 Sara L. Cook1
Sara L. Cook1 Christian Stout1Lindsey Kirkeby2
Christian Stout1Lindsey Kirkeby2 Noemi Vidal-Folch1Devin Oglesbee1
Noemi Vidal-Folch1Devin Oglesbee1 Linda Hasadsri1
Linda Hasadsri1 Duygu Selcen3
Duygu Selcen3 Margherita Milone3Daniel Anderson4
Margherita Milone3Daniel Anderson4 Nathan P. Staff3*
Nathan P. Staff3*- 1Department of Laboratory Medicine and Pathology, Mayo Clinic, Rochester, MN, United States
- 2Center for Regenerative Medicine, Mayo Clinic, Rochester, MN, United States
- 3Department of Neurology, Mayo Clinic, Rochester, MN, United States
- 4Department of Neurology, Mayo Clinic Health System, La Crosse, WI, United States
Introduction: Spinal muscular atrophy (SMA) is caused by homozygous loss of the SMN1 gene with SMN2 gene copy number correlating with disease severity. Rarely SMA is caused by a deletion on one allele and a pathogenic variant on the other. The pathogenic missense variant c.5C>G (p.Ala2Gly) correlates with a mild disease phenotype that does not correlate with SMN2 copy number. In a mouse model the c.5C>G transgene produces SMN that is thought to form partially functional SMN complexes, but levels in humans have not yet been investigated.
Methods: We identified two patients with mild SMA caused by a heterozygous deletion of SMN1 and the heterozygous variant, c.5C>G. Molecular findings were confirmed with deletion/duplication analysis and Sanger sequencing. Skin fibroblasts were collected and cultured, and SMN expression was analyzed using immunofluorescence.
Results: Two patients with slowly progressing mild weakness were confirmed to have heterozygous pathogenic missense variant c.5C>G and a heterozygous deletion of SMN1. Their clinical presentation revealed much milder disease progression than patients with matched SMN2 copy number. Analysis of the patients’ fibroblasts revealed much higher numbers of SMN nuclear complexes than a patient with a homozygous SMN1 deletion and matched SMN2 copy number.
Conclusions: These case reports reinforce that the rare c.5C>G variant causes mild disease. Furthermore, the analysis of SMA nuclear gems in patient samples supports the theory that the p.Ala2Gly SMN can form partially functional SMN complexes that may carry out essential cellular functions and result in mild disease.
Introduction
Spinal muscular atrophy (SMA) is an autosomal recessive neurodegenerative disease which occurs in one out of every 10,000 live births (Pearn, 1978; Sugarman et al., 2012). SMA is characterized by degeneration of the alpha motor neurons of the spinal cord leading to progressive muscle weakness, and in cases of severe SMA, progression to respiratory failure and death before the age of two if disease-modifying therapies are not initiated (Burghes and Beattie, 2009; Faravelli et al., 2015). However, SMA is a clinically heterogeneous disease, historically divided into 4 different types, with Type I being the most severe of the four types. Patients with Type II SMA present with marked delay in gross motor development by 6–18 months of life, never become ambulatory, and suffer from progressive weakness, scoliosis, and restrictive lung disease (Faravelli et al., 2015). Patients with Type III SMA are ambulatory initially, but often require wheelchair assistance by adolescence (Faravelli et al., 2015). The mildest type of SMA, Type IV, generally presents with mild proximal limb girdle weakness that progresses slowly (Faravelli et al., 2015). Rarely patients exhibit disease more severe than Type I SMA and are considered to have Type 0 SMA. Type 0 SMA is characterized by diminished fetal movements detected prenatally and subsequent presentation at birth with asphyxia and severe hypotonia (MacLeod et al., 1999).
Most commonly SMA, with its four types, is caused by pathogenic variants in the survival motor neuron 1 (SMN1) gene. Humans also have the survival motor neuron 2 (SMN2) gene, which differs from SMN1 gene by five nucleotides (Lefebvre et al., 1995; Burglen et al., 1996), all of which are in non-coding regions, except for the cysteine to thymine variation in exon 7 (Lefebvre et al., 1995; Burglen et al., 1996). Ultimately, this variation is translationally silent and the amino acid sequence of SMN2 is identical to SMN1; however, the variation affects the alternative splicing such that 90% of transcripts are missing exon 7 and results in an unstable protein that is rapidly degraded (Cartegni and Krainer, 2002; Vitte et al., 2007; Burnett et al., 2009; Cho and Dreyfuss, 2010; Singh and Singh, 2018). Accordingly, there is an inverse relationship between SMN2 copy number and disease severity. In general, Type 0 SMA have one copy of SMN2, Type I SMA patients have two copies of SMN2, Type II SMA patients have three copies of SMN2, Type III SMA patients have three or four copies of SMN2, and Type IV SMA patients have four or greater copies of SMN2 (Feldkotter et al., 2002; Wadman et al., 2020). A small percent of SMA patients (non-5q SMA patients) have pathogenic variants in other genes, such as BICD2, VRK1, and VAPB, among others (Peeters et al., 2014).
Approximately 95% of affected SMA patients, regardless of type, have a homozygous deletion of SMN1 exon 7 (Hahnen et al., 1995). Therefore, when a diagnosis of SMA is suspected, first-tier molecular analysis consists of deletion/duplication testing which utilizes techniques to quantify SMN1 exon 7. Such techniques include multiplex ligation-dependent probe amplification (MLPA), quantitative PCR (qPCR), or droplet digital PCR (ddPCR). Often, these testing strategies will simultaneously quantify SMN2 copy number. Five percent of SMA patients are compound heterozygotes and have a deletion on one allele and a more subtle pathogenic variant in the other allele (Wirth, 2000). Following this, if the results from MLPA, qPCR, or ddPCR methods return as negative in a patient with clinical and electrodiagnostic findings consistent with SMA, it is recommended to perform full gene analysis of SMN1. One percent of SMA patients have a loss of function SMN1 missense variant (McAndrew et al., 1997; Wirth, 2000). Of these loss of function missense variants, some are reported to cause disease more severe or milder than predicted by SMN2 copy number. The pathogenic missense variant c.5C>G (p.Ala2Gly) in compound heterozygosity with an SMN1 deletion is known to correlate with a mild disease phenotype that does not correlate with SMN2 copy number. These patients are indeed reported to be ambulatory into adolescence and adulthood even with low copy numbers of SMN2 (Parsons et al., 1998; Bai et al., 2014; Yamamoto et al., 2014; Kubo et al., 2015; Mendonca et al., 2020).
The missense variant c.5C>G occurs in exon 1 of SMN1. Exon 1 of SMN1 is identical to exon 1 of SMN2, making design of full gene analysis challenging. There are reports of efficient long-range PCR that can amplify exons 1-8 of SMN1 (Kubo et al., 2015; Miller et al., 2023). This method utilizes the variation in exon 8 for primer design to specifically amplify SMN1, not SMN2. However, most clinical tests utilizing long range PCR only amplify exons 2a-7. This strategy utilizes the variation in intron 7 for primer design to specifically amplify SMN1, not SMN2. SMN1 exon 1 is separately PCR-amplified. However, PCR primers will amplify both SMN1 exon 1 and SMN2 exon 1. Therefore, if a pathologic variant is detected in exon 1, a definitive molecular diagnosis cannot be made. In these cases, a clinician must use these molecular findings to supplement a clinical diagnosis of SMA.
