 Hanne M. Boen1,2*
Hanne M. Boen1,2* Maaike Alaerts3
Maaike Alaerts3 Lut Van Laer3
Lut Van Laer3 Johan B. Saenen1,2
Johan B. Saenen1,2 Inge Goovaerts2,3
Inge Goovaerts2,3 Jarl Bastianen3
Jarl Bastianen3 Pieter Koopman4
Pieter Koopman4 Philippe Vanduynhoven5Elke De Vuyst5Michael Rosseel5Hein Heidbuchel1,2
Philippe Vanduynhoven5Elke De Vuyst5Michael Rosseel5Hein Heidbuchel1,2 Emeline M. Van Craenenbroeck1,2
Emeline M. Van Craenenbroeck1,2 Bart Loeys3
Bart Loeys3- 1Research Group Cardiovascular Diseases, Genetics, Pharmacology and Physiopathology of Heart, Blood Vessels and Skeleton (GENCOR) Department, University of Antwerp, Antwerp, Belgium
- 2Department of Cardiology, Antwerp University Hospital, Antwerp, Belgium
- 3Center of Medical Genetics, Cardiogenomics, Genetics, Pharmacology and Physiopathology of Heart, Blood Vessels and Skeleton (GENCOR) Department, Antwerp University Hospital and University of Antwerp, Antwerp, Belgium
- 4Hartcentrum Hasselt, Jessa Hospital Hasselt, Hasselt, Belgium
- 5Department of Cardiology, ASZ Aalst, Aalst, Belgium
Background: Variants in the MYBPC3 gene are a frequent cause of hypertrophic cardiomyopathy (HCM) but display a large phenotypic heterogeneity. Founder mutations are often believed to be more benign as they prevailed despite potential negative selection pressure. We detected a pathogenic variant in MYBPC3 (del exon 23-26) in several probands. We aimed to assess the presence of a common haplotype and to describe the cardiac characteristics, disease severity and long-term outcome of mutation carriers.
Methods: Probands with HCM caused by a pathogenic deletion of exon 23-26 of MYBPC3 were identified through genetic screening using a gene panel encompassing 59 genes associated with cardiomyopathies in a single genetic center in Belgium. Cascade screening of first-degree relatives was performed, and genotype positive relatives were further phenotyped. Clinical characteristics were collected from probands and relatives. Cardiac outcomes included death, heart transplantation, life-threatening arrhythmia, heart failure hospitalization or septal reduction therapy. Haplotype analysis, using microsatellite markers surrounding MYBPC3, was performed in all index patients to identify a common haplotype. The age of the founder variant was estimated based on the size of the shared haplotype using a linkage-disequilibrium based approach.
Results: We identified 24 probands with HCM harbouring the MYBPC3 exon 23-26 deletion. Probands were on average 51 ± 16 years old at time of clinical HCM diagnosis and 62 ± 10 years old at time of genetic diagnosis. A common haplotype of 1.19 Mb was identified in all 24 probands, with 19 of the probands sharing a 13.8 Mb haplotype. The founder event was estimated to have happened five generations, or 175–200 years ago, around the year 1830 in central Flanders. Through cascade screening, 59 first-degree relatives were genetically tested, of whom 37 (62.7%) were genotype positive (G+) and 22 (37.3%) genotype negative (G-). They were on average 38 ± 19 years old at time of genetic testing. Subsequent clinical assessment revealed a HCM phenotype in 19 (51.4%) G+ relatives. Probands were older (63 ± 10 vs. 42 ± 21 years; p < 0.001) and had more severe phenotypes than G+ family members, presenting with more symptoms (50% vs. 13.5%; p = 0.002), arrhythmia (41.7% vs. 12.9%, p = 0.014), more overt hypertrophy and left ventricular outflow tract obstruction (43.5% vs. 3.0%; p < 0.001). Male G+ relatives more often had a HCM phenotype (78.6% vs. 34.8%; p = 0.010) and were more severely affected than females. At the age of 50, a penetrance of 78.6% was observed, defined as the presence of HCM in 11 of 14 G+ relatives with age ≥50 years. Overall, 20.3% of all variant carriers developed one of the predefined cardiac outcomes after a median follow-up of 5.5 years with an average age of 50 (±21) years.
Conclusion: A Belgian founder variant, an exon 23-26 deletion in MYBPC3, was identified in 24 probands and 37 family members. The variant is characterized by a high penetrance of 78.6% at the age of 50 years but has variable phenotypic expression. Adverse outcomes were observed in 20.3% of patients during follow-up.
1 Introduction
As the evidence of a genetic basis in hypertrophic cardiomyopathy (HCM) has grown in the last decades, genetic testing in all patients with HCM is now recommended by both the American Heart Association and the European Cardiac Society (Musunuru et al., 2020; Wilde et al., 2022). In about half of all patients a genetic diagnosis can be made, with a diagnostic yield of about 32%–40% in unselected patients and 70% in patients with a positive family history (Alfares et al., 2015; Wilcox and Hershberger, 2018). In an additional 9%–15% of patients a variant of uncertain significance (VUS) is identified (Alfares et al., 2015). Causal genetic variation in HCM patients is most often found in the sarcomeric genes (Van Driest et al., 2005; Wilcox and Hershberger, 2018), including MYBPC3, which is the most frequently affected gene, with up to 30%–57% of all identified variants (Richard et al., 2003; Alfares et al., 2015).
The myosin-binding protein C (MyBP-C) protein is part of the thick filament of the sarcomere (Winegrad, 1999). The specific cardiac isoform, cMyBP-C can be found in cardiomyocytes only and is encoded by the MYBPC3 gene, located on chromosome 11 and consisting of 35 exons (Carrier et al., 1997; Winegrad, 1999). The cMyBP-C-protein binds to myosin with its C-terminal module as well as to titin, another important structural protein of the sarcomere (Winegrad, 1999). Overall, cMyBP-C plays a regulatory role in cardiac contractility by altering myofilament sliding velocity (Winegrad, 1999).
Both missense and loss-of-function variants in MYBPC3 have been associated with HCM (Walsh et al., 2017; Ingles et al., 2019). Truncating variants in MYBPC3 can be considered pathogenic since they associate strongly with HCM and haploinsufficiency is a well-established disease mechanism (Walsh et al., 2017). Copy Number Variants (CNV) involving MYBPC3 are seen in 1.4% of HCM patients (Mates et al., 2020). Depending on the size and exact location, deletions can lead to a premature stop-codon, leading to truncation, or result in a shortened protein (Rottbauer et al., 1997; Andersen et al., 2004; Frank-Hansen et al., 2008).
In HCM, an extensive phenotypic variability in severity and outcomes is seen. The identification of a sarcomeric mutation seems to play a role in disease severity as genotype positive HCM patients have a two-fold higher risk for adverse outcomes (death, heart failure, malignant ventricular arrhythmias, and atrial fibrillation) than genotype negative HCM patients (Ho et al., 2018). Interestingly, also HCM patients with a VUS show an increased incidence of death and atrial fibrillation compared to completely genotype negative HCM patients (Ho et al., 2018). The specific affected gene also influences disease severity and outcome, with MYH7 carriers being in general more severely affected compared to MYBPC3 carriers (Ho et al., 2018).
In addition, genetic and environmental modifiers, such as arterial hypertension, likely play a role in this observed phenotypic variability (Daw et al., 2007; Claes et al., 2016; Walsh et al., 2017; Helms et al., 2020).
Within the group of MYBPC3-variant harbouring individuals a significant degree of phenotypic variability is observed, with variable penetrance and expressivity, depending on age and gender (Page et al., 2012; Helms et al., 2020). Founder variants, generally present in large cohorts, provide interesting opportunities to investigate genetic and non-genetic modifiers. This results in a better understanding of phenotype-genotype correlations which can aid in genetic counselling of patients and family members.
Here, we describe the phenotypic spectrum of an exon 23-26 deletion in MYBPC3, a Belgian founder variant.
2 Materials and methods
2.1 Proband selection and variant identification
In a total of 1,364 HCM probands who presented at the cardiogenetics clinic of the Antwerp University Hospital for genetic testing, from January 2015 until October 2023, a recurrent MYBPC3 (ENST00000545968; NM_000256.3) variant, a large heterozygous deletion of exon 23 to 26 (del23-26), was identified in 24 distinct families (1.8% of probands). Probands were defined as patients with a new diagnosis of HCM, presenting for cardiogenetic diagnosis, without prior knowledge of a genetic variant within their family. For each proband a family tree was drawn up to four generations. Through comparison of family trees, possible closely related probands could be identified, This was the case for two separately presenting individuals of family three, which were subsequently merged in 1 single family with the first identified patient serving as proband.
This cardiogenetic screening was performed using a cardiomyopathy (CM) next-generation sequencing (NGS) gene panel encompassing 59 genes associated with CMs. In short, genomic DNA of HCM patients was extracted from EDTA blood using standard procedures (Chemagic DNA bloodkit special, Perkin Elmer, Waltham, MA, United States). Genetic analysis was performed using either custom HaloPlex target enrichment followed by NGS (Illumina) as previously described (Proost et al., 2015) (before June 2021) or whole exome sequencing after enrichment of the exome (Twist Human Core Exome kit, Twist Bioscience with additional custom probes) (starting from June 2021). The deletion was identified through CNV analysis using a read-depth strategy (SeqPilot, JSI) and confirmed with a multiplex amplicon quantification (MAQ) assay. When applying a read-depth strategy for CNV analysis, the raw number of reads for each region of interest (ROI) is normalized against the number of reads of selected control regions within the sample and compared between all samples within the same sequencing run. A deletion is characterized by approximately 50% of the reads found when compared to controls, while a duplication will show approximately 150% of the reads.
