 Rafael Tamayo-Trujillo1†
Rafael Tamayo-Trujillo1† Rita Ibarra-Castillo2
Rita Ibarra-Castillo2 José Luis Laso-Bayas2
José Luis Laso-Bayas2 Patricia Guevara-Ramirez1
Patricia Guevara-Ramirez1 Santiago Cadena-Ullauri1
Santiago Cadena-Ullauri1 Elius Paz-Cruz1
Elius Paz-Cruz1 Viviana A. Ruiz-Pozo1
Viviana A. Ruiz-Pozo1 Nieves Doménech3
Nieves Doménech3 Adriana Alexandra Ibarra-Rodríguez4
Adriana Alexandra Ibarra-Rodríguez4 Ana Karina Zambrano1*†
Ana Karina Zambrano1*†- 1Centro de Investigación Genética y Genómica, Facultad de Ciencias de la Salud Eugenio Espejo, Universidad UTE, Quito, Ecuador
- 2Clinical Cardiac Electrophysiologist, Quito, Ecuador
- 3Instituto de Investigación Biomédica de A Coruña (INIBIC)-CIBERCV, Complexo Hospitalario Universitario de A Coruña (CHUAC), Sergas. Universidad da Coruña (UDC), Coruña, Spain
- 4Grupo de investigación identificación Genética-IdentiGEN, Facultad de Ciencias Exactas y Naturales (FCEN), Universidad de Antioquia, Medellín, Colombia
Introduction: Long QT syndrome (LQTS) is an autosomal dominant inherited cardiac condition characterized by a QT interval prolongation and risk of sudden death. There are 17 subtypes of this syndrome associated with genetic variants in 11 genes. The second most common is type 2, caused by a mutation in the KCNH2 gene, which is part of the potassium channel and influences the final repolarization of the ventricular action potential. This case report presents an Ecuadorian teen with congenital Long QT Syndrome type 2 (OMIM ID: 613688), from a family without cardiac diseases or sudden cardiac death backgrounds.
Case presentation: A 14-year-old girl with syncope, normal echocardiogram, and an irregular electrocardiogram was diagnosed with LQTS. Moreover, by performing Next-Generation Sequencing, a pathogenic variant in the KCNH2 gene p.(Ala614Val) (ClinVar ID: VCV000029777.14) associated with LQTS type 2, and two variants of uncertain significance in the AKAP9 p.(Arg1654GlyfsTer23) (rs779447911), and TTN p. (Arg34653Cys) (ClinVar ID: VCV001475968.4) genes were identified. Furthermore, ancestry analysis showed a mainly Native American proportion.
Conclusion: Based on the genomic results, the patient was identified to have a high-risk profile, and an implantable cardioverter defibrillator was selected as the best treatment option, highlighting the importance of including both the clinical and genomics aspects for an integral diagnosis.
1 Introduction
Long QT syndrome (LQTS) is a predominantly autosomal dominant inherited cardiac channelopathy characterized by QT interval prolongation and T-wave abnormalities (Schwartz et al., 2012; Wallace et al., 2019). LQTS occurs in young individuals without structural heart abnormalities and is associated with syncope, ventricular tachycardia (VT), ventricular fibrillation (VF), and sudden cardiac death (SCD) (Schwartz et al., 2012; Fonseca and Joaquim, 2018). Additionally, ethnic differences in LQTS have been described. For instance, Lopez. et al. (2018) found that Hispanic patients were more likely to carry complex polygenic mutations, have longer corrected QT (QTc), and be diagnosed at a younger age compared to Caucasians (Lopez et al., 2018). Moreover, specific alleles have been described in different ethnic groups, like an increased KCNH2 mutation frequency in Caucasians (Kong et al., 2017). Therefore, race and ancestral components could be risk factors associated with specific clinical phenotype.
LQTS has been classified into 17 subtypes according to genetic variations in 11 associated genes (Wilde et al., 2022a). Most of them affect potassium, sodium, or calcium channel function leading to cardiac channelopathies and arrhythmogenesis (Wallace et al., 2019). For instance, mutations in the KCNH2 gene have been associated with LQTS type 2 (LQT2) (OMIM ID: 613688) (Nagaoka et al., 2008; Sakaguchi et al., 2008). The KCNH2 gene encodes a potassium channel pore-forming alpha subunit, which forms a tetrameric ion channel composed of four copies of the KCNH2 protein and one copy of the KCNE2. Moreover, the protein is mainly expressed in tissues such as nerve cells and cardiac muscle (Safran et al., 2021).
The first-line treatment for symptomatic individuals with LQT2 is beta-blockers. However, patients with a high risk of SCD may require an implantable cardioverter defibrillator (ICD), but selecting those who will benefit from this therapy may be challenging, especially for the youngest (Cho, 2016a; Wilde et al., 2022b). We report the case of a female adolescent diagnosed with LQT2, in whom the identification of a mutation p.(Ala614Val) (ClinVar ID: VCV000029777.14) in the KCNH2 gene, the persistence of cardiac events under beta blocker treatment, and an increased SCD risk, allowed the configuration of a high-risk profile aiding the decision to implant an ICD.
