 Chen Jiali
Chen Jiali Peng Huifang
Peng Huifang Jiang Yuqing
Jiang Yuqing Zeng Xiantao
Zeng Xiantao Jiang Hongwei
Jiang Hongwei- 1Henan Key Laboratory of Rare Diseases, The First Affiliated Hospital, and College of Clinical Medicine of Henan University of Science and Technology, Luoyang, China
- 2Center for Evidence-Based and Translational Medicine, Zhongnan Hospital of Wuhan University, Wuhan, China
Differences/disorders of sex development (DSDs) in individuals with a 46, XY karyotype are a group of congenital disorders that manifest as male gonadal hypoplasia or abnormalities of the external genitalia. Approximately 50% of patients with 46, XY DSDs cannot obtain a molecular diagnosis. The aims of this paper were to review the most common causative genes and rare genes in patients with 46, XY DSDs, analyze global molecular diagnostic cohorts for the prevalence and geographic distribution of causative genes, and identify the factors affecting cohort detection results. Although the spectrum of genetic variants varies across regions and the severity of the clinical phenotype varies across patients, next-generation sequencing (NGS), the most commonly used detection method, can still reveal genetic variants and aid in diagnosis. A comparison of the detection rates of various sequencing modalities revealed that whole-exome sequencing (WES) facilitates a greater rate of molecular diagnosis of the disease than panel sequencing. Whole-genome sequencing (WGS), third-generation sequencing, and algorithm advancements will contribute to the improvement of detection efficiency. The most commonly mutated genes associated with androgen synthesis and action are AR, SR5A2, and HSD17B3, and the most commonly mutated genes involved in gonadal formation are NR5A1 and MAP3K1. Detection results are affected by differences in enrollment criteria and sequencing technologies.
Introduction
Sex differentiation in humans is a delicate process regulated by multiple genes and is divided into three stages: the bipotential gonad stage, sex determination, and gonadal differentiation. In the bipotential gonad stage, which lasts from fertilization to the sixth week of embryo formation, there are no significant morphological differences between female (XX) and male (XY) embryos. In the sex determination stage, the successful expression of the SRY gene on the Y chromosome in XY embryos causes a complex cascade of expression of dozens of genes (SOX9, SF1, CBX2, WT1, FOG2, GATA4, and other genes) in the testicular pathway (Sekido, 2010). In contrast to testicular determination, the absence of the SRY gene in an XX embryo coincides with the expression of pro-ovarian genes (RSPO1 and FOXL2) that dictate ovarian fate. During the gonadal differentiation stage, in XY embryos, Sertoli cells secrete anti-Müllerian hormone (AMH) to induce Müllerian duct regression, and Leydig cells secrete androgens to induce the Wolffian duct to differentiate into the paratestis, vas deferens, and seminal vesicles. Due to the absence of AMH and androgen, the Müllerian duct of XX embryos differentiates into the oviduct, uterus, and upper vagina. In all of these processes, molecular-level changes can lead to differences/disorders of sex development (DSDs), which result in discordance between an individual’s sex chromosomes, gonads, and/or anatomic sex (Hughes et al., 2006). DSDs are classified into three subtypes (sex chromosome DSDs, 46, XY DSDs, and 46, XX DSDs) according to the sex chromosomes; these subtypes are widely recognized internationally (Hughes et al., 2006). The clinical presentations of DSDs are complex and varied. Sex chromosome DSDs account for 15% of all DSDs, including Klinefelter syndrome (47, XXY), Turner syndrome variants with Y chromosome contributions (typically mosaic 45, X/46, XY, with or without isodicentric Y), segmental deletions of the X chromosome, or translocations of an SRY-containing Y fragment to the X chromosome (Délot and Vilain, 2021a).
Among 46, XX individuals with genital anomalies, 90%–95% of cases are associated with congenital adrenal hyperplasia (CAH) caused by CYP21A2 deficiency, and patients may present with varying degrees of external genital masculinization in infancy and early childhood and primary amenorrhea or infertility in adolescence or adulthood (Speiser et al., 2018). Some patients with DSDs possess a 46, XY chromosome complement and can present with variable degrees of virilization of their external genitalia, from a small penis or mild hypospadias to full female external genitalia. The 46, XY DSD subtype can be further subdivided into 46, XY gonadal dysgenesis (GD), disorders of androgen synthesis or action, and disorders of AMH synthesis or action. Gonadal development disorders include complete gonadal hypoplasia (previously known as Swyer syndrome), partial gonadal hypoplasia, gonadal degeneration, and ovotesticular DSD (King and Conway, 2014). The external masculinization score (EMS) is an objective and standardized tool for describing the external genitalia of male infants (Ahmed et al., 2000). The EMS is calculated from the following features: micropenis (0/3), fused scrotum (3/0), position of the urethral opening (3/2/1/0), and position of the right and left gonads (1.5/1.0/0.5/0). Regardless of its phenotypic variability, the etiology of 46, XY DSDs is ultimately impaired steroid synthesis or action or a defective/affected gonadal differentiation process.