2.2 Cascade screening
Cascade-screening of first-degree relatives was performed. Family members were invited to participate by family letter and genetic counselling by a multidisciplinary team was offered before and after variant testing. After family members underwent genetic testing (using MAQ) for the MYPBC3 del23-26 variant, genotype positive (G+) relatives were subsequently offered clinical evaluation. If the proband carried an additional variant, identified with the 59 CM-panel, this additional variant was sequenced in the family members as well, using sanger sequencing.
2.3 Clinical data
Demographic characteristics, cardiac assessment and outcome were collected from probands and relatives’ patient files. Composite cardiac outcome included death, heart transplantation (HTx), life-threatening arrhythmia, heart failure hospitalization or septal reduction therapy.
2.4 Identification of the deletion break point
To assess the exact localization of the breakpoints of the deletion a touchdown PCR was performed using a forward primer located at the end of exon 22 and a reverse primer at the beginning of exon 27 of the MYBPC3 gene (95°C 5′, 20× (95°C 30″, 65 (−0.5×)°C 30″, 72°C 1′), 15× (95°C 30″, 55°C 30″, 72°C 1′), 72°C 10′, 15°C 10′).
MYBPC3-E22F-M13F: GTTTTCCCAGTCACGACGAATAAGGCCCCAGCCAGGCCA
MYBPC3-E27R-M13R: CAGGAAACAGCTATGACATGTGTGCTCTGTCAGCCCCTGCA
The presence of a PCR product within the expected range was confirmed on Qiaxcel® capillary electrophoresis (Qiagen). For the non-affected allele, we did not expect a PCR product as this would be too big (>4,000 bp) for amplification. Sanger sequencing was used to sequence the resulting PCR product (3500 Dx Genetic Analyzer, Applied Biosystems) and sequence was compared to the reference sequence ENST00000545968.6 retrieved from Ensembl (Martin et al., 2022) using CLC DNA workbench software version 5.7.1 (CLC Bio, Aarhus, Denmark).
2.5 Haplotype analysis
First, a shared genetic background was identified in all index patients, based on intragenic SNPs within the MYBPC3 gene (Table 2). Next, haplotype analysis, using microsatellite markers surrounding MYBPC3, was performed to confirm the presence and assess the size of the common haplotype in all probands (Figure 1).
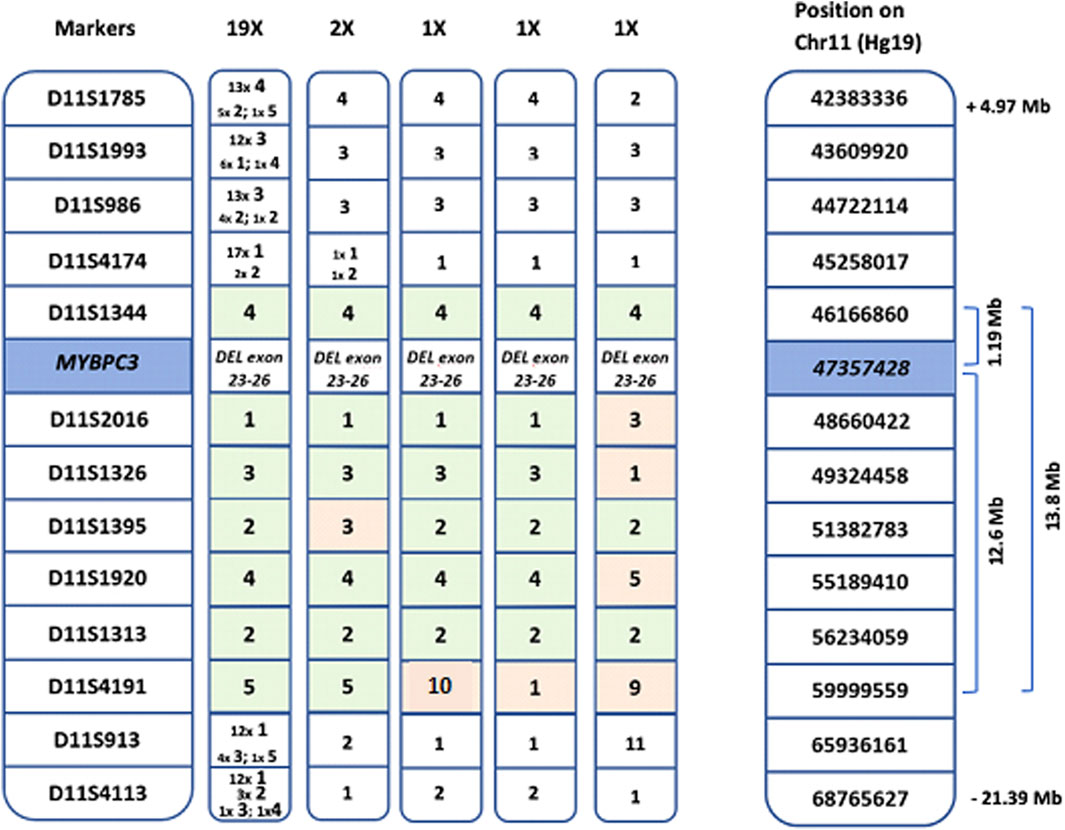
Figure 1. Length of the common haplotype, based on markers surrounding the MYBPC3 gene. Shared haplotypes spanning seven markers derived from the 24 probands. From left to right: common haplotype found in 19 probands, 4 haplotypes of the 5 probands with recombination events and genomic location of the markers on chromosome 11 (Hg19). The common haplotype shared by all probands is 1.19 Mb, 19 of the 24 probands share a common haplotype of 13.8 Mb.
Thirteen microsatellite markers surrounding MYBPC3 were analyzed in all selected probands as has been described before (Bergman et al., 2001) (D11S1785; D11S1993; D11S986; D11S4174; D11S1344; D11S2016; D11S1326; D11S1395; D11S1920; D11S1313; D11S4191; D11S913; D11S4113). Forward primers were labeled with 6-fluorescein amidite (FAM), and PCR products were analyzed in the presence of an internal sizing standard (ROX) using ABI3130XL (Applied Biosystems). The sizes of the amplicons were determined using ABI GeneMapper software v3.7 (Applied Biosystems).
After confirming a shared haplotype, the age of the variant was estimated based on the common microsatellite markers using a linkage-disequilibrium based approach (Bergman et al., 2001). The age of the variant (t, in number of generations) was estimated using the following formula:
Where Pd1 is the frequency of the founder marker allele in variant-carrying haplotypes and Pn2 is the frequency of the founder marker allele in non-variant carrying haplotypes. The founder marker allele was assumed to be the most frequent allele among the variant-carrying haplotypes. When the founder marker allele is present in all variant-carrying haplotypes (Pd1 = 1), but absent from all non-variant carrying haplotypes (Pn2 = 0), this formula can no longer be used as the estimated mutation age would equal 0. r is the recombination fraction between a marker and the variant and is calculated as:
2.6 Statistical analysis
Frequencies are expressed as numbers and percentages. Continuous variables are expressed as mean ± SD if normally or median ± interquartile range if non-normally distributed. Unpaired T-test or Mann-Whitney U test was used for comparisons of continuous variables. Chi-square test was used for comparisons of categorical variables. All analyses were performed using SPSS Statistics version 28 (IBM Corporation). A p-value of <0.05 was considered statistically significant.
3 Results
3.1 Identification of probands
From January 2015 till October 2023, we identified 24 probands with HCM carrying the MYBPC3 exon 23-26 deletion (Table 1), with a mean age 51 ± 16 years at time of clinical HCM diagnosis and 62 ± 10 years at time of genetic diagnosis. A positive family history of HCM was present in ten (41.7%) probands, sudden cardiac death (SCD) had occurred in nine families (37.5%) of which in two (8.3%) families before the age of 35.
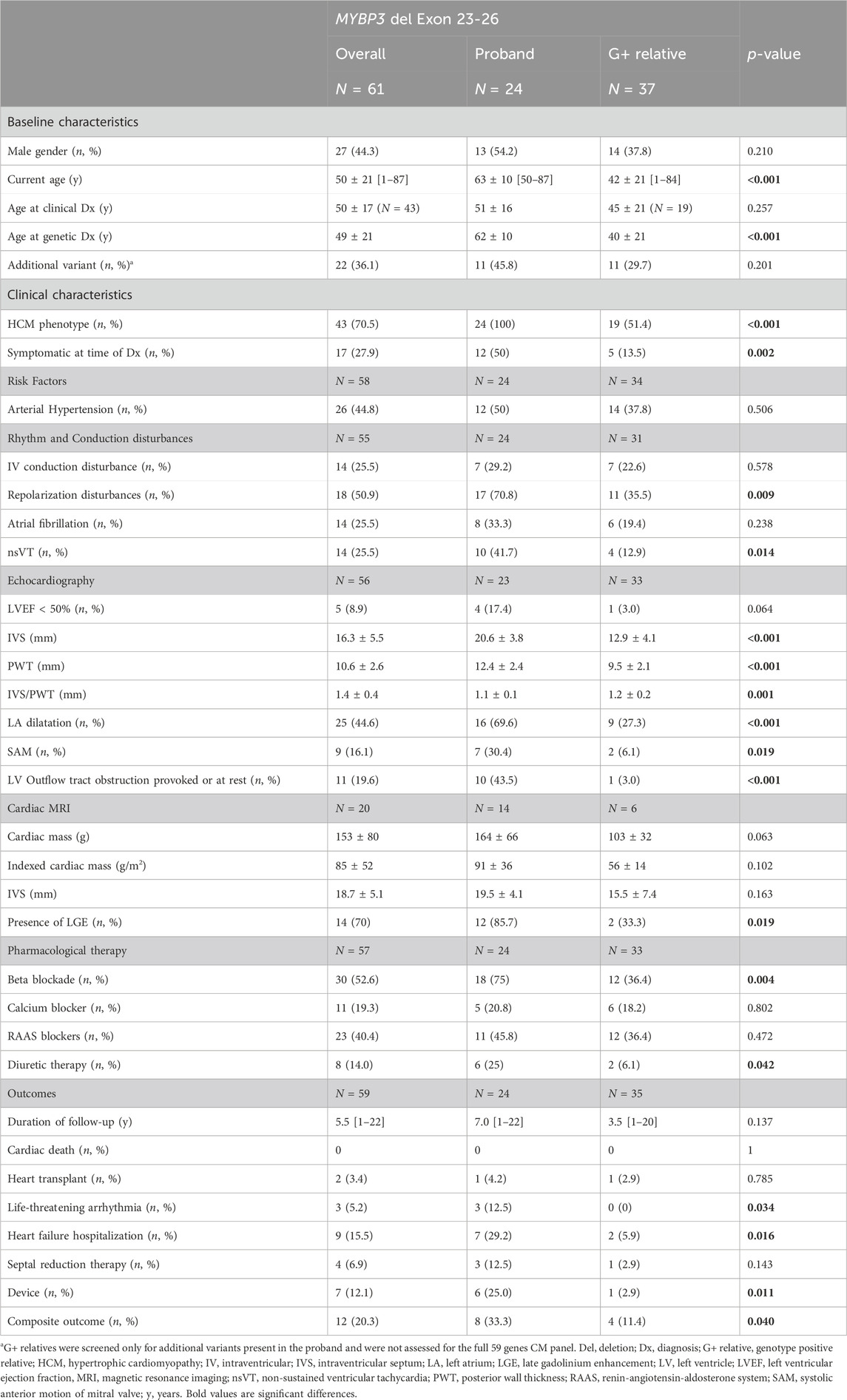
Table 1. Overview of patient characteristics and comparison between probands and G+ relatives.