2 Case presentation
2.1 Clinical phenotype
A 14-year-old Ecuadorian female was referred to the Arrhythmia Unit for evaluation after two episodes of loss of consciousness. The first event occurred 30 min after practicing sport; while resting, she described a brief period of dizziness before losing consciousness and falling unconscious from the bed. She recovered immediately, feeling dizzy with visual dimming, pale, and diaphoretic; micturition occurred during the event. The second episode was after getting out of bed in the morning; no prodromal symptoms were reported before losing consciousness and falling; the recovery was rapid, and micturition also was present. The subject’s father shows cardiac symptoms; however, he does not have a definitive diagnosis and has refused to participate in the study. Similarly, no other family members agreed to participate in the research. Her physical exam was unremarkable. The electrocardiogram showed sinus rhythm, abnormal T waves, and a prolonged QTc interval (528 msec) (Figure 1). In the stress test, at recovery, she developed sinus tachycardia conducted with aberrancy, and when the QRS normalized, the QTc was normal (390-411 msec). The echocardiogram confirmed a structurally normal heart. According to the Schwartz score, a high probability of LQTS was estimated (5 points) (Wilde et al., 2022b). Propranolol was prescribed; however, even under treatment, the patient suffered from cardiac symptoms with increased SCD risk. Approval by the Ethics Committee of Universidad UTE, the participant informed assent and legal guardian consent were obtained. A genomic test was performed in accordance with the Declaration of Helsinki and relevant guidelines (Supplementary Figure S1). A pathogenic variant in the KCNH2 gene p.(Ala614Val) was detected, which along with abnormal electrocardiogram confirm the diagnosis of LQT2 syndrome. The high SCD risk observed in this proband allows the ICD implantation as the best treatment option based on genomic results and lack of treatment response (persistent VT and prolonged QTc). After ICD implantation, lifestyle changes, adequate serum K+ levels maintenance, exercise restriction, and a reduction in auditory triggers were recommended (Cho, 2016b; Goldstein et al., 2017). Figure 2 presents a timeline with the episodes of care history.
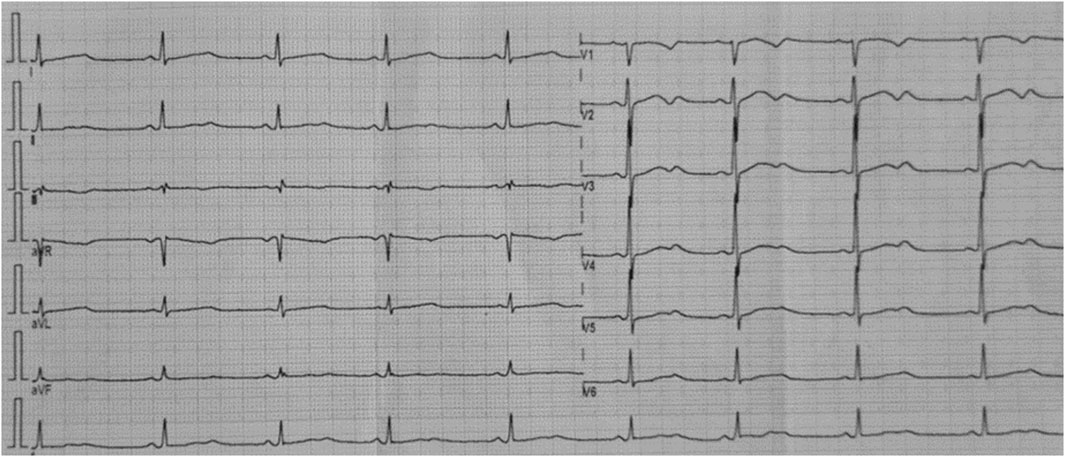
Figure 1. Twelve-lead rest electrocardiogram of the analyzed patient. Corrected QT interval 528 msec (Bazett formula). Flattened and notched T waves are diffusely present.
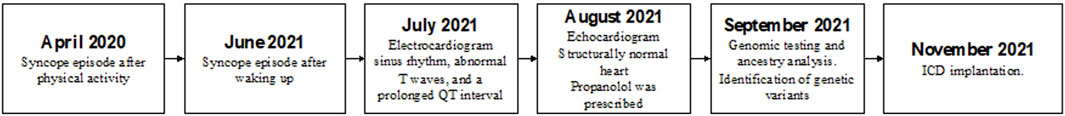
Figure 2. Participant’s episodes of care timeline. The disease associated episodes of care are presented in the figure.
2.2 Sampling and DNA extraction
The legal representative signed the informed consent, and the patient gave her informed assent to participate in the study.
A peripheral blood sample was collected in an EDTA tube. The DNA was extracted with PureLink Genomic DNA Mini Kit (Life Technologies) according to the manufacturer’s protocol (Invitrogen, 2024).