Exon 1 of SMN1 codes for a region just upstream of the Gemin-2 binding domain which is important for SMN complex assembly (Lorson and Androphy, 1998; Otter et al., 2007; Ogawa et al., 2009; Zhang et al., 2011). The SMN complex, which includes SMN, Gemin 2–8, and Unrip, plays an important role in small nuclear ribonucleoprotein (snRNP) assembly (Paushkin et al., 2002; Gubitz et al., 2004; Kolb et al., 2007). SMN complexes are thought to be involved in recycling and regeneration of spliceosomal U snRNPs (Pellizzoni et al., 1998). SMN complexes are also thought to be involved in pre-mRNA splicing by regulation of the Sm core protein composition of U snRNPs (Meister et al., 2001). This complex is observed in prominent nuclear dot-like structures termed gemini of coiled bodies, or “gems” (Liu and Dreyfuss, 1996).
Tissues collected from SMA patients, including lymphocytes, fibroblasts, and induced pluripotent stems cells, contain fewer gems (Coovert et al., 1997; Lefebvre et al., 1997; Ebert et al., 2009). The number of gems is inversely correlated with SMA severity (Coovert et al., 1997; Lefebvre et al., 1997; Patrizi et al., 1999). Furthermore, patient-specific fibroblasts treated with antisense oligonucleotides to correct aberrant SMN2 splicing to increase SMN levels resulted in a significant increase in the number of gems (Al-Hilal et al., 2024).
In a mouse model the c.5C>G transgene causes mild SMA, and motor neurons from transgene mice are positive for nuclear gems, suggesting that the p.Ala2Gly SMN can form partially functional higher order SMN complexes (Monani et al., 2003). Analysis of SMN levels and nuclear gem quantities in tissues collected from human SMA patients with the c.5C>G variant have not yet been investigated.
Our aim was to investigate the clinical progression in two patients with the rare mild missense c.5C>G variant. Furthermore, we aimed to collect tissues from these patients and investigate levels of nuclear gems within these tissues.
Materials and methods
Ethics declaration
Results and available clinical information were acquired from patients included in this study; approval was granted by the Mayo Clinic Investigational Review Board (IRB # 19-004374) and informed consent from patients was obtained.
SMA deletion/duplication analysis
The SMA deletion/duplication assay uses novel droplet digital PCR technology to accurately quantify SMN1 exon 7 NC_000005.9 (NM_022874.2) and SMN2 exon 7 NC_000005.9 (NM_022876.2) by counting copies of the c.744C in SMN1 and the c.744T in SMN2 (Vidal-Folch et al., 2018). QuantaSoft Analysis Pro version 1.0.596 (Bio-Rad) was used for droplet-cluster classification and Poisson function application was used for determination of absolute copy numbers (Vidal-Folch et al., 2018).
SMN1 full gene analysis
The SMN1 full gene sequencing assay utilizes long-range PCR of exons 2–8 of SMN1 gene NC_000005.9 (NM_000344.3) followed by bidirectional Sanger sequencing to identify variants. Exon 1 of SMN1 gene NC_000005.9 (NM_000344.3) is PCR-amplified and bidirectionally Sanger-sequenced to identify variants in exon 1 (Vidal-Folch et al., 2018). Variants detected in SMN1 exon 1 cannot be distinguished from variants in SMN2 exon 1.
Fibroblast isolation
Tissue was rinsed in collection media and placed in a 10 cm dish with media. The biopsy was minced and selected pieces were transferred to a dish and covered with a coverslip and covered in media. The plate was incubated for 4 days, and media was changed every 5–7 days. Once the coverslip became confluent, cultures were removed from the coverslip using trypsin and expanded in culture dishes.
Tissue culture
Cultures were grown in DMEM media supplemented with 5% FBS. Media was changed every 1–2 days. Cultures were passaged using trypsin. Trypsin was inactivated by 1:1 dilution with media and centrifugation at 500 g for 4 min. Cultures were resuspended in media and plated.
Immunofluorescence
Fibroblasts were fixed in 4.0% paraformaldehyde (Sigma Aldrich) solution in phosphate buffered saline (PBS), blocked and permeabilized using 0.1% Triton-X, 0.1% Tween-20 and 2.5% BSA in PBS. Primary antibody (BDBiosciences, at 1:100) to SMN was added, and counter stained with AlexaFluor secondary antibody (1:300). Nuclei were labelled with DAPI. Cells were imaged using Agilent BioTek Cytation.
Quantification of nuclear gems
Nuclear gems per nuclei were identified and counted from two 40x immunofluorescence photomicrographs from two patients with the c.5C>G SMN1 variant and an SMN1 deletion, an SMA patient with homozygous SMN1 deletion and SMN2 copy numbers equal to the patients (two copies of SMN2), and a carrier control individual (one copy of SMN1). The carrier control individual is the mother of the SMA patient with homozygous deletion of SMN1.
Results
Clinical presentation
We identified two SMA patients with the rare missense pathogenic variant c.5C>G. We confirmed these patients have heterozygous deletion of exon 7 and heterozygous c.5C>G variant. These patients are siblings and they presented to the neurology clinic in adolescence with progressive muscle weakness. The older patient, a 16-year-old boy designated as Patient 1, suffered from a delay in gross motor milestones; only gaining the ability to roll over at 12 months of life, sit independently by 24 months of life, and walk by 30 months of life. He was notably slower than his peers as a child. He began suffering from frequent falls by 11 years of age and required a wheelchair around the time of the clinical evaluation. The younger patient, a 15-year-old girl designated as Patient 2, had normal gross motor milestones, but developed progressive weakness by 11 years of age. She was still ambulatory at the time of the clinical evaluation but required a wheelchair a few months later.
Both patients exhibited proximal muscle weakness that was more prominent in their lower extremities. Patient 1 had 4/5 strength in his biceps, triceps, and brachioradialis bilaterally, 4+/5 strength in his wrist extensors and digit extensors and flexors bilaterally. In his lower extremities, he had 2/5 strength in his iliopsoas, hip adductors, quadriceps, and hamstrings. His deep tendon reflexes were normal in his upper extremities. In his lower extremities, he had reflexes of 1+ throughout. He exhibited mild fine tremor in his hands bilaterally. Patient 2 had 4+/5 strength in her triceps and biceps bilaterally. In her lower extremities, she had 4+/5 strength of her iliopsoas and quadriceps. Her deep tendon reflexes were normal in the upper extremities. In her lower extremities, reflexes were 1+ at patella and ankle.
A muscle biopsy from Patient 2 and EMGs from both patients were performed at an external institution. The muscle biopsy diagnosis was reported as abnormal muscle with features of neurogenic atrophy. EMGs were consistent with a diffuse anterior horn cell disease.
Spinal muscular atrophy was suspected, and molecular testing was pursued. Results from deletion/duplication analysis showed that both patients had one copy of SMN1, consistent with SMA carrier status and not diagnostic of SMA. It was suspected they had a non-deletion pathogenic variant in their single copy of SMN1, and they were clinically diagnosed with SMA.