3.2 Identification of the deletion breakpoint
Using a nested-PCR approach we delineated the exact breakpoints of the MYBPC3 deletion of exon 23 to 26. The complete deletion spans 3546 base pairs from intron 22 to intron 26: Chr11:g.47360721 – 47357175del (GRCh37/hg19). Exon 23 and part of exon 24 encode the last part of the C5 immunoglobulin-I domain whereas the remainder of exon 24, exon 25 and exon 26 encode the C6 fibronectin-3 domain of the cMyBP-C protein (Carrier et al., 1997).
3.3 Variant classification
The variant was classified as pathogenic (class 5) according to ACMG guidelines (Richards et al., 2015; Riggs et al., 2020) with the following arguments: 1. Copy number loss content 1A (contains protein-coding functionally important element; 0p); 2. Overlap with established haplo-insufficient gene 2E [both breakpoints are within the same gene; classification according to sequence variant interpretation criteria: PVS1 (null variant in a gene where loss of function is a known mechanism), PS4/PM2 (increased prevalence in affected individuals compared to controls/absent from controls); PP1 (co-segregation with disease in multiple affected family members); 0.9 p]; and 3. Evaluation of number of genes in the CNV: 3A (0-24 genes; 0p); 4. Detailed evaluation of genomic content 4F and 5D (7 or more observed segregations; 0.45 p). 66 informative meioses were present over all families. Of a total of 20 relatives with HCM, 19 carried the MYBPC3 variant. An overview of the pedigrees of all 24 families is available in the Supplementary Material.
3.4 Haplotyping
A common genetic background, defined by intragenic SNPs within the MYBPC3 gene was identified in all 24 probands (Table 2).
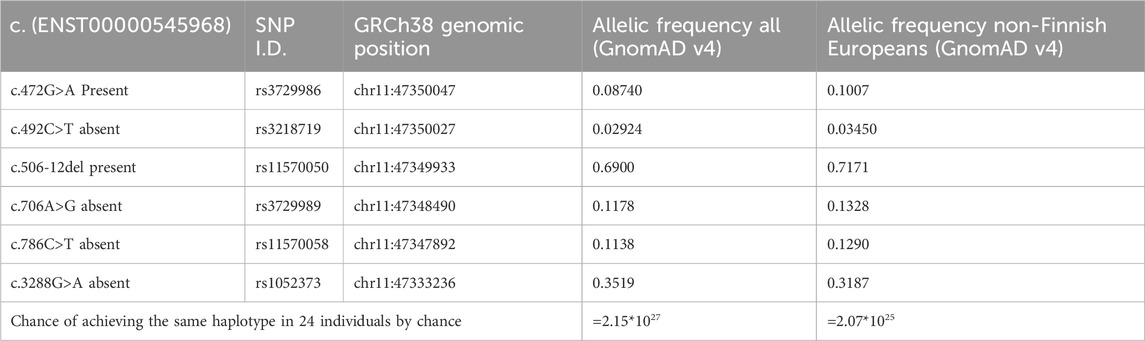
Table 2. Overview of shared genetic background in probands. Based on the allelic frequency of the intragenic SNPs, within the MYBPC3 gene. The chance of all 24 probands having the same haplotype by chance is calculated to be 2.07*1025, suggesting a founder event.
The length of the common haplotype was assessed in all probands by 13 microsatellite markers spanning 26.4 Mb. A common haplotype of 1.19 Mb was identified in all 24 probands, with 19 of the probands sharing a 13.8 Mb haplotype (Figure 1). The founder event was estimated to have happened five generations ago. Taking into account that probands were on average 63 years old (ranging 50–87 years) and assuming that each generation spans circa 25 years (5*25 + 63), the founder event presumably took place 175–200 years ago (around the year 1830). All probands now live in a well-defined region within central Flanders, presumably the founder lived close to the region of Aalst (Figure 2).

Figure 2. Overview of the location of the Belgian founders. The sizes of the dots represent the number of families identified in each city. All families live in a well-defined region between Brussels, Gent and Antwerp, centring around Aalst, except for one family identified in Tongeren in the East of Belgium.
3.5 Cardiac screening of relatives
Through cascade screening 59 first-degree relatives were genetically tested, of whom 37 (62.7%) were genotype positive (G+) and 22 (37.3%) genotype negative (G-). Screened family members were on average 38 ± 19 years old at the time of genetic testing. Differences between G+ and G- family members are displayed in Table 3.
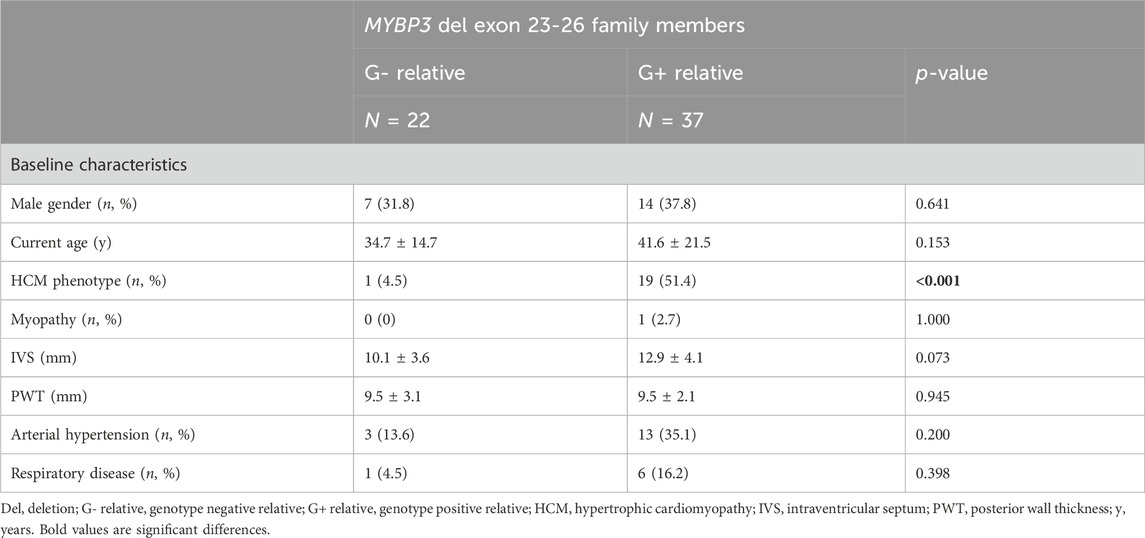
Table 3. Overview of baseline characteristics and comparison between G- and G+ relatives.
Subsequent clinical assessment revealed an HCM phenotype in 19 (51.4%) G+ relatives, of whom only 5 (26.3% of newly identified HCM phenotypes) were symptomatic (NYHA class >1, angina, or syncope) at the time of genetic diagnosis (Table 1). G+ relatives with an HCM phenotype were older than those without an HCM phenotype (52.7 ± 21.3 vs. 30.6 ± 15.3; p < 0.001).
Characteristics of probands and G+ relatives are displayed in Table 1. Probands were older than G+ relatives (current age 63 ± 10 years vs. 42 ± 21 years; p < 0.001) and had more severe phenotypes with more clinical symptoms, non-sustained ventricular tachycardia (nsVT), more overt hypertrophy and left ventricular outflow tract obstruction (LVOTO) on echocardiography. The average interventricular septum thickness (IVS) was significantly higher in probands than G+/phenotype positive family members (20.6 ± 3.8 vs. 15.3 ± 3.5 mm; p < 0.001). Probands also presented with worse outcomes: more life-threatening arrhythmias, more heart failure hospitalizations, and a higher need for device therapy.
One might hypothesize that the differences between probands and G+ relatives is related to the age difference but when probands were compared to an age matched subgroup of G+ relatives (n = 16) we could still observe significant differences in hypertrophy (IVS 20.6 ± 3.8 vs. 15.2 ± 3.4; p ≤ 0.001) prevalence of device implantation (25.0% vs. 0%; p = 0.035) and prevalence of LVOTO (43.5% vs. 6.7%; p = 0.014) (Figure 3). Gender distribution was similar between probands and matched relatives. At the age of 50, a penetrance of 78.6% was observed, defined as the presence of HCM in 11 of 14 G+ relatives with age ≥50 years.