Currently, there are few investigations of DSDs worldwide. To generalize the prevalence and geographic variability of 46, XY DSD pathogenic genes, we collected data from worldwide cohorts to identify patterns.
Diagnosis of DSDs
The incidence of newborns with ambiguous genitalia ranges from 1/5,000 to 1/4,500 (Lee et al., 2006; Thyen et al., 2006). Because patients with DSDs always have a complex etiology and clinically distinct phenotypes of abnormal internal or external genital development, overlapping phenotypes and genetic heterogeneity make diagnosis challenging (Délot and Vilain, 2021b). Currently, the diagnosis of DSDs relies on analyses complementary to physical examination, such as sex hormone testing, imaging, and molecular testing (Délot and Vilain, 2021b). Hormone tests for diagnosis and hormone replacement therapy require blood or urine samples, which can be used to analyze disorders of a part of the gonadal axis. Moreover, ordinary biochemical tests are unable to identify the cause of androgen abnormalities. Prenatal ultrasound (US) can detect signs of DSDs but is often limited by technology and gestational age (Wisniewski et al., 2019). Neonates often present with atypical external genitalia, children with bilateral inguinal hernias, adolescents with abnormal development of secondary sexual characteristics during puberty, and adults with infertility. Ultrasonography can reveal undescended unilateral or bilateral testes and even uterus- and ovary-like tissue. First, sex chromosome karyotyping is performed to determine whether there are abnormalities in the number or structure of sex chromosomes or a difference between genetics and gender identity. The next step is obtaining a molecular diagnosis (Délot and Vilain, 2021b).
Currently, the most utilized technology for diagnosing DSDs is next-generation sequencing (NGS), but many patients do not receive a molecular diagnosis (Baxter et al., 2015b). It is more difficult to obtain a molecular diagnosis for 46, XY DSD patients. Over the past decade, many cohorts have been studied worldwide to determine the cause of DSDs using NGS, but the diagnosis rate for most studies of 46, XY DSD patients does not exceed 50% (Arboleda et al., 2013; Baxter et al., 2015a; Eggers et al., 2016; Kim et al., 2017; Özen et al., 2017; Hughes et al., 2019; Globa et al., 2022; Zidoune et al., 2022). We analyzed common pathogenic mutations and novel genes, as well as possible causative genes, from various cohort studies of DSDs worldwide and observed whether there are geographical and ethnic differences in mutated genes or loci.
Worldwide cohorts
The cohorts mentioned in this paper were identified by searching PubMed for relevant articles using the keywords “46, XY DSD, next-generation sequencing (NGS), whole-exome sequencing (WES), and genetic diagnosis.” We excluded cohort studies targeting only a certain phenotype or a particular type of disease, such as a cohort that included patients with only hypospadias or androgen insensitivity syndrome (AIS). We categorized the countries where the study was conducted into Asia, Africa, North America, South America, Europe, and Oceania. The aim was to understand the main pathogenic genes of 46, XY DSDs worldwide and observe whether there are geographical and ethnic specificities. Information on the mutated genes detected in each study cohort, the mode of diagnosis, and the diagnosis rate are shown in Table 1.