The DNA quantity and purity were measured by spectrophotometry and fluorometric methods. The quality was analyzed with an agarose gel. The sample was then diluted to 5 ng/μL according to the sequencing protocol.
2.3 Next-Generation Sequencing (NGS)
The genomic procedure was performed in the Genetics and Genomics Research Center of UTE University. A commercially available kit was used to identify the possible presence of variants and their association with the clinical diagnosis. We used the TruSight Cardio kit (Illumina) covering 174 genes (575 kb of cumulative target region size) with known association of 17 Inherited Cardiac Conditions (between them fifteen genes for LQTS). The samples were processed according to the manufacturer’s protocol (Illumina, 2016b). In the last step, one pool was built, denatured, and diluted to load in a V2 cartridge. The cartridge and the flow cell were inserted into the MiSeq System (Illumina) at the laboratory and run for 315 cycles.
2.4 Ancestry proportion analysis
For ancestral proportion analyses, PCR amplification was done with 46 ancestral informative INDEL markers in one multiplex reaction, according to Zambrano, et al. (2019) (Zambrano et al., 2019). Fragment separation and detection were executed on the 3500 Genetic Analyzer (Thermo Fisher Scientific). The results were collected on Data Collection v 3.3 and analyzed in Gene Mapper v. 5 (Thermo Fisher Scientific).
2.5 Genomic data analysis
For data analyses, Dragen Enrichment (v. 3.9.5) was used to perform a workflow starting with: 1) alignment to the reference genome (hg38), 2) identifying genetic variants from the sequencing reads, 3) annotating those variants with relevant information, and 4) filtering and prioritizing variants that are likely to cause the patient condition. Then, the data generated were analyzed with Variant Interpreter Software (Illumina) that integrates Sorting Intolerant From Tolerant (SIFT), Polymorphism Phenotyping (PolyPhen), ClinVar, 1000 Genomes Project, ExAC, EVS, COSMIC, dbSNP, and OMIM (Illumina, 2016a).
Data was filtered in Variant Interpreter Software and Excel to correlate if there were differences.
2.6 Diagnostic assessment
Ancestry proportion analysis showed an 8.3%, 40.3% and 51.3% of African, European, and Native American components, respectively.
Genomic data shown that quality score was a Q30 (probability of one incorrect base call in 1.000 bases) of 97.23%. Moreover, 1′680.470 passing filter reads with 979.287 aligned reads (78′436.355 aligned bases). Besides, the length of targeted reference (hg38) was 571.899 bp, and the depth coverage was 90.14% for 30X or above. The fragment length median was 74 bp, minimum 35 bp, and maximum 219 bp (SD = 29 pb).
The genomic pipeline of variant filter analysis started with 357 variants; the first variants that passed the quality control resulted in 350 variants. The subsequent filter was the consequence, leaving the variants that showed to be pathogenic, likely pathogenic, and uncertain significance (US); as a result, three variants were found (Table 1). Lastly, the patient was heterozygous for all the variants presented.
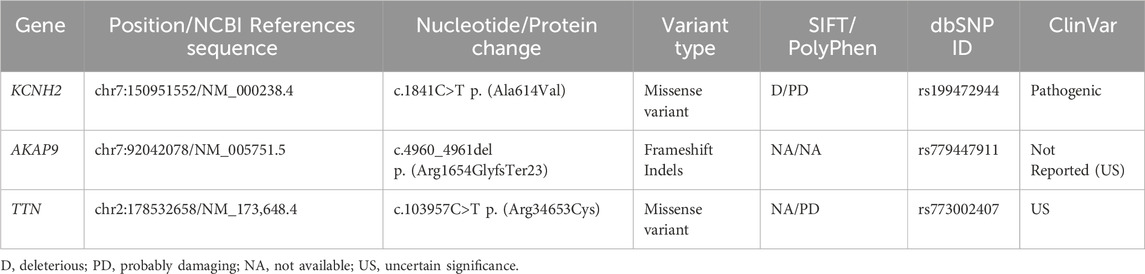
Table 1. Genetic variants detected in the patient diagnosed with congenital long QT syndrome.
The genomic test detected a missense variant c.1841C>T; p. (Ala614Val) in the KCNH2 gene, which was a known pathogenic variant associated with LQT2. This result was thoughtfully discussed with the patient and her parents, and an ICD implantation was decided based on: the presence of syncope while resting, which is considered an ominous symptom, female gender, location of the variant, and lack of treatment response.