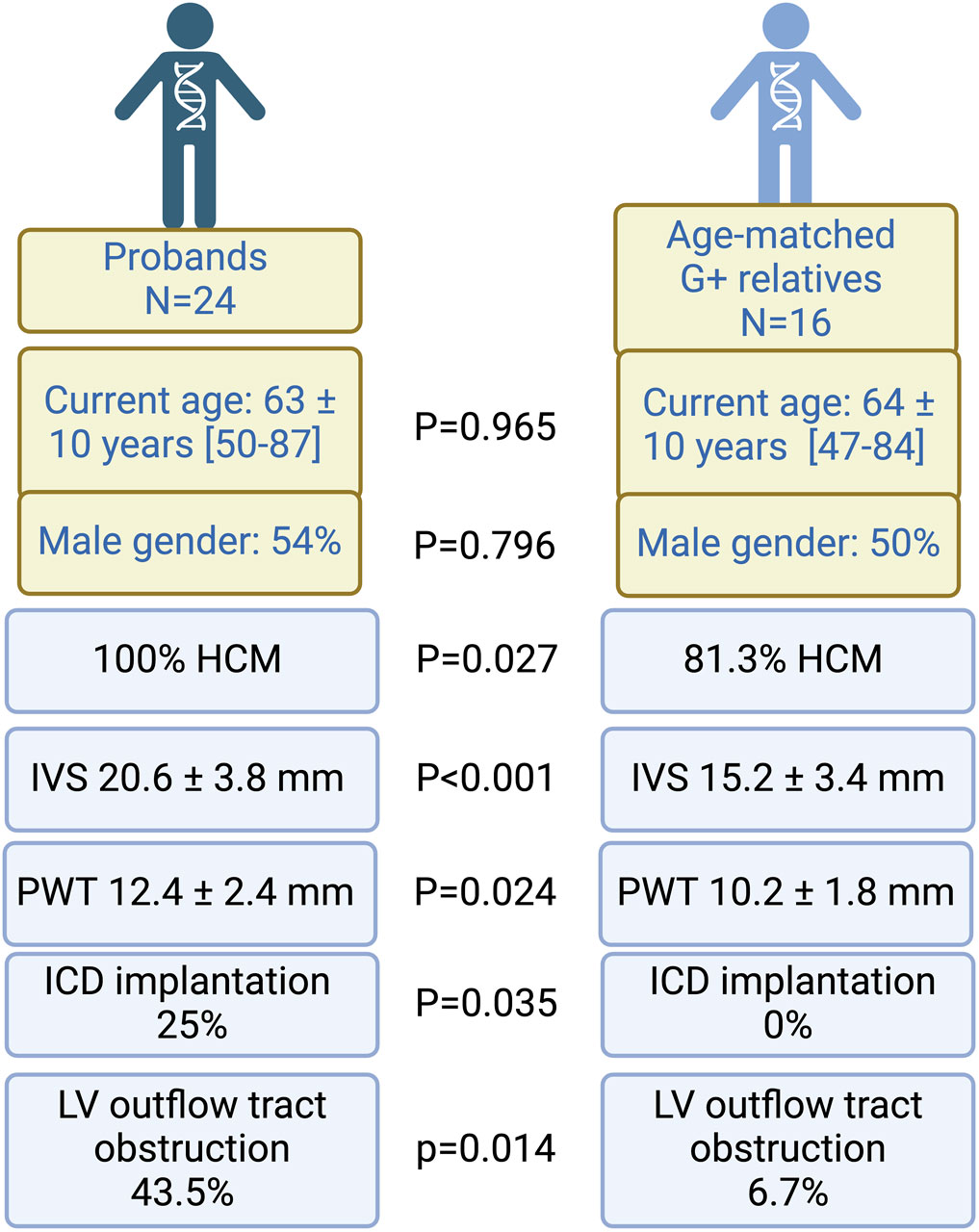
Figure 3. Comparison between probands and age-matched relatives. Probands have worse phenotypes, characterized by thicker left ventricles, a higher need for ICD implantation and more LV outflow tract obstruction. Proportions are displayed as percentages (%) of the comparison group (n = 24 for probands and N = 16 for G+ relatives). Echocardiography was performed in all individuals, however, a measurement of wall thickness was only available for 14 G+ relatives. G+, genotype positive; HCM, hypertrophic cardiomyopathy; ICD, implantable cardiac defibrillator; IVS, interventricular septum; LV, left ventricular; PWT, posterior wall thickness. Figure created with Biorender.
3.5.1 Gender differences
Male G+ relatives more often had a HCM phenotype (78.6% vs. 34.8%; p = 0.010) and were more severely affected than females. They had thicker ventricles evidenced by an increased IVS thickness (15.5 ± 3.5 mm vs. 11.2 ± 3.6 mm; p = 0.003), increased posterior wall thickness (PWT, 11.0 ± 0.9 mm vs. 8.4 ± 2.0 mm; p < 0.001), a higher frequency of left atrial dilatation (58.3% of males vs. 9.5% of females; p = 0.002) and mitral valve regurgitation (75% of males vs. 20% of females; p = 0.002). Males also presented more frequently with repolarization disturbances on their rest ECG (58.3% of males vs. 21.1% of females; p = 0.035). However, when we corrected Left ventricular wall thickness (IVS and PWT) and Left atrial posterior-anterior diameter (LAAP) for body surface area (BSA), indexed measurements were similar between males and females (IVS 9.3 ± 2.2 mm/m2 vs. 9.2 ± 3.8 mm/m2; p = 0.441; LAAP 2.3 ± 0.3 cm/m2 vs. 2.2 ± 0.3 cm/m2; p = 0.560) and PWT was smaller in men compared to women (PWT 5.6 ± 0.7 vs. 6.5 ± 2.3 mm/m2; p = 0.049). In probands similar findings were present, with smaller IVS and PWT in men compared to women, when corrected for BSA (IVS: 10.6 ± 1.9 vs. 11.5 ± 3.6; p = 0.468 and PWT: 5.5 ± 0.8 vs. 7.5 ± 1.8 p = 0.02).
3.5.2 Influence of environmental factors
G+ relatives with arterial hypertension (AHT) were more likely to have a HCM phenotype compared to G+ relatives without AHT (78.6% vs. 35%; p = 0.012). Smokers were not more likely to develop the HCM phenotype (57.1% of smokers vs. 51.9% of non-smoker had a HCM phenotype; p = 0.571). BMI tended to be higher in G+ relatives with a HCM phenotype (25.7 kg/m² ± 3.4 vs. 23.9 kg/m² ± 2.9; p = 0.201).
3.5.3 Additional phenotypic characteristics
Unexpectedly, as MYBPC3 encodes a cardiac specific isoform of MyBP-C, skeletal myopathy has been exceptionally described in association with MYBPC3 variants (Tajsharghi et al., 2010; Song et al., 2023). In the current cohort, one G+ individual also had evidence of skeletal myopathy. No association with a respiratory phenotype (assessed as the presence of asthma or COPD) was seen in the current cohort.
3.6 Outcomes of all MYBPC3 variant harbouring individuals
3.6.1 Overall
Overall, 20.3% of all G+ patients have reached the composite cardiac outcome after a median follow-up of 5.5 years with an average age of 50 (±21) years (Table 1). Of carriers ≥50 years of age (n = 38), 11 (28.9%) had reached the composite endpoint.
3.6.2 Effect of additional variant
A total of eleven (45.8%) probands carried an additional variant of uncertain significance (n = 10) or likely pathogenic variant (n = 1; MIB1 c.1A>C) (Table 1). An overview of the additional variants in the proband population is given in Supplementary Table S1.
There were no differences in family history for HCM, SCD or disease severity in probands with an additional variant compared to the probands without.
In one family with eight del23-26 carriers, an additional MYBPC3 VUS was identified in one individual (c.3763G>A; p.Ala1255Thr). In this family two patients had received a heart transplant and five patients experienced premature (sudden) cardiac death [at ages 24 (after HTx), 28, 50, 52 and 58 years]. Unfortunately, no DNA was available for three of the deceased patients, for two of the deceased patients (death at 50 and 52) presence of the founder variant could be confirmed based on the family tree. No individuals carrying only the VUS were identified. The patient who received a HTx and is still alive, is the family member who carries the additional MYBPC3 VUS. Eleven G+-family members carried the additional variant identified in their respective probands. They were more likely to be symptomatic at time of diagnosis (57.1% vs. 38.5%; p = 0.021) and to reach the composite endpoint (27.3% vs. 4.2%; p = 0.082) than family members without the additional variant identified in the proband. No significant difference in degree of left ventricular hypertrophy was observed (IVS 13.1 ± 4.2 vs. 12.8 ± 4.1 mm; p = 0.847).
Of note, a HCM phenotype was identified In 1 G- family member carrying two variants of uncertain significance (JUP c.1096 C>G (p.Pro366Ala) and CALR3 c.564delT (p.Gln189Serfs*8) (Identified with the full CM gene panel). Although JUP has so far only been linked to arrhythmogenic cardiomyopathy (Groeneweg et al., 2015) and the association of CALR3 to HCM is disputed (Verhagen et al., 2018), we cannot exclude that these variants play a role in the presence and severity of HCM in this family. The CALR3 variant was present in four additional family members, including the proband, the JUP variant was identified in two other family members, but was absent in the proband (Supplementary Material).
4 Discussion
The deletion of exon 23-26 of MYBPC3 is a pathogenic Belgian founder mutation. Within the cohort of 61 mutation carriers we collected, the mutation leads to a composite cardiac outcome in >29% of individuals ≥50 years and shows a penetrance of >75% in G+ relatives above age 50.