At age 36 and 34, they re-presented to the neurology clinic as new disease-modifying therapies had become available. At that time, both patients required a wheelchair and suffered from proximal weakness of the upper extremities. Neither patient was experiencing dysphagia, dyspnea, orthopnea, and neither required noninvasive ventilator support (Table 1).
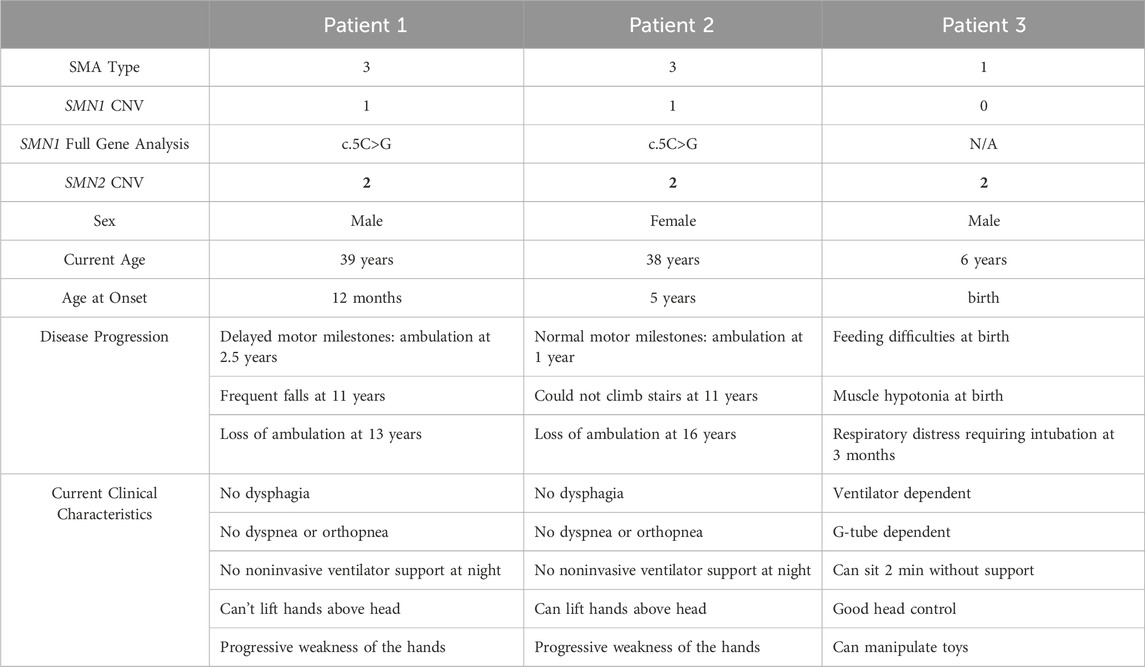
Table 1. Comparison of c.5C>G Patients to SMA Patient with Matched SMN2 Copy Number.
Molecular testing–SMN1 full gene analysis
SMN1 full gene analysis was pursued in an external diagnostic laboratory to search for the second variant. The patients were found to have one copy of SMN1 and the missense variant c.5C>G in exon 1. However, the molecular testing could not distinguish SMN1 exon 1 from SMN2 exon 1. Both patients were found to have two copies of SMN2.
Of note, their clinical course is not consistent with the classic clinical course of SMA patients with two copies of SMN2. This difference in clinical phenotype is striking when compared to an SMA patient with the same copy number of SMN2, designated as Patient 3 (zero copies of SMN1, two copies of SMN2). This patient is a 6-year-old boy who presented at birth with feeding difficulties and hypotonia (Table 1). He exhibited respiratory distress requiring intubation at 3 months of age. Currently, he is ventilator and G-tube dependent (Table 1).
Diagnostic testing was repeated at our internal reference laboratory. Patient 3 was tested with Patient 1 and 2 as a control. ddPCR deletion/duplication analysis of Patients 1 and 2 revealed SMN1 positive clusters, as compared to Patient 3 which did not have an SMN1 positive cluster (Figures 1A–C). Absolute copy number analysis revealed that Patients 1 and 2 had one copy of SMN1 and two copies of SMN2 (Figure 1D) as compared to Patient 3 which had zero copies of SMN1 and two copies of SMN2 (Figure 1D).
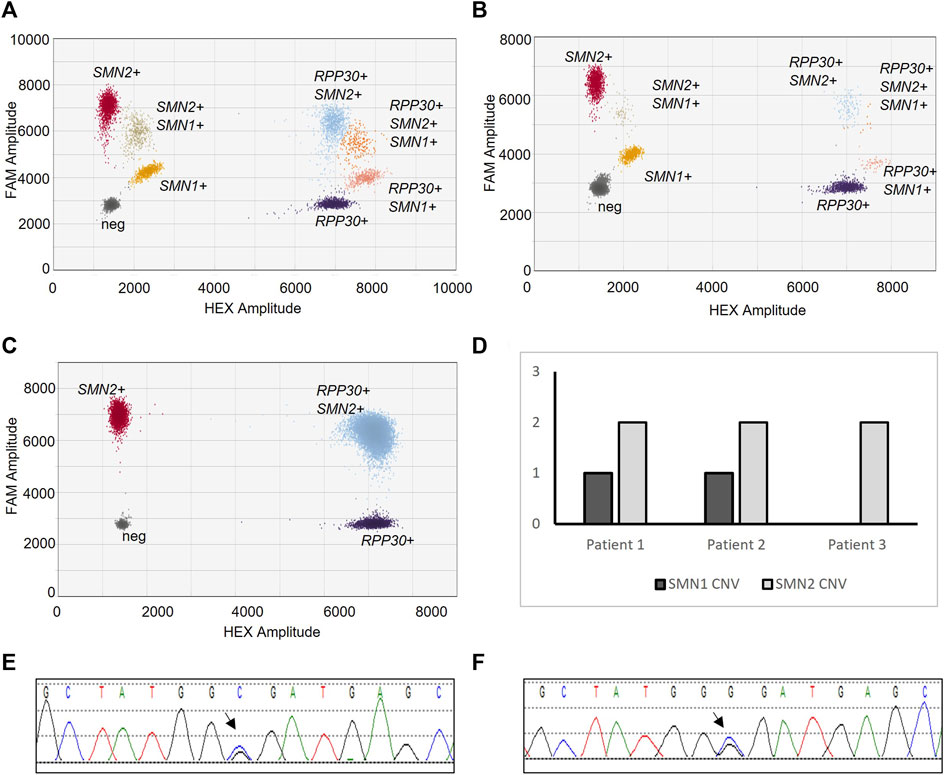
Figure 1. SMA Molecular Diagnostic Testing. SMA deletion/duplication analysis via ddPCR from Patient 1 (A), Patient 2 (B), and Patient 3 (C). Droplet clusters correspond to populations of nucleotide targets; SMN1, SMN2, or RPP30 (control). Resulting CNV of SMN1 and SMN2 for each sample (D). Sanger sequencing electropherogram of SMN1 from Patient 1 (E) and Patient 2 (F). Arrow notes c.5C>G missense variant.