3 Discussion
The LQTS is a primary inherited cardiac arrhythmia syndrome that may cause SCD. An estimate of its prevalence is 1:2000 (Schwartz et al., 2009). The usual mode of inheritance is autosomal dominant except for the autosomal recessive Jervell–Lange–Nielsen type (Wallace et al., 2019). LQTS has been described in all ethnic groups and its genotype depends on the harbored genetic variant. Also, each ethnic group with LQTS is associated with a specific more prevalent genetic variant. For example, genetic variants in the SCN5A gene of individuals with LQTS are more prevalent in African American people, increasing the risk of sudden death (Killen et al., 2010), while Brugada syndrome (BrS) associated with SCN5A variants have been reported more frequently in Caucasians and Asians individuals (Milman et al., 2019; Chen et al., 2020; Nakano and Shimizu, 2022), although BrS phenotype is more prevalent in East and Southeast Asia (Nakano and Shimizu, 2022; Khawaja et al., 2023). Instead, genetic variants in the KCNQ1 and KCNH2 genes have been described to be more prevalent in people with European and Japanese ancestry (Lahrouchi et al., 2020; Winbo et al., 2020). A missense variant p. (Ala614Val) in the KCNH2 gene (also known as hERG gene) have been described in several ethnic groups as a cause of LQT2. The clinical presentation of LQT2 harboring this variant is abnormal echocardiogram (prolonged QTc >500 msec), seizures, arrhythmic events and SCD (Moss et al., 2002; Tenenbaum et al., 2008). The subject described in the present case report harbors: a pathogenic variant p. (Ala614Val) in the KCNH2 gene, prolonged corrected QT interval (528 msec), propranolol resistance, and high SCD risk, which allows the diagnosis of LQT2. However, we could not evaluate the genomic data of the proband’s family, therefore we are not able to determine if this pathogenic variant is inherited.
In Ecuador, few studies have reported ancestry analyses and cardiovascular diseases (Cadena-Ullauri et al., 2022; Guevara-Ramírez et al., 2023). In the present case report, the proband shows a high European proportion (40.3%) in ancestry proportion analysis. This high European proportion could be a risk factor, due to increased KCNH2 mutation rates reported in European-descent population (Kong et al., 2017). However further studies should be performed to understand the ancestry role in LQTS development in the mestizo Ecuadorian population.
The various types of LQTS are associated with pathogenic variants in genes encoding distinct cardiac ion channels. The most common types are LQT1, LQT2, and LQT3, accounting for 85% of all genotyped LQTS cases (Wallace et al., 2019). LQT2 affects 25%-30% of LQTS individuals and is caused by heterozygous pathogenic variants in the KCNH2 gene. This gene encodes the voltage-gated K+ (Kv) channel α-subunit Kv11.1. Four Kv11.1 α-subunits co-assemble into a tetrameric ion channel that conducts the rapid delayed rectifier K+ current (IKr) on the heart. Loss-of-function KCNH2 mutations decrease IKr in LQT2. The impact on IKr in LQT2 depends on the variant. If the coding sequence involves amino acid residues located in the S5-loop-S6 region (552 through 657), these are considered pore variants that have been linked to an increased risk for arrhythmic events (Moss et al., 2002; Platonov et al., 2018). The pathogenic variant p. (Ala614Val) in the KCNH2 gene shows a dominant negative effect (Anderson et al., 2014; ClinVar, 2024). Hence, in this case, the function of the potassium channel is affected by the mutant protein. The subtypes of LQTS are associated with pathogenic variants in genes involved in sodium or calcium channel proteins, as well as proteins that interact with these ionic channels (Wallace et al., 2019). Therefore, the p. (Ala614Val) variant in the pore region of the KCNH2 gene, along with the prolonged QTc interval observed in this proband, allowed the diagnosis of LQT2 (Figures 1, 3).
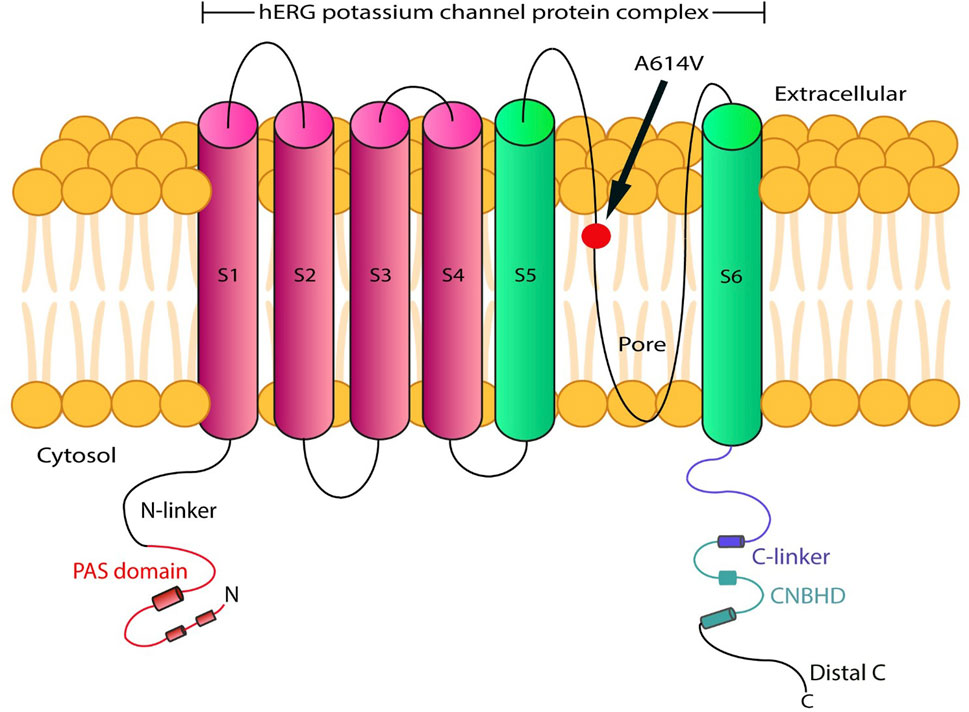
Figure 3. Schematic representation of the hERG protein variant (p.(Ala614Val)), located at the transmembrane pore region.
