G. (2010). Ainsliaea asaroides sp. nov. (Asteraceae) from guangdong, China. Nord. J. Bot. 28, 196–198. doi:10.1111/j.1756-1051.2009.00590.x
Wang, Y., Zhan, D. F., Jia, X., Mei, W. L., Dai, H. F., Chen, X. T., et al. (2016). Complete chloroplast genome sequence of Aquilaria sinensis (Lour.) Gilg and evolution analysis within the Malvales order. Front. Plant Sci. 7, 280. doi:10.3389/fpls.2016.00280
Wick, R. R., Schultz, M. B., Zobel, J., and Holt, K. E. (2015). Bandage: interactive visualization of de novo genome assemblies. Bioinformatics 31, 3350–3352. doi:10.1093/bioinformatics/btv383
Wicke, S., Schneeweiss, G. M., dePamphilis, C. W., Muller, K. F., and Quandt, D. (2011). The evolution of the plastid chromosome in land plants: gene content, gene order, gene function. Plant Mol. Biol. 76, 273–297. doi:10.1007/s11103-011-9762-4
Yu, S. H., Yang, X. C., Tian, X. Y., Liu, X. F., Lu, X., Huang, C. P., et al. (2022). The complete chloroplast genome sequence of the monotypic and enigmatic genus Cavea (tribe Gymnarrheneae) and a comparison with other species in Asteraceae. J. Genet. 101, 20. doi:10.1007/s12041-022-01360-3
Yun, S., and Kim, S. C. (2022). Comparative plastomes and phylogenetic analysis of seven Korean endemic Saussurea (Asteraceae). BMC Plant Biol. 22, 550. doi:10.1186/s12870-022-03946-6
Zeng, R. T., Fang, X., Tian, X. H., Shi, Z. R., Dong, X. Y., Zhuo, Z. G., et al. (2016). Triterpenoids from Ainsliaea henryi and their cytotoxic activities. Phytochem. Lett. 15, 174–179. doi:10.1016/j.phytol.2016.01.002
Zhang, C. F., Tian, J., Cheng, Y. H., Peng, S., Chen, Y. S., Gao, T. G., et al. (2024). An updated phylogeny of Ainsliaea (Asteraceae: Pertyoideae) and its implications for classification and habit evolution. Taxon, 1–23. doi:10.1002/tax.13202
Zhang, G., Yang, J., Zhang, C., Jiao, B., Panero, J. L., Cai, J., et al. (2024). Nuclear phylogenomics of Asteraceae with increased sampling provides new insights into convergent morphological and molecular evolution. Plant Commun. 5, 100851. doi:10.1016/j.xplc.2024.100851
Zhang, M. J., Su, X. X., An, C., Li, H. Q., and Zhang, Z. (2021). Ainsliaea polystachya (Asteraceae), a new species from Fujian, China based on morphological and molecular evidence. Phytotaxa 497, 277–284. doi:10.11646/phytotaxa.497.3.6
Zhang, M. J., Yu, W. J., and Li, H. Q. (2019a). Ainsliaea simplicissima (Asteraceae), a new species from southeast China and its phylogenetic position. Phytotaxa 424, 243–252. doi:10.11646/phytotaxa.424.4.4
Zhang, X., Deng, T., Moore, M. J., Ji, Y., Lin, N., Zhang, H., et al. (2019b). Plastome phylogenomics of Saussurea (Asteraceae: Cardueae). BMC Plant Biol. 19, 290. doi:10.1186/s12870-019-1896-6
Zhang, Y. Y., Liu, F., Wang, X. Q., Shi, X. B., Tian, N., Lai, Z. X., et al. (2019c). Characterization of the complete chloroplast genome of Gerbera jamesonii Bolus in China and phylogenetic relationships. Mitochondrial DNA B Resour. 4, 2706–2707. doi:10.1080/23802359.2019.1644230
Keywords: Ainsliaea, Pertyeae, chloroplast genome, genome comparative analysis, phylogeny
Citation: Chen X, Feng Y, Qu T, Chen H, Liu X, Pang L, Chen M and Fu Z (2024) Complete chloroplast genomes of two Ainsliaea species and the phylogenetic analysis in the tribe Pertyeae. Front. Genet. 15:1408114. doi: 10.3389/fgene.2024.1408114
Received: 27 March 2024; Accepted: 08 July 2024;
Published: 23 July 2024.
Edited by:
Yan Cheng, Fujian Agriculture and Forestry University, ChinaReviewed by:
Liang Liu, University of Georgia, United StatesCopyright © 2024 Chen, Feng, Qu, Chen, Liu, Pang, Chen and Fu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ming Chen, NDUwMzE2NDAyQHFxLmNvbQ==; Zhixi Fu, ZnV6eDIwMTdAc2ljbnUuZWR1LmNu
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.