SMN1 full gene sequencing analysis was performed. Sanger sequencing detected the c.5C>G variant in Patients 1 and 2 (Figures 1E, F). The C allele was also detected. Importantly, the sequenced PCR reaction amplified both SMN1 exon 1 and SMN2 exon 1.
The mother of Patient 1 and 2 was also tested, and results from an external reference laboratory revealed two copies of SMN1 and the missense variant c.5C>G in exon 1, also unable to distinguish SMN1 exon 1 from SMN2 exon 1.
Using this additional information, a family pedigree was constructed (Supplemental Figure S1). It is assumed that the patient’s father (not tested) is a traditional SMA carrier with an SMN1 deletion, while their mother is likely an SMA carrier with two copies of SMN1, one copy with the missense variant c.5C>G. The affected patients inherited the allele with the deletion from their father and the allele with the missense variant from their mother (Supplemental Figure S1). Molecular testing and clinical information regarding the patients’ siblings were unavailable. Ultimately, the molecular tests could not render a definitive diagnosis. The patients were clinically diagnosed with SMA.
SMA patients with two copies of SMN2 typically have severe disease (Type 1 SMA). However, patients with the c.5C>G missense variant and an SMN1 deletion are known to have disease milder than their SMN2 copy number would predict (Table 2). This variant is rare with scattered reports. All reported cases have one or two copies of SMN2 (Table 2). All but three patients have mild disease and remain ambulatory into adolescence or even adulthood (Table 2).
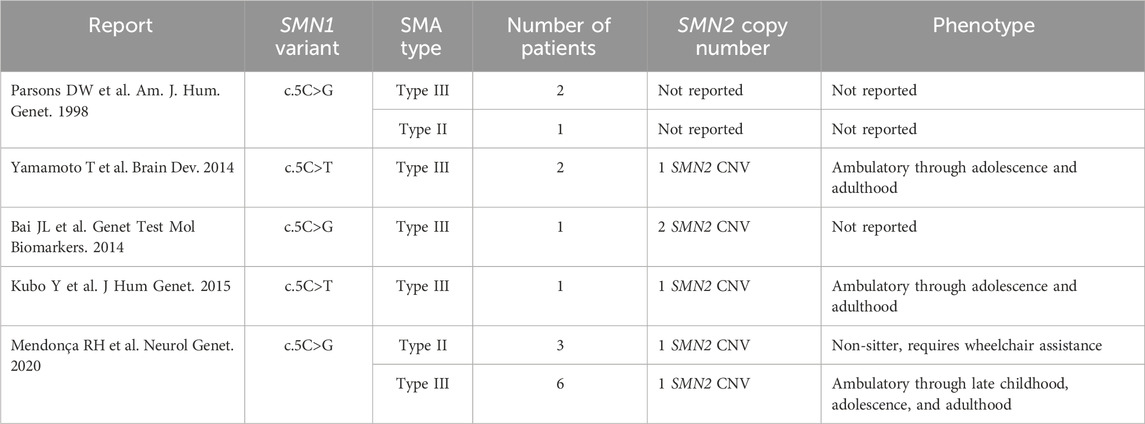
Table 2. Reported Cases of SMN1 c.5C>G.
Patient-specific analysis of nuclear gems
SMN immunofluorescence staining of skin fibroblasts was performed on Patients 1-3, along with a healthy SMA carrier control (one copy of SMN1). The SMA carrier control is the mother of Patient 3.
SMN levels in fibroblasts from Patient 1 and Patient 2 are higher than SMN levels in fibroblasts from Patient 3 (Figures 2A–D). Furthermore, like the fibroblasts from the SMA carrier control, the fibroblasts from Patient 1 and Patient 2 are positive for multiple nuclear gems per nuclei (Figures 2A–D). Quantification of gems revealed that nuclei of fibroblasts from Patient 1 contained a mean of 3.1 gems per nuclei, and fibroblasts from Patient 2 contained a mean of 3.0 gems per nuclei. This is compared to a mean of 5.2 gems per nuclei in the SMA carrier control fibroblasts and 1.4 gems per nuclei in Patient 3 fibroblasts (Figure 2E).
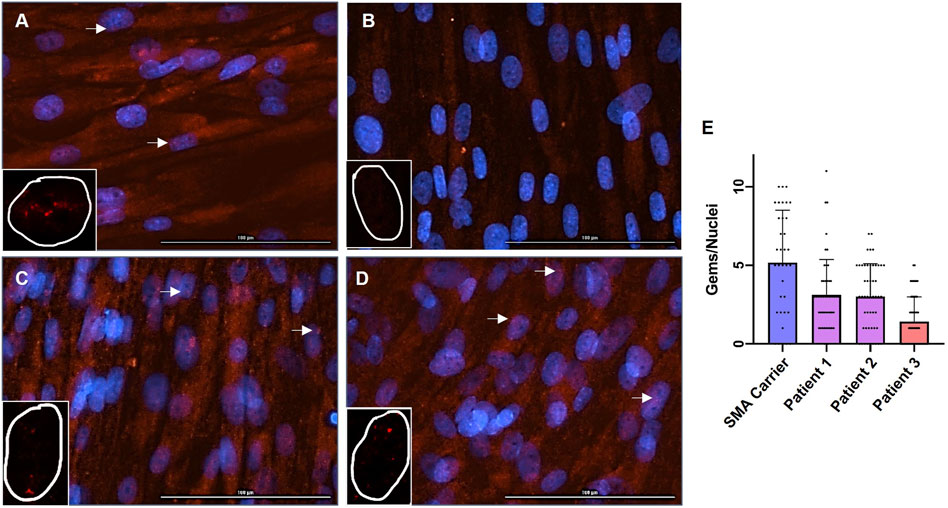
Figure 2. Immunofluorescence Staining of SMN in Fibroblasts and Quantification of Nuclear Gems. Immunofluorescence staining for SMN (red) and DAPI (blue) in patient-specific fibroblasts; (A) SMA Carrier, (B) Patient 3, (C) Patient 1, and (D) Patient 2. The arrow denotes gems. ×20 magnification. Bar = 100 uM. Inset with representative SMN staining (red) of single nuclei. Identified gems were quantified and normalized to total nuclei (E); SMA Carrier: mean = 5.17, n = 35, standard error from the mean (SEM) = ± 0.56, Patient 1: n = 52, mean = 3.11, SEM = ± 0.30, Patient 2: mean = 3.00, n = 50, SEM = ± 0.21, Patient 3: mean = 1.41, n = 58, SEM = ± 0.31. A one-way ANOVA was performed to reveal there was a statistically significant difference in mean gems per nuclei between the fibroblast cultures [F(3, 191) = (19.84), p-value < 0.00001].
A one-way ANOVA revealed that there was a statistically significant difference in mean gems per nuclei between the fibroblast cultures [F(3, 191) = (19.84), p-value < 0.00001]. Tukey’s HSD Test for comparisons found that the mean gems per nuclei was significantly different between the SMA carrier control and Patient 3 (p < 0.00001). The mean gems per nuclei was significantly different between the SMA carrier control and Patient 1 and Patient 2; Patient 1 and Patient 2 (p = 0.00012 and p = 0.00004 respectively). The mean gems per nuclei was significantly different between Patient 3 and Patient 1 and Patient 2; Patient 1 and Patient 2 (p = 0.00220 and p = 0.00509 respectively). There was no statistically significant difference in mean gems per nuclei between Patient 1 and Patient 2 (p = 0.99483).