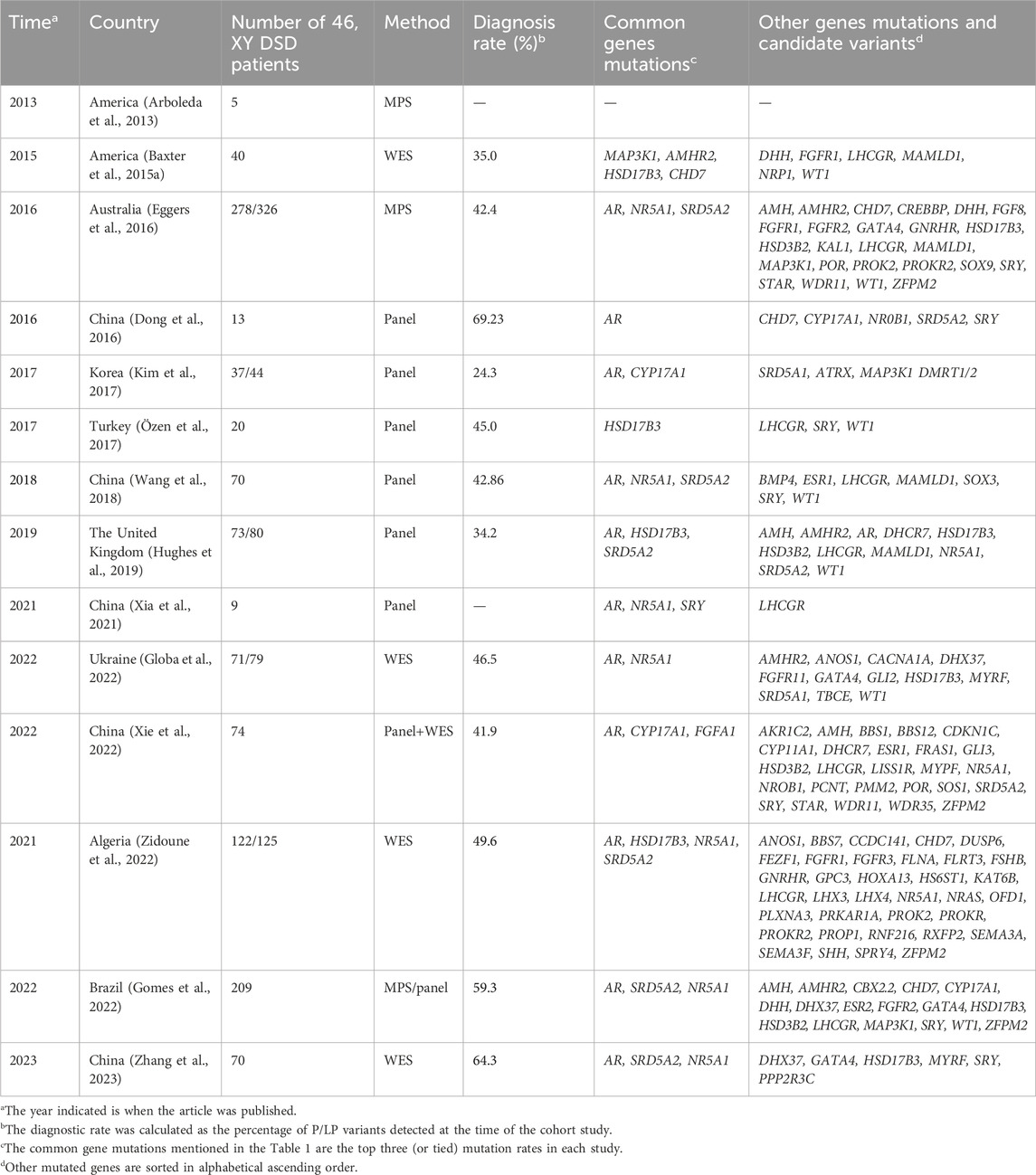
Table 1. Worldwide cohorts study for molecular diagnosis of DSD.
Asia
China
Studies on the molecular detection of 46, XY DSD patients in China included a total of six cohorts. A study published in 2016 by Southwest Hospital, Third Military Medical University, Chongqing, China, included 21 Han Chinese DSD patients: 13 patients with 46, XY DSDs and 8 patients with 46, XX DSDs, and no clear inclusion criteria were described (Dong et al., 2016). A total of 9 (69.23%, 9/13) of the 13 patients with 46, XY DSDs received a molecular genetic diagnosis. NGS-based targeted sequencing technology was used for the detection of comprehensive DSD-related genes (a total of 2,972 exons and 1,078,042 bases of 219 genes). Mutations in the AR gene, which was the most common pathogenic gene in this cohort, were detected in five (55.56%) of nine probands. The other mutations detected were located in SRD5A2 (22.22%, 2/9), NR0B1 (22.22%, 2/9), CYP17A1 (11.11%, 1/9), GK (11.11%, 1/9), and SRY (11.1%, 1/9) (Dong et al., 2016). The detected mutations were limited to a few common DSD genes due to the small sample size of this study, but this study also provides new evidence for the molecular diagnosis of DSDs and supports subsequent studies.