Platovov et al. also showed that LQT2 female carriers with abnormal T-wave morphology had a significantly higher risk of cardiac events compared to LQT2 female carriers with normal T waves (HR = 3.31; 95% CI: 1.68–6.52; p = 0.001) (Platonov et al., 2018). These electrocardiographic changes were observed in our patient (Figure 1).
In addition to the KCNH2 gene variant, the patient had a mutation in the AKAP9 gene c.4960_4961delAG; p. (Arg1654GlyfsTer23) (rs779447911) classified as a variant of uncertain significance (VUS) but the in silico predictions indicate likely pathogenic effects. This gene encodes for an isoform of kinase-A anchoring protein (AKAP9), which forms a complex with the α subunit of voltage-gated potassium channel (KCNQ1) and associates with ß subunits (KCNE1), that control the slowly activating delayed rectifier potassium channels (IKs). Pathogenic variants in this gene have been associated with the progress of long QT syndrome type 1 (de Villiers et al., 2015).
The variant c.4960_4961delAG; p. (Arg1654GlyfsTer23) reported in the present study has not been previously associated with LQTS. Further studies should be performed to understand the role of this variant and its influence on the patient’s risk of fatal events. Notably, the AKAP9 missense variant p. (Ser1570Leu) has been linked with LQTS through its involvement on IKs dysregulation. This mutation disrupts the KCNQ1-AKAP9 interaction and reduces KCNQ1 phosphorylation by cAMP induction (Chen et al., 2007). Additionally, the nonsense mediated decay (NMD) pathway could be implicated in the AKAP9 mRNA degradation, due to earlier stop codon detected in this gene (Lindeboom et al., 2019); hence, due to the heterozygous pattern of this gene variant, the lack of AKAP9 protein could also disrupts the KCNQ1-AKAP9 interaction (Chen et al., 2007). The reported premature stop codon in the AKAP9 gene could be implicated in the disruption of this interaction, promoting the IKs prolongation in this patient and worsening the proband’s phenotype (loss of consciousness). Therefore, KCNH2 and AKAP9 pathogenic variants observed in this patient, could explain the persistent cardiac events (IKr-IKs dysfunction, VT, prolonged QTc) under Propranolol therapy. Some studies have suggested that the combination of genetic variants and the additive effect of mutations in different genes can probably trigger a more aggressive phenotype in cardiac pathologies (Coll et al., 2018). Hence, all these risk factors aided the decision of ICD implantation. Recently, the link between AKAP9 gene pathogenic variants and LQTS development has been questioned (Adler et al., 2020). However, this report could provide evidence about the role of AKAP9 gene in potassium channelopathies.
Similarly, a missense variant in the TTN gene c.103957C>T p. (Arg34653Cys) (ClinVar ID: VCV001475968.4), classified as VUS, has not been previously described in LQT2. However, several nonsynonymous variants in this gene have been associated with QT interval variation and act as a risk factor for cardiac arrhythmias and SCD (Kapoor et al., 2016).
The use of genomic tools allows the identification of rare variants in a specific population. The role of these rare variants could influence in the subject clinical phenotype, due to its possible effects in cellular functional processes like signaling pathways. Moreover, the detection of rare variants could encourage the development of pharmaceutical strategies that lead to the personalized medicine approach (Momozawa and Mizukami, 2021). Therefore, the presence of the genetic variants in the AKAP9 and TTN genes described in this case report, could be associated with an increased pathogenicity of the LQT2 syndrome detected in this proband. However, functional analysis of these variants must be performed to assess its effects in congenital cardiovascular diseases of Ecuadorian population.
4 Conclusion
Genomic and genetic tests constitute powerful tools to evaluate the predisposition and diagnosis of LQTS and other inheriting cardiac disorders associated with SCD risk and may contribute to the treatment decision-making process, especially when this process involves complex and risky procedures.
Data availability statement
The data presented in the study are deposited in NCBI Sequence Read Archive (SRA) with the BioProject accession number PRJNA917705. (https://www.ncbi.nlm.nih.gov/bioproject/PRJNA917705/). For more information, please contact the corresponding author AZ (YW5hemFtYnJhbm8xN0Bob3RtYWlsLmNvbQ==).