Discussion
The patients described here have the heterozygous c.5C>G variant, a heterozygous deletion of exon 7, and two copies of SMN2, however, they exhibit a mild clinical course that is not consistent with patients with two copies of SMN2. This is highlighted by direct comparison to an SMA patient with homozygous deletion of exon 7 and two copies of SMN2 and a typical clinical course (Table 2). Of note, this patient is not genetically related to the patients with the c.5C>G variant and their different genetic backgrounds may contribute to some variability in phenotype.
Interestingly, the clinical presentation of the two patients is mildly discordant. The female patient presented with a later age of onset and remained ambulatory longer than the male patient. Sex-specific phenotypic variability has been reported in SMA (Singh et al., 2021; Zhao et al., 2021). Non-clinical and clinical reports have shown that SMA may affect the male reproductive system (Ottesen et al., 2016; Howell et al., 2017; Lipnick et al., 2019; Bar-Chama et al., 2024). Furthermore, many known disease modifiers are encoded by genes on the X chromosome, and overexpression of modifiers results in milder phenotypes in female patients (Oprea et al., 2008; Han et al., 2012; Powis et al., 2016).
The missense variant c.5C>G occurs in exon 1 of SMN1. Exon 1 of SMN1 is identical to exon 1 of SMN2, making the design of full gene analysis challenging. Most clinical tests utilizing long range PCR only amplify exons 2a-7. The patients investigated here did not receive a definitive diagnosis from an outside reference laboratory, and our internal diagnostic testing has the same limitation. Most clinical diagnostic tests cannot definitively molecularly diagnose exon 1 variants that cause SMA. This is a significant limitation in SMA diagnostic testing.
The mutation spectrum of SMN1 includes pathogenic alterations in exon 1 of SMN1, including missense variants, duplications, insertions, and deletions (Fokkema et al., 2011; Qu et al., 2016). In a review of cases referred to our laboratory in the last six years, we have detected nine SMN1 variants, three of which occurred in exon 1; a known pathogenic frameshift variant, a novel pathogenic deletion, and missense variant classified as a variant of unknown significance (data not shown). Now, with new disease-modifying therapies available to patients, it is critical to accurately detect all pathogenic alterations in SMN1.
Methods utilizing long-range PCR that can amplify exons 1–8 of SMN1 have been developed (Kubo et al., 2015). In fact, this method successfully detected the c.5 > G variant in exon 1 of SMN1 (Kubo et al., 2015). Furthermore, some clinical assays have been optimized to use long range PCR to differentiate the full coding region of SMN1 from SMN2 (Miller et al., 2023). This method effectively detected the c.5C>G variant in exon 1 of SMN1 (Miller et al., 2023). There is a need for molecular diagnostic labs to optimize current diagnostic methods to differentiate SMN1 exons 1–8 from SMN2 exons 1–8.
Long-read sequencing is a potential future method for clinical diagnostic testing. Long-read PacBio HiFi sequencing and subsequent analysis with Paraphase, an informatics method that identifies SMN1 and SMN2 haplotypes, detects pathogenic variants, determines the copy number variation, and detects phased variants (Chen et al., 2023). Another HiFi long read sequencing approach effectively identified SMN1 and SMN2 single nucleotide variants, small insertions and deletions, and large SMN1 deletions. This method achieved 100% accuracy for SMN1 copy number and 99.5% accuracy for SMN2 copy number (Bai et al., 2023). Long read technology will ultimately lead to more comprehensive SMA molecular diagnostic testing.
Both optimized long-range PCR based sequencing and long read sequencing technology detects the SMN1 c.5C>G variant. This variant is significant as it is known to cause less severe disease than SMN2 copy number would predict (Table 2). Our data reinforces this discordant phenotype (Table 1). Furthermore, mice with the c.5C>G (p.Ala2Gly) transgene have a mild SMA (Monani et al., 2003). A mild phenotype indicates that p.Ala2Gly SMN is partially functional, thereby preventing the development of a severe phenotype.
Nuclei of motor neurons collected from p.Ala2Gly transgenic mice contain nuclear SMN gems, indicating that p.Ala2Gly SMN can form nuclear complexes. To the best of our knowledge, levels of SMN and nuclear gems in tissues collected from patients with the c.5C>G variant have not yet been previously reported. Our novel data from human tissues are in keeping with the data obtained from p.Ala2Gly transgenic mice. Our data revealed that SMN levels in fibroblasts from Patient 1 and Patient 2 are higher than the SMN levels in fibroblasts from Patient 3, and are comparable to SMN levels in fibroblasts from a carrier control. This data is consistent with a prior study showing that SMN1 transcript levels isolated from a patient with the c.5C>G variant were not reduced compared to a healthy carrier control (Bai et al., 2014). Furthermore, nuclei of fibroblasts from Patient 1 and Patient 2 contained nuclear gems. The gem number is higher than that observed in Patient 3, but fewer than that observed in the carrier control. Of note, SMN2 copy number of the carrier control is unknown. In addition, Patient 3 and the healthy carrier control are not genetically related to Patient 1 and Patient 2, and their different genetic backgrounds may contribute to differences in SMN levels and numbers of nuclear gems. Even so, this suggests that the p.Ala2Gly SMN cannot form the same quantity of gems as wild-type SMN. This is consistent with prior reports that the quantity of gems correlates to disease severity (Coovert et al., 1997; Lefebvre et al., 1997; Patrizi et al., 1999). Overall, such findings indicates that the p.Ala2Gly affects SMN interactions and ultimately fully functional gem formation.
Exon 1 of SMN1 codes for the N-terminus of SMN and is just proximal to the Gemin-2 binding domain which is important for SMN complex assembly (Lorson and Androphy, 1998; Otter et al., 2007; Ogawa et al., 2009; Zhang et al., 2011). However, the utmost N-terminal is not known to be responsible for vital interactions needed for oligomerization or higher order complex formation. Experiments performed using an immortalized mouse embryonic fibroblast (iMEF) line in which Smn can be conditionally deleted using Cre recombinase revealed that the p.Ala2Gly SMN did not rescue survival in the conditionally deleted iMEFs, suggesting the SMN function that is lost with this variant is essential for cell function and survival (Blatnik et al., 2020). When the p.Ala2Gly SMN was co-expressed with many other missense variants, iMEF survival and snRNP assembly was rescued. The only variant tested that did not rescue survival is the p.Thr274Ile (Blatnik et al., 2020). p.Thr274Ile is coded by exon 6 and is in the YG-box oligomerization domain (Lorson et al., 1998). Like p.Ala2Gly, p.Thr274Ile is also a missense variant that causes disease milder than SMN2 copy number would predict (Hahnen et al., 1997). Collectively, this suggests a role that is shared by the N-terminus and C-terminus of SMN. This shared interaction and function does not preclude partially functional complex formation but does limit SMN function by limiting fully functional complex formation to wild-type levels.
In addition to SMN’s role in RNA metabolism, SMN has been known to be involved in multiple cellular functions, including macromolecular trafficking, stress granule formation, cell signaling, and cytoskeleton maintenance, and not all of these functions require the canonical SMN complex (Singh et al., 2017). The N-terminus may have an important role in multiple SMN-dependent cellular functions and may have important SMN complex-independent interactions. As such, the c.5C>G variant may alter these functions and interactions.