In this work we described the presence of a larger intragenic deletion of four exons in MYBPC3. CNVs are a rare cause of cardiomyopathy and are present in up to 1.9% of HCM patients (Mates et al., 2020). Although copy number variants (CNVs) can be missed by standard genetic testing using Sanger or some next-generation sequencing techniques (Mates et al., 2020), multi-exon deletions can be detected based on NGS data, by comparing the normalized sequencing depth of exons between cases (Fromer et al., 2012). The MYBPC3 exon 23-26 deletion in our population was identified based on NGS sequencing data using a read-depth strategy, showing it is feasible to pick these up. As data is already available and added time and cost is limited, we advise that such CNV analysis of the MYBPC3 gene is routinely implemented in genetic analysis of HCM patients. We were able to pinpoint the exact breakpoints of this deletion using long-range PCR. To our knowledge, this is the first MYBPC3 CNV described as a founder variant. We could identify a common haplotype of 1.19 Mb (including the 2 intragenic SNPs) in all 24 probands and a larger common haplotype of 13.8 Mb in 19 of the 24 assessed probands. This allowed us to estimate the founder event to have happened around the year 1830.
The variant was classified according the ACMG guidelines for CNVs (Riggs et al., 2020). Argument 2E “Both breakpoints are within the same gene” requires further assessment with additional standard ACMG criteria (PVS1or PM4) which were both present for the MYBPC3 variant (Richards et al., 2015). However, these criteria are general and do not take gene-specific characteristics into account. Several re-assessments of these standard criteria have been proposed for specific genes, including MYH7, the second most important HCM-related gene (Kelly et al., 2018). For MYBPC3 however, no gene-specific criteria have been proposed yet.
The deletion of exon 23-26 of MYBPC3 is predicted to result in an unstable transcript due to the deletion of 589 nucleotides, which is expected to undergo nonsense-medited mRNA decay. Therefore, the effect of the variant is expected to be similar to that of other loss-of-function (haploinsufficient) variants in MYBPC3. Whereas pathogenic loss-of-function (haploinsufficient) variants are evenly distributed throughout the whole MYBPC3 gene, pathogenic missense variants cluster in specific domains. Most pathogenic missense variants in MYBPC3 result in absence or altering of the myosin binding site, although disruption of the titin binding site and phosphorylation sites can also result in HCM (Winegrad, 1999; Helms et al., 2020). It is likely that, opposed to a dominant-negative effect of missense mutations, nonsense-mediated decay and subsequent haploinsufficiency cause a HCM phenotype in this patient population (Rottbauer et al., 1997; Andersen et al., 2004; Frank-Hansen et al., 2008).
4.1 Phenotype-genotype correlations of the MYBPC3 gene
A large phenotypic variability has been reported (Page et al., 2012; Helms et al., 2020) in MYPBC3-related HCM and this hinders the assignment of prognostic significance to specific disease-associated mutations or groups of mutations, complicating (genetic) counselling of family members.
Based on literature, the disease penetrance of MYBPC3 variants was 58%–83% at age 50 years (Charron et al., 1998; Niimura et al., 1998; Maron et al., 2001), which resembles our founder population’s disease penetrance of 78.6% at age 50 (in family members). Average age of clinical onset for MYBPC3-harbouring individuals is around 40 years (Charron et al., 1998; Niimura et al., 1998; Van Driest et al., 2004; Sedaghat-Hamedani et al., 2018; Lorenzini et al., 2020b). In our MYBPC3 founder population, age of clinical diagnosis was considerably later, with an average of 51 ± 17 years (probands and family members). Considering that the presence of HCM leads to a loss in quality of life, and HCM is often progressive, an earlier onset can be seen as a more severe phenotypic presentation when compared to late onset.
After the initial discovery of MYBPC3, several studies reported on a less severe phenotype in MYBPC3 variants compared to other known HCM-related genes, especially MYH7. The degree of hypertrophy in our probands (IVS 20.6 mm) was similar to the described MYBPC3 literature [20.6 (19-22)mm, current age not known (Sedaghat-Hamedani et al., 2018)]. However, when all G+ individuals were considered, IVS was lower (16.3 ± 5.5 mm). In a large meta-analysis of 31 studies with 6123 HCM patients, MYBPC3 variant carriers (20% of all HCM patients) had less cardiac conduction disease, less ventricular arrythmia and less HTx (present in 15%, 20% and 0.6% of all MYBPC3 variant carriers respectively) compared to MYH7 variant carriers (Sedaghat-Hamedani et al., 2018). We observed intraventricular conduction disturbances in 25.5% and ventricular arrhythmia in 25.5% of the G+ individuals in our cohort which was not significantly higher than the observation in the MYBPC3 group described above (p = 0.1 for conduction disease and p = 0.47 for ventricular arrythmia). None of the patients who were followed in our cohort died during follow-up (5.5 years [1-22]), but three (5.2%) did experience a life-threatening arrhythmia.
A large study stratified MYBPC3 families according to prognosis, from benign (no death or HTx before the age of 60 years in the family, including the proband) to intermediate (1 event in the family) and malignant (2 or more events in the family). Overall, 90% of families with a MYBPC3 variant were stratified as having a benign or intermediate prognosis (Richard et al., 2003). According to this classification three families with the founder variant can be classified as malignant (12.5%), one family as intermediate (4.2%) and the other 20 families as benign (83.3%). This inter-familial variability indicates that even a single specific variant can be identified with a range of disease severities.
4.2 Benign phenotype of founder variants?
Most variants identified in MYBPC3 are “private” variants, meaning that they have been identified in a single family. In a large cohort, up to 56% of variants could be considered private (Alfares et al., 2015). When a variant is identified in several families and a shared haplotype is characterized, it represents a founder variant. The proportion of founder variants in HCM patients differs from region to region with higher proportions in isolated populations. Most strikingly, in Iceland up to 58% of the complete HCM population has the same specific MYBPC3 founder variant (MYBPC3 c.927-2A>G), with an overall population prevalence of 0.36% (Adalsteinsdottir et al., 2014). Founder variants are suggested to be associated with a more benign phenotype, as they have withstood negative selection pressure (Jääskeläinen et al., 2004; Thompson and Day, 2017). In several populations, however, no difference in LV hypertrophy severity or prevalence of adverse events between founders and non-founders were observed (Christiaans et al., 2010a; Christiaans et al., 2010b; Ross et al., 2017; Sabater-Molina et al., 2017; van Velzen et al., 2017; Helms et al., 2020).
Our current cohort is characterized by a higher age of onset, but an overall high penetrance at the age of 50 years (78.6% of G+ family members, in comparison to an average penetrance of 50% at this age in genotype positive HCM (Lorenzini et al., 2020b). Despite this late onset, negative outcomes were frequent, suggesting a stronger contribution of this specific founder variant on the HCM phenotype penetrance.
Therefore, the presence of any (MYBPC3) genetic variant seems to be the most important predictor of phenotype and severity, rather than the nature of the exact MYBPC3 variant itself. These MYBPC3 founder variants could withstand negative selection pressure as overall HCM mortality is low and the mean age of HCM related mortality is around 68 years which is long after the age of reproduction (compared to 81 years for non-HCM related mortality, p = 0.02) (Adalsteinsdottir et al., 2014).
4.3 Difference in disease severity between probands and G+ relatives
In our study cohort, probands were significantly more affected than G+ relatives and some of these differences stayed significant after correction for age by comparison with an age-matched cohort. Probands had thicker left ventricles compared to age- and gender matched G+ family members with a HCM phenotype, had more outflow tract obstruction and worse outcomes. It should be noted however, that overall, probands were more extensively investigated than family members, with a higher proportion of probands undergoing cardiac MRI and longer follow-up periods.
A similar observation was present in other MYBPC3 variant carrying populations, regardless of this being a founder variant or not. Overall, G+ relatives with a clinical HCM phenotype had less hypertrophy, less adverse remodeling and had lower risk of complications and mortality compared to probands (van Velzen et al., 2016; van Velzen et al., 2017). Age at time of genetic diagnosis was lower in G+ family members, but age at time of clinical HCM diagnosis was similar between probands and their family members (van Velzen et al., 2016; van Velzen et al., 2017). Likewise in our cohort, G+ family members were younger at the time of clinical diagnosis, which likely is explained by the active screening of individuals through cascade screening, whereas probands only present when symptoms are obvious, or complications have occurred.
4.4 Genetic and non-genetic influencing factors
Male sex is a significant predictor for the development of an HCM phenotype in G+ carriers who were phenotype negative at initial investigation. This is true for genotype positive HCM in general, MYBPC3 carriers and MYBPC3 founder carriers, including the founder population presented in the current work (Christiaans et al., 2010a; Page et al., 2012; Terauchi et al., 2015; Sabater-Molina et al., 2017; van Velzen et al., 2017; Lorenzini et al., 2020b). Additionally, female patients with HCM are diagnosed later than men (Adalsteinsdottir et al., 2014; Sabater-Molina et al., 2017; van Velzen et al., 2017; Lakdawala et al., 2021). However, women experience more heart failure symptoms than men, have more LV outflow tract obstruction and more frequently have a need for medical treatment (Adalsteinsdottir et al., 2014; Terauchi et al., 2015; Lakdawala et al., 2021). Several studies, including a large meta-analysis, on a total of 9,427 HCM patients (39.5% women), confirmed that during follow-up, women have a higher risk for the development of heart failure and all-cause mortality even after correction for differences present at baseline (Terauchi et al., 2015; Geske et al., 2017; Lorenzini et al., 2020a; Lakdawala et al., 2021; Trongtorsak et al., 2021).