Ethics statement
The studies involving humans were approved by Human Research Ethics Committee (CEISH) of UTE University (CEISH-2021-016). The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants’ legal guardians/next of kin. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author contributions
RT-T: Methodology, Writing–original draft, Formal Analysis. RI: Resources, Writing–review and editing. JL-B: Resources, Writing–review and editing. PG-R: Formal Analysis, Methodology, Writing–review and editing. SC-U: Formal Analysis, Methodology, Writing–original draft. EP-C: Methodology, Writing–review and editing. VR-P: Methodology, Writing–review and editing. ND: Conceptualization, Writing–review and editing. AI-R: Conceptualization, Writing–review and editing. AZ: Conceptualization, Formal Analysis, Funding acquisition, Project administration, Supervision, Writing–original draft.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. The publication fee of this article was funded by Universidad UTE.
Acknowledgments
The authors are grateful to all the volunteer students that were part of Centro de Investigación Genética y Genómica while the present project was performing; specially to Gabriela Salazar-Navas.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2024.1395012/full#supplementary-material
References
Adler, A., Novelli, V., Amin, A. S., Abiusi, E., Care, M., Nannenberg, E. A., et al. (2020). An international, multicentered, evidence-based reappraisal of genes reported to cause congenital long QT syndrome. Circulation. 141 (6), 418–428. doi:10.1161/CIRCULATIONAHA.119.043132
Anderson, C. L., Kuzmicki, C. E., Childs, R. R., Hintz, C. J., Delisle, B. P., and January, C. T. (2014). Large-scale mutational analysis of Kv11.1 reveals molecular insights into type 2 long QT syndrome. Nat. Commun. 5 (5535), 1–13. doi:10.1038/ncomms6535
Cadena-Ullauri, S., Guevara-Ramirez, P., Ruiz-Pozo, V., Tamayo-Trujillo, R., Paz-Cruz, E., Sánchez Insuasty, T., et al. (2022). Case report: genomic screening for inherited cardiac conditions in Ecuadorian mestizo relatives: improving familial diagnose. Front. Cardiovasc Med. 9 (1037370), 1037370–1037377. doi:10.3389/fcvm.2022.1037370
Chen, C., Tan, Z., Zhu, W., Fu, L., Kong, Q., Xiong, Q., et al. (2020). Brugada syndrome with SCN5A mutations exhibits more pronounced electrophysiological defects and more severe prognosis: a meta-analysis. Clin. Genet. 97 (1), 198–208. doi:10.1111/cge.13552
Chen, L., Marquardt, M. L., Tester, D. J., Sampson, K. J., Ackerman, M. J., and Kass, R. S. (2007). Mutation of an A-kinase-anchoring protein causes long-QT syndrome. Proc. Natl. Acad. Sci. U. S. A. 104 (52), 20990–20995. doi:10.1073/pnas.0710527105
Cho, Y. (2016a). Left cardiac sympathetic denervation: an important treatment option for patients with hereditary ventricular arrhythmias. J. Arrhythmia 32 (5), 340–343. doi:10.1016/j.joa.2015.08.002
Cho, Y. (2016b). Management of patients with long QT syndrome. Korean Circ. J. 46 (6), 747–752. doi:10.4070/kcj.2016.46.6.747
ClinVar (2024). NCBI NL of M. VCV000029777.13 - ClinVar - NCBI ClinVar. Available at: https://www.ncbi.nlm.nih.gov/clinvar/variation/29777/(Accessed May 1, 2024).
Coll, M., Pérez-Serra, A., Mates, J., del Olmo, B., Puigmulé, M., Fernandez-Falgueras, A., et al. (2018). Incomplete penetrance and variable expressivity: hallmarks in channelopathies associated with sudden cardiac death. Biol. (Basel). 7 (3), 3–10. doi:10.3390/biology7010003
de Villiers, C., van der Merwe, L., Crottu, L., Goosen, A., George, Jr. A., Schwartz, P. J., et al. (2015). AKAP9 is a genetic modifier of congenital long-QT syndrome type 1. Circ. Cardiovasc Genet. 61 (6), 515–525. doi:10.1161/CIRCGENETICS.113.000580
Fonseca, D. J., and Joaquim, M. (2018). Cardiac channelopathies: the role of sodium channel mutations. Rev. Port. Cardiol. 37 (2), 179–199. doi:10.1016/j.repc.2017.11.007
Goldstein, S., Ward, C., and Al-Khatib, S. (2017). The use of implantable cardioverter-defibrillators in the prevention of sudden cardiac death: a focus on congenital heart disease and inherited arrhythmia syndromes. J. Innov. Card. Rhythm Manag. 9 (1), 2996–3005. doi:10.19102/icrm.2018.090103
Guevara-Ramírez, P., Cadena-Ullauri, S., Ibarra-Castillo, R., Laso-Bayas, J. L., Paz-Cruz, E., Tamayo-Trujillo, R., et al. (2023). Genomic analysis of a novel pathogenic variant in the gene LMNA associated with cardiac laminopathies found in Ecuadorian siblings: a case report. Front. Cardiovasc Med. 10 (1141083), 1141083–1141086. doi:10.3389/fcvm.2023.1141083
Illumina (2016a). BaseSpace variant interpreter beta illumina. Available at: https://www.illumina.com/content/dam/illumina-marketing/documents/products/datasheets/basespace-variant-interpreter-data-sheet-970-2016-004.pdf.