It is also important to consider the effects of the c.5C>G variant on the transcription of SMN1. Exon 1 variants proximal to the promoter may affect transcription initiation and transcript stability, and possibly translation and protein stability. Future studies include measuring SMN1 transcript levels and SMN protein levels isolated from the patient’s fibroblasts. Recently, an SMN2 super minigene was generated to investigate the regulation of SMN2 transcription (Ottesen et al., 2024). This super minigene was utilized to recapitulate the splicing pattern of endogenous SMN1 with a pathogenic variant (Ottesen et al., 2024). Using such a construct may address questions regarding the effects of the c.5C>G variant on transcription.
Taken together, this data suggests there are discrete functions of SMN that can regulate motor neuron survival and function and investigating the p.Ala2Gly SMN will lead to a greater understanding of SMN function. Investigating the c.5C>G variant, including protein and transcript levels resulting from the c.5C > G variant, differential components of p.Ala2Gly SMN complex, and differential gene expression resulting from p.Ala2Gly SMN complexes, will reveal novel SMN complex interacting partners with discrete functions crucial to SMN function and subsequent altered gene expression that is critical for motor neuron function and survival.
Data availability statement
The datasets presented in this article are not readily available because of ethical and privacy restrictions. Requests to access the datasets should be directed to the corresponding author.
Ethics statement
The studies involving humans were approved by the Mayo Clinic Institutional Review Board. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants’ legal guardians/next of kin. Written informed consent was obtained from the individual(s), and minor(s)’ legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
Author contributions
SC: Conceptualization, Data curation, Investigation, Writing–original draft, Writing–review and editing. CS: Data curation, Writing–review and editing. LK: Project administration, Writing–review and editing. NV: Data curation, Writing–review and editing. DO: Writing–review and editing. LH: Writing–review and editing. DS: Writing–review and editing. MM: Writing–review and editing. DA: Writing–review and editing. NS: Resources, Supervision, Writing–review and editing.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Acknowledgments
The authors thank the patients and their families for their participation in this study.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2024.1406819/full#supplementary-material
Supplementary Figure S1 | Family Pedigree of Patients. Arrows designate SMA patients. I2, father of the proband, indicates SMA carrier status, heterozygous deletion of SMN1, and I3, mother of proband indicates SMA carrier status, heterozygous c.5C>G pathogenic missense variant in exon 1.
References
Al-Hilal, H., Maretina, M., Egorova, A., Glotov, A., and Kiselev, A. (2024). Assessment of nuclear gem quantity for evaluating the efficacy of antisense oligonucleotides in spinal muscular atrophy cells. Methods Protocols 7 (1), 9. doi:10.3390/mps7010009
Bai, J., Qu, Y., OuYang, S., Jiao, H., Wang, Y., Li, J., et al. (2023). Novel Alu-mediated deletions of the SMN1 gene were identified by ultra-long read sequencing technology in patients with spinal muscular atrophy. Neuromuscul. Disord. 33 (5), 382–390. doi:10.1016/j.nmd.2023.03.001
Bai, J. L., Qu, Y. J., Cao, Y. Y., Li, E. Z., Wang, L. W., Li, Y., et al. (2014). Subtle mutation detection of SMN1 gene in Chinese spinal muscular atrophy patients: implication of molecular diagnostic procedure for SMN1 gene mutations. Genet. Test. Mol. Biomarkers 18 (8), 546–551. doi:10.1089/gtmb.2014.0002
Bar-Chama, N., Elsheikh, B., Hewamadduma, C., Guittari, C. J., Gorni, K., and Mueller, L. (2024). Male reproduction in spinal muscular atrophy (SMA) and the potential impact of oral survival of motor neuron 2 (SMN2) pre-mRNA splicing modifiers. Neurol. Ther. doi:10.1007/s40120-024-00626-5
Blatnik, A. J., McGovern, V. L., Iyer, C. C., Kaspar, B. K., and Burghes, A. H. M. (2020). Conditional deletion of SMN in cell culture identifies functional SMN alleles. Hum. Mol. Genet. 29 (21), 3477–3492. doi:10.1093/hmg/ddaa229
Burghes, A. H., and Beattie, C. E. (2009). Spinal muscular atrophy: why do low levels of survival motor neuron protein make motor neurons sick? Nat. Rev. Neurosci. 10 (8), 597–609. doi:10.1038/nrn2670
Burglen, L., Lefebvre, S., Clermont, O., Burlet, P., Viollet, L., Cruaud, C., et al. (1996). Structure and organization of the human survival motor neurone (SMN) gene. Genomics 32 (3), 479–482. doi:10.1006/geno.1996.0147
Burnett, B. G., Muñoz, E., Tandon, A., Kwon, D. Y., Sumner, C. J., and Fischbeck, K. H. (2009). Regulation of SMN protein stability. Mol. Cell Biol. 29 (5), 1107–1115. doi:10.1128/MCB.01262-08
Cartegni, L., and Krainer, A. R. (2002). Disruption of an SF2/ASF-dependent exonic splicing enhancer in SMN2 causes spinal muscular atrophy in the absence of SMN1. Nat. Genet. 30 (4), 377–384. doi:10.1038/ng854
Chen, X., Harting, J., Farrow, E., Thiffault, I., Kasperaviciute, C., Hoischen, A., et al. (2023). Comprehensive SMN1 and SMN2 profiling for spinal muscular atrophy analysis using long-read PacBio HiFi sequencing. Am. J. Hum. Genet. 110 (2), 240–250. doi:10.1016/j.ajhg.2023.01.001
Cho, S., and Dreyfuss, G. (2010). A degron created by SMN2 exon 7 skipping is a principal contributor to spinal muscular atrophy severity. Genes Dev. 24 (5), 438–442. doi:10.1101/gad.1884910
Coovert, D. D., Le, T. T., McAndrew, P. E., Strasswimmer, J., Crawford, T. O., Mendell, J. R., et al. (1997). The survival motor neuron protein in spinal muscular atrophy. Hum. Mol. Genet. 6 (8), 1205–1214. doi:10.1093/hmg/6.8.1205
Ebert, A. D., Yu, J., Rose, F. F., Mattis, V. B., Lorson, C. L., Thomson, J. A., et al. (2009). Induced pluripotent stem cells from a spinal muscular atrophy patient. Nature 457 (7227), 277–280. doi:10.1038/nature07677