Part of the explanation of a smaller portion of women in (MYBPC3-related) HCM could be the routine use of non-indexed measurements of wall thickness for the diagnosis of HCM. As men are on average larger and have thicker ventricle walls, they are more likely to fall above the threshold for hypertrophy (Lang et al., 2015). This is supported by the observation that differences in wall thickness were reversed after indexation for body surface area (van Velzen et al., 2017; Lakdawala et al., 2021). Gender-specific cut-offs, or the routine implementation of BSA indexed cut-offs is therefore warranted. This is further supported by the higher uptake of testing with a genetic panel in female HCM patients compared to male HCM patients (36% vs. 30% p < 0.001 and 51% vs. 43% p < 0.001) (Alfares et al., 2015; Lakdawala et al., 2021). All clinicians should be aware of this gender bias, as the delayed diagnosis in women seems to be related to worse outcomes.
In literature, the presence of an additional (likely) pathogenic variant in MYBPC3 or another HCM-related gene seems to result in worse phenotypes. Homozygosity or compound heterozygosity is present in 0.6%–7.5% of HCM patients and is associated with earlier onset and more severe phenotype (Van Driest et al., 2004; Ingles et al., 2005; Girolami et al., 2010; Marziliano et al., 2012; Page et al., 2012; Teirlinck et al., 2012; Alfares et al., 2015; Lorenzini et al., 2020b). The presence of a VUS next to a (likely) pathogenic variant has been observed in 7.4%–9.8% of HCM patients, but the effect of an additional VUS on the phenotype and disease severity has not been well defined (Alfares et al., 2015; Lakdawala et al., 2021).
In our cohort, 45.8% of probands carried one (or more) additional variants in a gene associated with cardiomyopathy, most of these were VUS, but in one family an additional likely pathogenic/pathogenic MIB1 c.1A>C variant (4.2%) was present. We could not observe any clear differences between probands with or without an additional variant. It should be taken into account that VUS encompasses a broad range of variants, and some VUS might be leaning more to likely pathogenic (hot) whereas others are more likely to be benign after all.
As family members only underwent testing of the MYBPC3 deletion, the presence of additional variants within the 59 CM genes, functioning as genetic modifiers cannot be excluded. As the probands were the most severely affected individuals, we expect genetic modifiers that worsen the phenotype to be, preferentially, identified in these probands. We showed that the G+ family members carrying the additional variant of the proband presented with a more severe phenotype than G+ family members without the additional variant, suggesting that some of the additional variants could act as aggravating modifiers having a synergistic effect on the MYBPC3 founder variant. Identification in more individuals and experiments investigating the actual functional effect of these variants should be performed to prove this hypothesis. Concerning potential protective genetic modifiers that lead to a less severe phenotype, we would expect to detect them in the family members, but we did not perform the full panel screening in these individuals and as such have no data to investigate this. We did not collect genetic data outside of the 59 CM genes, so cannot investigate protective or aggravating modifiers in other parts of the genome. Genome-wide linkage analysis based on short tandem repeat markers or genome-wide associations studies (GWAS) using common SNPs can aid to identify genetic modifiers. However, large families for linkage analysis and large cohorts of patients for GWAS are needed. Additionally, a large number of SNPs or markers need to be assessed, rendering this time- and cost consuming (Ott et al., 2015).
Overall, large differences in penetrance and disease severity are present in patients with the same genotype. Differences in the genetic background can be part of the explanation. Besides compound heterozygosity, more frequent genetic variation based on single nucleotide polymorphisms may play a role. Indeed, it has been shown that a polygenic risk score for HCM could stratify the HCM risk in family members of G+ probands (Zheng et al., 2023). Furthermore, this score aided the prediction of adverse outcomes in HCM patients (Zheng et al., 2023). However, the use of a polygenic risk score requires additional sequencing in all individuals, requiring extra time- and cost investment, which might not be feasible for all patients at the moment. For the current cohort, the data on the SNPs included in the risk score were not available to further stratify HCM risk.
Additionally, environmental factors may play a role. In our cohort G+ relatives with AHT were more likely to present with a HCM phenotype, whereas smoking or BMI did not significantly influenced disease penetrance. Interestingly, a marked difference in disease severity has been observed in three pairs of monogenic twins with a MYBPC3 (c.787G>T; p.Gly263ter) founder variant (Rodríguez Junquera et al., 2022). As they are genetically identical, these differences cannot be explained by genetic background. The authors could not find any clear differences in known environmental risk factors for HCM such as AHT or physical activities (including pregnancy) (Rodríguez Junquera et al., 2022). Other, currently undiscovered, influencing factors (e.g., epigenetics) are therefore likely at play.
5 Limitations and future perspectives
A deletion of exon 23-26 of the MYBPC3 gene has been reported before in one patient based in the UK and another patient based in the Netherlands, both were HCM patients (Nannenberg et al., 2011; Walsh et al., 2017). Although the exact breaking points of the deletion in these patients has not been identified, they most likely represent the same variant. Therefore, the fact that the haplotype in these two patients was not assessed and these individuals were not included in the current cohort is a limitation of the study.
Even though 24 probands with the MYBPC3 del exon 23-26 variant have been identified, the overall sample size is limited. For follow-up of patients with limited disease, not all clinical tests (24 rhythm monitoring and cardiac MRI) were performed, further limiting our sample size and statistical power. Additionally, median follow-up is 5.5 years (7.0 [1-22] in probands, 3.5 [1-20] in G+ family members and 5.7 [1-20] in G-family members). Further follow-up of the current cohort is needed to assess long-term health outcomes in variant carriers and disease penetrance, especially in young G+ family members in whom a phenotype is currently absent.
6 Conclusion
We describe a Belgian founder variant, the deletion of exon 23-26 in MYBPC3 associated with HCM. We identified 24 probands and subsequent cascade screening of 59 relatives identified 37 additional carriers. A common intragenic haplotype was confirmed in all 24 probands and a haplotype of 13.8 Mb in 19 probands could date the founder event to around 1830. This founder variant seems to be associated with a later onset (average age of clinical diagnosis around 50 years), nonetheless, above the age of 50 years a penetrance of 78.6% was observed. Despite this late onset, conduction disorders, outflow tract obstruction and ventricular arrhythmias were frequent, and the variant is associated with a high prevalence (20.3% overall) of adverse outcomes (HTx, life threatening arrhythmia, heart failure hospitalization or the need for septal ablation).
Data availability statement
The data presented in the study are deposited in the Clinvar repository https://www.ncbi.nlm.nih.gov/clinvar/, accession number VCV000525134.1.
Ethics statement
The studies involving humans were approved by the Ethical Committee of the University Hospital Antwerp. The studies were conducted in accordance with the local legislation and institutional requirements. The ethics committee/institutional review board waived the requirement of written informed consent for participation from the participants or the participants’ legal guardians/next of kin because of retrospective analysis of existing clinical dataset.
Author contributions
HB: Data curation, Formal Analysis, Investigation, Methodology, Project administration, Visualization, Writing–original draft. MA: Conceptualization, Data curation, Formal Analysis, Methodology, Supervision, Writing–review and editing. LVL: Conceptualization, Data curation, Formal Analysis, Methodology, Writing–review and editing. JS: Data curation, Supervision, Writing–review and editing. IG: Data curation, Investigation, Writing–review and editing. JB: Formal Analysis, Investigation, Writing–review and editing. PK: Data curation, Writing–review and editing. PV: Data curation, Writing–review and editing. EDV: Data curation, Writing–review and editing. MR: Data curation, Supervision, Writing–review and editing. HH: Supervision, Writing–review and editing. EVC: Data curation, Investigation, Methodology, Supervision, Visualization, Writing–review and editing. BL: Data curation, Funding acquisition, Methodology, Supervision, Writing–review and editing.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This work was supported by the Flanders Research Foundation- FWO (research grant G055821N, doctoral research grant 1192420N to HB and senior clinical investigator grant 1804320N to EC). BL holds a consolidator grant from the European Research Council (Genomia–ERC-COG-2017-771945).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2024.1392527/full#supplementary-material
Abbreviations
AHT, arterial hypertension; CNV, copy number variant; G+/G-, genotype positive/negative; HCM, hypertrophic cardiomyopathy; HTx, heart transplantation; ICD, implantable cardiac defibrillator; IVS, interventricular septum; LAAP, left atrial anterior-posterior diameter; LV, left ventricle/Left ventricular; MYBPC3, Coding gene for myosin binding protein C; MYH7, Coding gene for myosin heavy chain 7 protein; PWT, posterior wall thickness; SCD, sudden cardiac death; Y, year(s).