Illumina (2016b). TruSight TM Cardio sequencing kit illumina. Available at: https://support.illumina.com/content/dam/illumina-support/documents/documentation/chemistry_documentation/samplepreps_trusight/trusight-cardio/trusight-cardio-sequencing-kit-reference-guide-15063774-01.pdf.
Invitrogen (2024). PureLink ® genomic DNA kits for purification of genomic DNA. Carlsbad, CA, United States: Invitrogen, 1–48. Available at: https://tools.thermofisher.com/content/sfs/manuals/purelink_genomic_man.pdf.
Kapoor, A., Bakshy, K., Xu, L., Nandakumar, P., Lee, D., Boerwinkle, E., et al. (2016). Rare coding TTN variants are associated with electrocardiographic QT interval in the general population. Sci. Rep. 6 (28356), 28356–28357. doi:10.1038/srep28356
Khawaja, M., Qadeer, Y. K., Siddiqui, R., Chelu, M. G., Aiumtrakul, N., Pickett, J. K., et al. (2023). Brugada syndrome within asian populations: state-of-the-art review. Cardiogenetics 13 (2), 61–74. doi:10.3390/cardiogenetics13020007
Killen, S. A. S., Kunic, J., Wang, L., Lewis, A., Levy, B. P., Ackerman, M. J., et al. (2010). SCN5A allelic expression imbalance in African-Americans heterozygous for the common variant p.Ser1103Tyr. BMC Med. Genet. 11 (1), 74–4. doi:10.1186/1471-2350-11-74
Kong, T., Feulefack, J., Ruether, K., Shen, F., Zheng, W., Chen, X. Z., et al. (2017). Ethnic differences in genetic ion channelopathies associated with sudden cardiac death: a systematic review and meta-analysis. Ann. Clin. Lab. Sci. 47 (4), 481–490.
Lahrouchi, N., Tadros, R., Crotti, L., Mizusawa, Y., Postema, P. G., Beekman, L., et al. (2020). Transethnic genome-wide association study provides insights in the genetic architecture and heritability of long QT syndrome. Circulation. 142 (4), 324–338. doi:10.1161/CIRCULATIONAHA.120.045956
Lindeboom, R. G. H., Vermeulen, M., Lehner, B., and Supek, F. (2019). The impact of nonsense-mediated mRNA decay on genetic disease, gene editing and cancer immunotherapy. Nat. Genet. 51 (11), 1645–1651. doi:10.1038/s41588-019-0517-5
Lopez, K. N., Nunez-Gallegos, F., Wang, Y., Cannon, B., Kim, J., and Valdes, S. (2018). LONG QT SYNDROME IN THE HISPANIC POPULATION: A COMPARATIVE STUDY. J. Am. Coll. Cardiol. 71 (11), A430. doi:10.1016/s0735-1097(18)30971-9
Milman, A., Andorin, A., Postema, P. G., Gourraud, J. B., Sacher, F., Mabo, P., et al. (2019). Ethnic differences in patients with Brugada syndrome and arrhythmic events: new insights from survey on arrhythmic events in Brugada syndrome. Hear Rhythm 16 (10), 1468–1474. doi:10.1016/j.hrthm.2019.07.003
Momozawa, Y., and Mizukami, K. (2021). Unique roles of rare variants in the genetics of complex diseases in humans. J. Hum. Genet. 66 (1), 11–23. doi:10.1038/s10038-020-00845-2
Moss, A. J., Zareba, W., Kaufman, E. S., Gartman, E., Peterson, D. R., Benhorin, J., et al. (2002). Increased risk of arrhythmic events in long-QT syndrome with mutations in the pore region of the human ether-a-go-go-related gene potassium channel. Circulation 105 (7), 794–799. doi:10.1161/hc0702.105124
Nagaoka, I., Shimizu, W., Itoh, H., Yamamoto, S., Sakaguchi, T., Oka, Y., et al. (2008). Mutation site dependent variability of cardiac events in Japanese LQT2 form of congenital long-QT syndrome. Circ. J. 72 (5), 694–699. doi:10.1253/circj.72.694
Nakano, Y., and Shimizu, W. (2022). Brugada syndrome as a major cause of sudden cardiac death in Asians. JACC Asia 2 (4), 412–421. doi:10.1016/j.jacasi.2022.03.011
Platonov, P. G., McNitt, S., Polonsky, B., Rosero, S., Kutyifa, V., Huang, A., et al. (2018). Risk stratification of type 2 long QT syndrome mutation carriers with normal QTc interval: the value of gender, T-wave morphology and mutation type. Circ. Arrhythm. Electrophysiol. 11 (7), 1–9. doi:10.1161/circep.117.005918