Faravelli, I., Nizzardo, M., Comi, G. P., and Corti, S. (2015). Spinal muscular atrophy--recent therapeutic advances for an old challenge. Nat. Rev. Neurol. 11 (6), 351–359. doi:10.1038/nrneurol.2015.77
Feldkotter, M., Schwarzer, V., Wirth, R., Wienker, T. F., and Wirth, B. (2002). Quantitative analyses of SMN1 and SMN2 based on real-time lightCycler PCR: fast and highly reliable carrier testing and prediction of severity of spinal muscular atrophy. Am. J. Hum. Genet. 70 (2), 358–368. doi:10.1086/338627
Fokkema, I. F., Taschner, P. E. M., Schaafsma, G. C. P., Celli, J., Laros, J. F. J., and den Dunnen, J. T. (2011). LOVD v.2.0: the next generation in gene variant databases. Hum. Mutat. 32 (5), 557–563. doi:10.1002/humu.21438
Gubitz, A. K., Feng, W., and Dreyfuss, G. (2004). The SMN complex. Exp. Cell Res. 296 (1), 51–56. doi:10.1016/j.yexcr.2004.03.022
Hahnen, E., Forkert, R., Marke, C., Rudnik-Schöneborn, S., Schönling, J., Zerres, K., et al. (1995). Molecular analysis of candidate genes on chromosome 5q13 in autosomal recessive spinal muscular atrophy: evidence of homozygous deletions of the SMN gene in unaffected individuals. Hum. Mol. Genet. 4 (10), 1927–1933. doi:10.1093/hmg/4.10.1927
Hahnen, E., Schönling, J., Rudnik-Schöneborn, S., Raschke, H., Zerres, K., and Wirth, B. (1997). Missense mutations in exon 6 of the survival motor neuron gene in patients with spinal muscular atrophy (SMA). Hum. Mol. Genet. 6 (5), 821–825. doi:10.1093/hmg/6.5.821
Han, K. J., Foster, D. G., Zhang, N. Y., Kanisha, K., Dzieciatkowska, M., Sclafani, R. A., et al. (2012). Ubiquitin-specific protease 9x deubiquitinates and stabilizes the spinal muscular atrophy protein-survival motor neuron. J. Biol. Chem. 287 (52), 43741–43752. doi:10.1074/jbc.M112.372318
Howell, M. D., Ottesen, E. W., Singh, N. N., Anderson, R. L., and Singh, R. N. (2017). Gender-specific amelioration of SMA phenotype upon disruption of a deep intronic structure by an oligonucleotide. Mol. Ther. 25 (6), 1328–1341. doi:10.1016/j.ymthe.2017.03.036
Kolb, S. J., Battle, D. J., and Dreyfuss, G. (2007). Molecular functions of the SMN complex. J. Child. Neurol. 22 (8), 990–994. doi:10.1177/0883073807305666
Kubo, Y., Nishio, H., and Saito, K. (2015). A new method for SMN1 and hybrid SMN gene analysis in spinal muscular atrophy using long-range PCR followed by sequencing. J. Hum. Genet. 60 (5), 233–239. doi:10.1038/jhg.2015.16
Lefebvre, S., Bürglen, L., Reboullet, S., Clermont, O., Burlet, P., Viollet, L., et al. (1995). Identification and characterization of a spinal muscular atrophy-determining gene. Cell 80 (1), 155–165. doi:10.1016/0092-8674(95)90460-3
Lefebvre, S., Burlet, P., Liu, Q., Bertrandy, S., Clermont, O., Munnich, A., et al. (1997). Correlation between severity and SMN protein level in spinal muscular atrophy. Nat. Genet. 16 (3), 265–269. doi:10.1038/ng0797-265
Lipnick, S. L., Agniel, D. M., Aggarwal, R., Makhortova, N. R., Finlayson, S. G., Brocato, A., et al. (2019). Systemic nature of spinal muscular atrophy revealed by studying insurance claims. PLoS One 14 (3), e0213680. doi:10.1371/journal.pone.0213680
Liu, Q., and Dreyfuss, G. (1996). A novel nuclear structure containing the survival of motor neurons protein. EMBO J. 15 (14), 3555–3565. doi:10.1002/j.1460-2075.1996.tb00725.x
Lorson, C. L., and Androphy, E. J. (1998). The domain encoded by exon 2 of the survival motor neuron protein mediates nucleic acid binding. Hum. Mol. Genet. 7 (8), 1269–1275. doi:10.1093/hmg/7.8.1269
Lorson, C. L., Strasswimmer, J., Yao, J. M., Baleja, J. D., Hahnen, E., Wirth, B., et al. (1998). SMN oligomerization defect correlates with spinal muscular atrophy severity. Nat. Genet. 19 (1), 63–66. doi:10.1038/ng0598-63
MacLeod, M. J., Taylor, J. E., Lunt, P. W., Mathew, C. G., and Robb, S. A. (1999). Prenatal onset spinal muscular atrophy. Eur. J. Paediatr. Neurol. 3 (2), 65–72. doi:10.1053/ejpn.1999.0184
McAndrew, P. E., Parsons, D. W., Simard, L. R., Rochette, C., Ray, P. N., Mendell, J. R., et al. (1997). Identification of proximal spinal muscular atrophy carriers and patients by analysis of SMNT and SMNC gene copy number. Am. J. Hum. Genet. 60 (6), 1411–1422. doi:10.1086/515465
Meister, G., Bühler, D., Pillai, R., Lottspeich, F., and Fischer, U. (2001). A multiprotein complex mediates the ATP-dependent assembly of spliceosomal U snRNPs. Nat. Cell Biol. 3 (11), 945–949. doi:10.1038/ncb1101-945
Mendonca, R. H., Matsui, C., Polido, G. J., Silva, A. M. S., Kulikowski, L., Torchio Dias, A., et al. (2020). Intragenic variants in the SMN1 gene determine the clinical phenotype in 5q spinal muscular atrophy. Neurol. Genet. 6 (5), e505. doi:10.1212/NXG.0000000000000505
Miller, C. R., Fang, J., Snyder, P., Long, S. E., Prior, T. W., Jones, D., et al. (2023). Clinical SMN1 and SMN2 gene-specific sequencing to enhance the clinical sensitivity of spinal muscular atrophy diagnostic testing. Hum. Mutat. 2023, 7. doi:10.1155/2023/6436853
Monani, U. R., Pastore, M. T., Gavrilina, T. O., Jablonka, S., Le, T. T., Andreassi, C., et al. (2003). A transgene carrying an A2G missense mutation in the SMN gene modulates phenotypic severity in mice with severe (type I) spinal muscular atrophy. J. Cell Biol. 160 (1), 41–52. doi:10.1083/jcb.200208079
Ogawa, C., Usui, K., Ito, F., Itoh, M., Hayashizaki, Y., and Suzuki, H. (2009). Role of survival motor neuron complex components in small nuclear ribonucleoprotein assembly. J. Biol. Chem. 284 (21), 14609–14617. doi:10.1074/jbc.M809031200
Oprea, G. E., Kröber, S., McWhorter, M. L., Rossoll, W., Müller, S., Krawczak, M., et al. (2008). Plastin 3 is a protective modifier of autosomal recessive spinal muscular atrophy. Science 320 (5875), 524–527. doi:10.1126/science.1155085
Otter, S., Grimmler, M., Neuenkirchen, N., Chari, A., Sickmann, A., and Fischer, U. (2007). A comprehensive interaction map of the human survival of motor neuron (SMN) complex. J. Biol. Chem. 282 (8), 5825–5833. doi:10.1074/jbc.M608528200