References
Adalsteinsdottir, B., Teekakirikul, P., Maron, B. J., Burke, M. A., Gudbjartsson, D. F., Holm, H., et al. (2014). Nationwide study on hypertrophic cardiomyopathy in Iceland: evidence of a MYBPC3 founder mutation. Circulation 130, 1158–1167. doi:10.1161/CIRCULATIONAHA.114.011207
Alfares, A. A., Kelly, M. A., Mcdermott, G., Funke, B. H., Lebo, M. S., Baxter, S. B., et al. (2015). Results of clinical genetic testing of 2,912 probands with hypertrophic cardiomyopathy: expanded panels offer limited additional sensitivity. Genet. Med. 17, 880–888. doi:10.1038/gim.2014.205
Andersen, P. S., Havndrup, O., Bundgaard, H., Larsen, L. A., Vuust, J., Pedersen, A. K., et al. (2004). Genetic and phenotypic characterization of mutations in myosin-binding protein C (MYBPC3) in 81 families with familial hypertrophic cardiomyopathy: total or partial haploinsufficiency. Eur. J. Hum. Genet. 12, 673–677. doi:10.1038/sj.ejhg.5201190
Bergman, A., Einbeigi, Z., Olofsson, U., Taib, Z., Wallgren, A., Karlsson, P., et al. (2001). The western Swedish BRCA1 founder mutation 3171ins5; a 3.7 cM conserved haplotype of today is a reminiscence of a 1500-year-old mutation. Eur. J. Hum. Genet. 9, 787–793. doi:10.1038/sj.ejhg.5200704
Carrier, L., Bonne, G., Bahrend, E., Yu, B., Richard, P., Niel, F., et al. (1997). Organization and sequence of human cardiac myosin binding protein C gene (MYBPC3) and identification of mutations predicted to produce truncated proteins in familial hypertrophic cardiomyopathy. Circulation Res. 80, 427–434. doi:10.1161/01.res.0000435859.24609.b3
Charron, P., Dubourg, O., Desnos, M., Bennaceur, M., Carrier, L., Camproux, A.-C., et al. (1998). Clinical features and prognostic implications of familial hypertrophic cardiomyopathy related to the cardiac myosin-binding protein C gene. Circulation 97, 2230–2236. doi:10.1161/01.cir.97.22.2230
Christiaans, I., Birnie, E., Van Langen, I. M., Van Spaendonck-Zwarts, K. Y., Van Tintelen, J. P., Van Den Berg, M. P., et al. (2010a). The yield of risk stratification for sudden cardiac death in hypertrophic cardiomyopathy myosin-binding protein C gene mutation carriers: focus on predictive screening. Eur. Heart J. 31, 842–848. doi:10.1093/eurheartj/ehp539
Christiaans, I., Nannenberg, E. A., Dooijes, D., Jongbloed, R. J., Michels, M., Postema, P. G., et al. (2010b). Founder mutations in hypertrophic cardiomyopathy patients in The Netherlands. Neth Heart J. 18, 248–254. doi:10.1007/BF03091771
Claes, G. R., Van Tienen, F. H., Lindsey, P., Krapels, I. P., Helderman-Van Den Enden, A. T., Hoos, M. B., et al. (2016). Hypertrophic remodelling in cardiac regulatory myosin light chain (MYL2) founder mutation carriers. Eur. Heart J. 37, 1815–1822. doi:10.1093/eurheartj/ehv522
Daw, E. W., Chen, S. N., Czernuszewicz, G., Lombardi, R., Lu, Y., Ma, J., et al. (2007). Genome-wide mapping of modifier chromosomal loci for human hypertrophic cardiomyopathy. Hum. Mol. Genet. 16, 2463–2471. doi:10.1093/hmg/ddm202
Frank-Hansen, R., Page, S. P., Syrris, P., Mckenna, W. J., Christiansen, M., and Andersen, P. S. (2008). Micro-exons of the cardiac myosin binding protein C gene: flanking introns contain a disproportionately large number of hypertrophic cardiomyopathy mutations. Eur. J. Hum. Genet. 16, 1062–1069. doi:10.1038/ejhg.2008.52
Fromer, M., Moran, J. L., Chambert, K., Banks, E., Bergen, S. E., Ruderfer, D. M., et al. (2012). Discovery and statistical genotyping of copy-number variation from whole-exome sequencing depth. Am. J. Hum. Genet. 91, 597–607. doi:10.1016/j.ajhg.2012.08.005
Geske, J. B., Ong, K. C., Siontis, K. C., Hebl, V. B., Ackerman, M. J., Hodge, D. O., et al. (2017). Women with hypertrophic cardiomyopathy have worse survival. Eur. Heart J. 38, 3434–3440. doi:10.1093/eurheartj/ehx527
Girolami, F., Ho, C. Y., Semsarian, C., Baldi, M., Will, M. L., Baldini, K., et al. (2010). Clinical features and outcome of hypertrophic cardiomyopathy associated with triple sarcomere protein gene mutations. J. Am. Coll. Cardiol. 55, 1444–1453. doi:10.1016/j.jacc.2009.11.062
Groeneweg, J. A., Bhonsale, A., James, C. A., Riele, A. S. T., Dooijes, D., Tichnell, C., et al. (2015). Clinical presentation, long-term follow-up, and outcomes of 1001 arrhythmogenic right ventricular dysplasia/cardiomyopathy patients and family members. Circ. Cardiovasc. Genet. 8, 437–446. doi:10.1161/CIRCGENETICS.114.001003
Helms, A. S., Thompson, A. D., Glazier, A. A., Hafeez, N., Kabani, S., Rodriguez, J., et al. (2020). Spatial and functional distribution of MYBPC3 pathogenic variants and clinical outcomes in patients with hypertrophic cardiomyopathy. Circ. Genom Precis. Med. 13, 396–405. doi:10.1161/CIRCGEN.120.002929
Ho, C. Y., Day, S. M., Ashley, E. A., Michels, M., Pereira, A. C., Jacoby, D., et al. (2018). Genotype and lifetime burden of disease in hypertrophic cardiomyopathy: insights from the sarcomeric human cardiomyopathy registry (SHaRe). Circulation 138, 1387–1398. doi:10.1161/CIRCULATIONAHA.117.033200
Ingles, J., Doolan, A., Chiu, C., Seidman, J., Seidman, C., and Semsarian, C. (2005). Compound and double mutations in patients with hypertrophic cardiomyopathy: implications for genetic testing and counselling. J. Med. Genet. 42, e59. doi:10.1136/jmg.2005.033886
Ingles, J., Goldstein, J., Thaxton, C., Caleshu, C., Corty, E. W., Crowley, S. B., et al. (2019). Evaluating the clinical validity of hypertrophic cardiomyopathy genes. Circ. Genom Precis. Med. 12, e002460. doi:10.1161/CIRCGEN.119.002460
Jääskeläinen, P., Miettinen, R., Kärkkäinen, P., Toivonen, L., Laakso, M., and Kuusisto, J. (2004). Genetics of hypertrophic cardiomyopathy in eastern Finland: few founder mutations with benign or intermediary phenotypes. Ann. Med. 36, 23–32. doi:10.1080/07853890310017161
Kelly, M. A., Caleshu, C., Morales, A., Buchan, J., Wolf, Z., Harrison, S. M., et al. (2018). Adaptation and validation of the ACMG/AMP variant classification framework for MYH7-associated inherited cardiomyopathies: recommendations by ClinGen's Inherited Cardiomyopathy Expert Panel. Genet. Med. 20, 351–359. doi:10.1038/gim.2017.218
Lakdawala, N. K., Olivotto, I., Day, S. M., Han, L., Ashley, E. A., Michels, M., et al. (2021). Associations between female sex, sarcomere variants, and clinical outcomes in hypertrophic cardiomyopathy. Circulation Genomic Precis. Med. 14, e003062. doi:10.1161/CIRCGEN.120.003062
Lang, R. M., Badano, L. P., Mor-Avi, V., Afilalo, J., Armstrong, A., Ernande, L., et al. (2015). Recommendations for cardiac chamber quantification by echocardiography in adults: an update from the American society of echocardiography and the European association of cardiovascular imaging. Eur. Heart J. - Cardiovasc. Imaging 16, 233–270. doi:10.1093/ehjci/jev014
Lorenzini, M., Anastasiou, Z., O'Mahony, C., Guttman, O. P., Gimeno, J. R., Monserrat, L., et al. (2020a). Mortality among referral patients with hypertrophic cardiomyopathy vs the general European population. JAMA Cardiol. 5, 73–80. doi:10.1001/jamacardio.2019.4534
Lorenzini, M., Norrish, G., Field, E., Ochoa, J. P., Cicerchia, M., Akhtar, M. M., et al. (2020b). Penetrance of hypertrophic cardiomyopathy in sarcomere protein mutation carriers. J. Am. Coll. Cardiol. 76, 550–559. doi:10.1016/j.jacc.2020.06.011
Maron, B. J., Niimura, H., Casey, S. A., Soper, M. K., Wright, G. B., Seidman, J. G., et al. (2001). Development of left ventricular hypertrophy in adults in hypertrophic cardiomyopathy caused by cardiac myosin-binding protein C gene mutations. J. Am. Coll. Cardiol. 38, 315–321. doi:10.1016/s0735-1097(01)01386-9
Martin, F. J., Amode, M. R., Aneja, A., Austine-Orimoloye, O., Azov, ANDREY G., Barnes, I., et al. (2022). Ensembl 2023. Nucleic Acids Res. 51, D933–D941. doi:10.1093/nar/gkac958
Marziliano, N., Merlini, P. A., Vignati, G., Orsini, F., Motta, V., Bandiera, L., et al. (2012). A case of compound mutations in the MYBPC3 gene associated with biventricular hypertrophy and neonatal death. Neonatology 102, 254–258. doi:10.1159/000339847
Mates, J., Mademont-Soler, I., Fernandez-Falgueras, A., Sarquella-Brugada, G., Cesar, S., Arbelo, E., et al. (2020). Sudden cardiac death and copy number variants: what do we know after 10 Years of genetic analysis? Forensic Sci. Int. Genet. 47, 102281. doi:10.1016/j.fsigen.2020.102281
Musunuru, K., Hershberger, R. E., Day, S. M., Klinedinst, N. J., Landstrom, A. P., Parikh, V. N., et al. (2020). Genetic testing for inherited cardiovascular diseases: a scientific statement from the American heart association. Circulation Genomic Precis. Med. 13, e000067. doi:10.1161/HCG.0000000000000067