Safran, M., Rosen, N., Twik, M., BarShir, R., Stein, T. I., Dahary, D., et al. (2021). The GeneCards suite. Pract Guid Life Sci Databases, 27–56. doi:10.1007/978-981-16-5812-9_2
Sakaguchi, T., Itoh, H., Ding, W. G., Tsuji, K., Nagaoka, I., Oka, Y., et al. (2008). Hydroxyzine, a first generation H1-receptor antagonist, inhibits human ether-a-go-go-related gene (HERG) current and causes syncope in a patient with the HERG mutation. J. Pharmacol. Sci. 108 (4), 462–471. doi:10.1254/jphs.08178fp
Schwartz, P. J., Crotti, L., and Insolia, R. (2012). Long-QT syndrome from genetics to management. Circ. Arrhythmia Electrophysiol. 5 (4), 868–877. doi:10.1161/CIRCEP.111.962019
Schwartz, P. J., Stramba-Badiale, M., Crotti, L., Pedrazzini, M., Besana, A., Bosi, G., et al. (2009). Prevalence of the congenital long QT syndrome. Circulation 120 (18), 1761–1767. doi:10.1161/CIRCULATIONAHA.109.863209
Tenenbaum, M., Lavi, S., Magal, N., Halpern, G. J., Bolocan, I., Boulos, M., et al. (2008). Identification of the gene causing long QT syndrome in an Israeli family. Isr. Med. Assoc. J. 10 (11), 809–811.
Wallace, E., Howard, L., Liu, M., O’Brien, T., Ward, D., Shen, S., et al. (2019). Long QT syndrome: genetics and future perspective. Pediatr. Cardiol. 40 (7), 1419–1430. doi:10.1007/s00246-019-02151-x
Wilde, A. A. M., Amin, A. S., and Postema, P. G. (2022b). Diagnosis, management and therapeutic strategies for congenital long QT syndrome. Heart 108 (5), 332–338. doi:10.1136/heartjnl-2020-318259
Wilde, A. A. M., Semsarian, C., Márquez, M. F., Shamloo, A. S., Ackerman, M. J., Ashley, E. A., et al. (2022a). European heart rhythm association (EHRA)/heart rhythm society (HRS)/Asia pacific heart rhythm society (APHRS)/Latin American heart rhythm society (LAHRS) expert consensus statement on the state of genetic testing for cardiac diseases. Europace 24 (8), 1307–1367. doi:10.1093/europace/euac030
Winbo, A., Earle, N., Marcondes, L., Crawford, J., Prosser, D. O., Love, D. R., et al. (2020). Genetic testing in Polynesian long QT syndrome probands reveals a lower diagnostic yield and an increased prevalence of rare variants. Hear Rhythm 17 (8), 1304–1311. doi:10.1016/j.hrthm.2020.03.015
Zambrano, A. K., Gaviria, A., Cobos-Navarrete, S., Gruezo, C., Rodríguez-Pollit, C., Armendáriz-Castillo, I., et al. (2019). The three-hybrid genetic composition of an Ecuadorian population using AIMs-InDels compared with autosomes, mitochondrial DNA and Y chromosome data. Sci. Rep. 9 (9247), 9247–9248. doi:10.1038/s41598-019-45723-w
Keywords: implantable defibrillator, long QT syndrome, NGS, kcnh2, case report
Citation: Tamayo-Trujillo R, Ibarra-Castillo R, Laso-Bayas JL, Guevara-Ramirez P, Cadena-Ullauri S, Paz-Cruz E, Ruiz-Pozo VA, Doménech N, Ibarra-Rodríguez AA and Zambrano AK (2024) Identifying genomic variant associated with long QT syndrome type 2 in an ecuadorian mestizo individual: a case report. Front. Genet. 15:1395012. doi: 10.3389/fgene.2024.1395012
Received: 02 March 2024; Accepted: 27 May 2024;
Published: 18 June 2024.
Edited by:
Jared C. Roach, Institute for Systems Biology (ISB), United StatesReviewed by:
Oscar Campuzano, University of Girona, SpainChai Ann Ng, Victor Chang Cardiac Research Institute, Australia
Copyright © 2024 Tamayo-Trujillo, Ibarra-Castillo, Laso-Bayas, Guevara-Ramirez, Cadena-Ullauri, Paz-Cruz, Ruiz-Pozo, Doménech, Ibarra-Rodríguez and Zambrano. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ana Karina Zambrano, YW5hemFtYnJhbm8xN0Bob3RtYWlsLmNvbQ==
†These authors have contributed equally to this work