Ottesen, E. W., Howell, M. D., Singh, N. N., Seo, J., Whitley, E. M., and Singh, R. N. (2016). Severe impairment of male reproductive organ development in a low SMN expressing mouse model of spinal muscular atrophy. Sci. Rep. 6, 20193. doi:10.1038/srep20193
Ottesen, E. W., Seo, J., Luo, D., Singh, N. N., and Singh, R. N. (2024). A super minigene with a short promoter and truncated introns recapitulates essential features of transcription and splicing regulation of the SMN1 and SMN2 genes. Nucleic Acids Res. 52 (7), 3547–3571. doi:10.1093/nar/gkad1259
Parsons, D. W., McAndrew, P. E., Iannaccone, S. T., Mendell, J. R., Burghes, A. H., and Prior, T. W. (1998). Intragenic telSMN mutations: frequency, distribution, evidence of a founder effect, and modification of the spinal muscular atrophy phenotype by cenSMN copy number. Am. J. Hum. Genet. 63 (6), 1712–1723. doi:10.1086/302160
Patrizi, A. L., Tiziano, F., Zappata, S., Donati, M. A., Neri, G., and Brahe, C. (1999). SMN protein analysis in fibroblast, amniocyte and CVS cultures from spinal muscular atrophy patients and its relevance for diagnosis. Eur. J. Hum. Genet. 7 (3), 301–309. doi:10.1038/sj.ejhg.5200286
Paushkin, S., Gubitz, A. K., Massenet, S., and Dreyfuss, G. (2002). The SMN complex, an assemblyosome of ribonucleoproteins. Curr. Opin. Cell Biol. 14 (3), 305–312. doi:10.1016/s0955-0674(02)00332-0
Pearn, J. (1978). Incidence, prevalence, and gene frequency studies of chronic childhood spinal muscular atrophy. J. Med. Genet. 15 (6), 409–413. doi:10.1136/jmg.15.6.409
Peeters, K., Chamova, T., and Jordanova, A. (2014). Clinical and genetic diversity of SMN1-negative proximal spinal muscular atrophies. Brain 137 (Pt 11), 2879–2896. doi:10.1093/brain/awu169
Pellizzoni, L., Kataoka, N., Charroux, B., and Dreyfuss, G. (1998). A novel function for SMN, the spinal muscular atrophy disease gene product, in pre-mRNA splicing. Cell 95 (5), 615–624. doi:10.1016/s0092-8674(00)81632-3
Powis, R. A., Karyka, E., Boyd, P., Côme, J., Jones, R. A., Zheng, Y., et al. (2016). Systemic restoration of UBA1 ameliorates disease in spinal muscular atrophy. JCI Insight 1 (11), e87908. doi:10.1172/jci.insight.87908
Qu, Y. J., Bai, J. L., Cao, Y. Y., Wang, H., Jin, Y. W., Du, J., et al. (2016). Mutation spectrum of the survival of motor neuron 1 and functional analysis of variants in Chinese spinal muscular atrophy. J. Mol. Diagn 18 (5), 741–752. doi:10.1016/j.jmoldx.2016.05.004
Singh, N. N., Hoffman, S., Reddi, P. P., and Singh, R. N. (2021). Spinal muscular atrophy: broad disease spectrum and sex-specific phenotypes. Biochim. Biophys. Acta Mol. Basis Dis. 1867 (4), 166063. doi:10.1016/j.bbadis.2020.166063
Singh, R. N., Howell, M. D., Ottesen, E. W., and Singh, N. N. (2017). Diverse role of survival motor neuron protein. Biochim. Biophys. Acta Gene Regul. Mech. 1860 (3), 299–315. doi:10.1016/j.bbagrm.2016.12.008
Singh, R. N., and Singh, N. N. (2018). Mechanism of splicing regulation of spinal muscular atrophy genes. Adv. Neurobiol. 20, 31–61. doi:10.1007/978-3-319-89689-2_2
Sugarman, E. A., Nagan, N., Zhu, H., Akmaev, V. R., Zhou, Z., Rohlfs, E. M., et al. (2012). Pan-ethnic carrier screening and prenatal diagnosis for spinal muscular atrophy: clinical laboratory analysis of >72,400 specimens. Eur. J. Hum. Genet. 20 (1), 27–32. doi:10.1038/ejhg.2011.134
Vidal-Folch, N., Gavrilov, D., Raymond, K., Rinaldo, P., Tortorelli, S., Matern, D., et al. (2018). Multiplex droplet digital PCR method applicable to newborn screening, carrier status, and assessment of spinal muscular atrophy. Clin. Chem. 64 (12), 1753–1761. doi:10.1373/clinchem.2018.293712
Vitte, J., Fassier, C., Tiziano, F. D., Dalard, C., Soave, S., Roblot, N., et al. (2007). Refined characterization of the expression and stability of the SMN gene products. Am. J. Pathol. 171 (4), 1269–1280. doi:10.2353/ajpath.2007.070399
Wadman, R. I., Jansen, M. D., Stam, M., Wijngaarde, C. A., Curial, C. A. D., Medic, J., et al. (2020). Intragenic and structural variation in the SMN locus and clinical variability in spinal muscular atrophy. Brain Commun. 2 (2), fcaa075. doi:10.1093/braincomms/fcaa075
Wirth, B. (2000). An update of the mutation spectrum of the survival motor neuron gene (SMN1) in autosomal recessive spinal muscular atrophy (SMA). Hum. Mutat. 15 (3), 228–237. doi:10.1002/(SICI)1098-1004(200003)15:3<228::AID-HUMU3>3.0.CO;2-9
Yamamoto, T., Sato, H., Lai, P. S., Nurputra, D. K., Harahap, N. I. F., Morikawa, S., et al. (2014). Intragenic mutations in SMN1 may contribute more significantly to clinical severity than SMN2 copy numbers in some spinal muscular atrophy (SMA) patients. Brain Dev. 36 (10), 914–920. doi:10.1016/j.braindev.2013.11.009
Zhang, R., So, B. R., Li, P., Yong, J., Glisovic, T., Wan, L., et al. (2011). Structure of a key intermediate of the SMN complex reveals Gemin2's crucial function in snRNP assembly. Cell 146 (3), 384–395. doi:10.1016/j.cell.2011.06.043
Keywords: spinal muscular atrophy, exon 1, SMN1, c.5C>G, p.Ala2Gly, nuclear gems, diagnostic testing
Citation: Cook SL, Stout C, Kirkeby L, Vidal-Folch N, Oglesbee D, Hasadsri L, Selcen D, Milone M, Anderson D and Staff NP (2024) SMN1 c.5C>G (p.Ala2Gly) missense variant, a challenging molecular SMA diagnosis associated with mild disease, preserves SMN nuclear gems in patient-specific fibroblasts. Front. Genet. 15:1406819. doi: 10.3389/fgene.2024.1406819
Received: 26 March 2024; Accepted: 25 June 2024;
Published: 30 July 2024.
Edited by:
Irene Bottillo, Sapienza University of Rome, ItalyReviewed by:
Dmitry Vlodavets, Veltischev Research and Clinical Institute for Pediatrics of the Pirogov Russian National Research Medical University, RussiaRavindra N. Singh, Iowa State University, United States
Copyright © 2024 Cook, Stout, Kirkeby, Vidal-Folch, Oglesbee, Hasadsri, Selcen, Milone, Anderson and Staff. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nathan P. Staff, c3RhZmYubmF0aGFuQG1heW8uZWR1