Nannenberg, E. A., Michels, M., Christiaans, I., Majoor-Krakauer, D., Hoedemaekers, Y. M., Van Tintelen, J. P., et al. (2011). Mortality risk of untreated myosin-binding protein C-related hypertrophic cardiomyopathy: insight into the natural history. J. Am. Coll. Cardiol. 58, 2406–2414. doi:10.1016/j.jacc.2011.07.044
Niimura, H., Bachinski, L. L., Sangwatanaroj, S., Watkins, H., Chudley, A. E., Mckenna, W., et al. (1998). Mutations in the gene for cardiac myosin-binding protein C and late-onset familial hypertrophic cardiomyopathy. N. Engl. J. Med. 338, 1248–1257. doi:10.1056/NEJM199804303381802
Ott, J., Wang, J., and Leal, S. M. (2015). Genetic linkage analysis in the age of whole-genome sequencing. Nat. Rev. Genet. 16, 275–284. doi:10.1038/nrg3908
Page, S. P., Kounas, S., Syrris, P., Christiansen, M., Frank-Hansen, R., Andersen, P. S., et al. (2012). Cardiac myosin binding protein-C mutations in families with hypertrophic cardiomyopathy: disease expression in relation to age, gender, and long term outcome. Circ. Cardiovasc Genet. 5, 156–166. doi:10.1161/CIRCGENETICS.111.960831
Proost, D., Vandeweyer, G., Meester, J. A., Salemink, S., Kempers, M., Ingram, C., et al. (2015). Performant mutation identification using targeted next-generation sequencing of 14 thoracic aortic aneurysm genes. Hum. Mutat. 36, 808–814. doi:10.1002/humu.22802
Richard, P., Charron, P., Carrier, L., Ledeuil, C., Cheav, T., Pichereau, C., et al. (2003). Hypertrophic cardiomyopathy: distribution of disease genes, spectrum of mutations, and implications for a molecular diagnosis strategy. Circulation 107, 2227–2232. doi:10.1161/01.CIR.0000066323.15244.54
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genet. Med. 17, 405–424. doi:10.1038/gim.2015.30
Riggs, E. R., Andersen, E. F., Cherry, A. M., Kantarci, S., Kearney, H., Patel, A., et al. (2020). Technical standards for the interpretation and reporting of constitutional copy-number variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics (ACMG) and the Clinical Genome Resource (ClinGen). Genet. Med. 22, 245–257. doi:10.1038/s41436-019-0686-8
Rodríguez Junquera, M., Salgado, M., González-Urbistondo, F., Alén, A., Rodríguez-Reguero, J. J., Silva, I., et al. (2022). Different phenotypes in monozygotic twins, carriers of the same pathogenic variant for hypertrophic cardiomyopathy. Life (Basel) 12, 1346. doi:10.3390/life12091346
Ross, S. B., Bagnall, R. D., Ingles, J., Tintelen, J. P. V., and Semsarian, C. (2017). Burden of recurrent and ancestral mutations in families with hypertrophic cardiomyopathy. Circ. Cardiovasc. Genet. 10, e001671. doi:10.1161/CIRCGENETICS.116.001671
Rottbauer, W., Gautel, M., Zehelein, J., Labeit, S., Franz, W. M., Fischer, C., et al. (1997). Novel splice donor site mutation in the cardiac myosin-binding protein-C gene in familial hypertrophic cardiomyopathy. Characterization of cardiac transcript and protein. J. Clin. Invest. 100, 475–482. doi:10.1172/JCI119555
Sabater-Molina, M., Saura, D., García-Molina Sáez, E., González-Carrillo, J., Polo, L., Pérez-Sánchez, I., et al. (2017). A novel founder mutation in MYBPC3: phenotypic comparison with the most prevalent MYBPC3 mutation in Spain. Rev. Esp. Cardiol. Engl. Ed. 70, 105–114. doi:10.1016/j.rec.2016.06.020
Sedaghat-Hamedani, F., Kayvanpour, E., Tugrul, O. F., Lai, A., Amr, A., Haas, J., et al. (2018). Clinical outcomes associated with sarcomere mutations in hypertrophic cardiomyopathy: a meta-analysis on 7675 individuals. Clin. Res. Cardiol. 107, 30–41. doi:10.1007/s00392-017-1155-5
Song, T., Landim-Vieira, M., Ozdemir, M., Gott, C., Kanisicak, O., Pinto, J. R., et al. (2023). Etiology of genetic muscle disorders induced by mutations in fast and slow skeletal MyBP-C paralogs. Exp. Mol. Med. 55, 502–509. doi:10.1038/s12276-023-00953-x
Tajsharghi, H., Leren, T. P., Abdul-Hussein, S., Tulinius, M., Brunvand, L., Dahl, H. M., et al. (2010). Unexpected myopathy associated with a mutation in MYBPC3 and misplacement of the cardiac myosin binding protein C. J. Med. Genet. 47, 575–577. doi:10.1136/jmg.2009.072710
Teirlinck, C. H., Senni, F., Malti, R. E., Majoor-Krakauer, D., Fellmann, F., Millat, G., et al. (2012). A human MYBPC3 mutation appearing about 10 centuries ago results in a hypertrophic cardiomyopathy with delayed onset, moderate evolution but with a risk of sudden death. BMC Med. Genet. 13, 105. doi:10.1186/1471-2350-13-105
Terauchi, Y., Kubo, T., Baba, Y., Hirota, T., Tanioka, K., Yamasaki, N., et al. (2015). Gender differences in the clinical features of hypertrophic cardiomyopathy caused by cardiac myosin-binding protein C gene mutations. J. Cardiol. 65, 423–428. doi:10.1016/j.jjcc.2014.07.010
Thompson, A. D., and Day, S. M. (2017). Founder mutations in myosin-binding protein C: maybe not so benign after all. Circ. Cardiovasc. Genet. 10, e001872. doi:10.1161/CIRCGENETICS.117.001872
Trongtorsak, A., Polpichai, N., Thangjui, S., Kewcharoen, J., Yodsuwan, R., Devkota, A., et al. (2021). Gender-related differences in hypertrophic cardiomyopathy: a systematic review and meta-analysis. Pulse (Basel) 9, 38–46. doi:10.1159/000517618
Van Driest, S. L., Ommen, S. R., Tajik, A. J., Gersh, B. J., and Ackerman, M. J. (2005). Yield of genetic testing in hypertrophic cardiomyopathy. Mayo Clin. Proc. 80, 739–744. doi:10.1016/S0025-6196(11)61527-9
Van Driest, S. L., Vasile, V. C., Ommen, S. R., Will, M. L., Tajik, A. J., Gersh, B. J., et al. (2004). Myosin binding protein C mutations and compound heterozygosity in hypertrophic cardiomyopathy. J. Am. Coll. Cardiol. 44, 1903–1910. doi:10.1016/j.jacc.2004.07.045
Van Velzen, H. G., Schinkel, A. F. L., Oldenburg, R. A., Van Slegtenhorst, M. A., Frohn-Mulder, I. M. E., Van Der Velden, J., et al. (2017). Clinical characteristics and long-term outcome of hypertrophic cardiomyopathy in individuals with a MYBPC3 (Myosin-Binding protein C) founder mutation. Circ. Cardiovasc Genet. 10, e001660. doi:10.1161/CIRCGENETICS.116.001660
Van Velzen, H. G., Vriesendorp, P. A., Oldenburg, R. A., Van Slegtenhorst, M. A., Van Der Velden, J., Schinkel, A. F. L., et al. (2016). Value of genetic testing for the prediction of long-term outcome in patients with hypertrophic cardiomyopathy. Am. J. Cardiol. 118, 881–887. doi:10.1016/j.amjcard.2016.06.038
Verhagen, J. M. A., Veldman, J. H., Van Der Zwaag, P. A., Von Der Thüsen, J. H., Brosens, E., Christiaans, I., et al. (2018). Lack of evidence for a causal role of CALR3 in monogenic cardiomyopathy. Eur. J. Hum. Genet. 26, 1603–1610. doi:10.1038/s41431-018-0208-1
Walsh, R., Thomson, K. L., Ware, J. S., Funke, B. H., Woodley, J., Mcguire, K. J., et al. (2017). Reassessment of Mendelian gene pathogenicity using 7,855 cardiomyopathy cases and 60,706 reference samples. Genet. Med. 19, 192–203. doi:10.1038/gim.2016.90
Wilcox, J. E., and Hershberger, R. E. (2018). Genetic cardiomyopathies. Curr. Opin. Cardiol. 33, 354–362. doi:10.1097/HCO.0000000000000512
Wilde, A. A. M., Semsarian, C., Márquez, M. F., Shamloo, A. S., Ackerman, M. J., Ashley, E. A., et al. (2022). European heart rhythm association (EHRA)/Heart rhythm society (HRS)/Asia pacific heart rhythm society (APHRS)/Latin American heart rhythm society (LAHRS) expert consensus statement on the state of genetic testing for cardiac diseases. Europace 24, e1–e60. doi:10.1016/j.hrthm.2022.03.1225
Winegrad, S. (1999). Cardiac myosin binding protein C. Circ. Res. 84, 1117–1126. doi:10.1161/01.res.84.10.1117
Keywords: MYBPC3, founder variant, cardiogenetic, phenotyping, hypertrophic cardiomyopathy
Citation: Boen HM, Alaerts M, Van Laer L, Saenen JB, Goovaerts I, Bastianen J, Koopman P, Vanduynhoven P, De Vuyst E, Rosseel M, Heidbuchel H, Van Craenenbroeck EM and Loeys B (2024) Phenotypic spectrum of the first Belgian MYBPC3 founder: a large multi-exon deletion with a varying phenotype. Front. Genet. 15:1392527. doi: 10.3389/fgene.2024.1392527
Received: 27 February 2024; Accepted: 22 April 2024;
Published: 21 May 2024.
Edited by:
Zahurul A. Bhuiyan, Centre Hospitalier Universitaire Vaudois (CHUV), SwitzerlandReviewed by:
Sitara Roy, Harvard Medical School, United StatesPauline Arnaud, Assistance Publique Hopitaux De Paris, France
Copyright © 2024 Boen, Alaerts, Van Laer, Saenen, Goovaerts, Bastianen, Koopman, Vanduynhoven, De Vuyst, Rosseel, Heidbuchel, Van Craenenbroeck and Loeys. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hanne M. Boen, aGFubmUuYm9lbkB1YW50d2VycGVuLmJl