Although the cohort was small and the paper did not provide clear inclusion criteria, the molecular diagnostic results indicated that AR mutations were most common in this cohort. In contrast, the Department of Endocrinology of the Ninth People’s Hospital of Shanghai Jiao Tong University School of Medicine published another study of 46, XY DSD patients in 2018 (Wang et al., 2018); patients with classical complete AIS (CAIS) were excluded, and AR mutations remained in the top three in this cohort (Wang et al., 2018). A study in Shanghai, China, excluded patients with CAIS, CAH, or syndromic DSD and those who underwent virilization or sex changes after puberty. They recruited 70 patients with 46, XY DSDs who could not be diagnosed on the basis of typical clinical presentation and conventional candidate genes (including 33 DSD candidate genes and 47 potential disease-causing genes). The final diagnostic rate obtained by panel sequencing was 42.7% (30/70). Patients with micropenis, cryptorchidism, and hypospadias had the highest diagnosis rate (58%). The most frequently mutated gene was SRD5A2 (15.04%, 17/113), followed by NR5A1 (8.84%, 10/113) and AR (7.96%, 9/113). Even after excluding patients with CAIS, the probability of an AR mutation was still high, which demonstrates the importance of AR mutations in the common 46, XY DSD etiology and in sexual development. Clinically, female patients often have primary amenorrhea in adolescence or insufficient masculinization of male internal and external genitalia. Among 9 patients with 46, XY DSDs whose gender identity was female, studied by Xia et al. (2021), more than half (55.6%) of the adolescent or adult patients had primary amenorrhea, 3 had external genital abnormalities, and 2 had masses in the bilateral inguinal region. The molecular diagnoses of the three patients with external genital anomalies were the NR5A1 mutations c.937C>T and c.398delC and LHCGR mutation, c.265A>T from the father and c.422T>C from the mother (Xia et al., 2021). Because this cohort of women with 46, XY DSDs was not randomized, they were not included in this analysis. However, this study still reveals that girls with primary amenorrhea should be aware of the possibility of 46, XY sex chromosomes.
In two studies of 46, XY DSD patients from Peking Union Medical College in Beijing, China, the most common mutated genes were AR, NR5A1, and SRD5A2 (Yu et al., 2021; Zhang et al., 2023). The two studies had the same inclusion criteria: patients with a sex chromosome karyotype of 46, XY and an external genital malformation. The most common pathogenic genes in both cohorts were SRD5A2, NR5A1, and AR (Yu et al., 2021; Zhang et al., 2023). The first article was published in 2021, and a total of 87 patients were eventually enrolled in the study. The final diagnosis rate was 42.5% by panel NGS (including 32 reported 46, XY DSD pathogenic genes and 51 genes related to gonadal development or differentiation). Patients with CYP17A1 mutations who presented with abnormal gonadal development combined with hypertension and hypokalemia were excluded. However, CYP17A1 mutations were still found in three patients in this cohort (Yu et al., 2021). The second article was published in 2023 and included 70 patients in total. A molecular diagnosis was identified in 64.3% (45/70) of patients in this cohort. The higher diagnosis rate in this cohort compared to other Chinese cohorts may be because this cohort had more patients with androgen synthesis or action and more severe clinical phenotypes, and the patients were sequenced using WES technology (Zhang et al., 2023).
In a 2022 study from Guangzhou, China, the overall diagnostic rate using a panel (108 candidate genes) or WES was 41.9% (31/74) in 74 patients with 46, XY DSDs (Xie et al., 2022). The diagnostic rates were 43.6% (24/55) for panel sequencing and 40.9% (9/22) for WES; in total, 52 patients were sequenced by panels alone, 19 patients by WES alone, and 3 patients with both panels and WES. Variants associated with steroid hormone synthesis and activation, such as AR (4.05%), CYP17A1 (4.05%), and FGFR1 (4.05%), were the most common in this cohort (Xie et al., 2022). There was little difference in the number of gene mutations detected in this cohort.
Korea
After excluding patients with sex chromosome abnormalities and 46, XX DSDs with SRY (+), a Korean cohort included 37 patients with 46, XY DSDs and 7 patients with 46, XX DSDs (Kim et al., 2017). In the 46, XY DSD cohort, pathogenic genes were identified in nine patients through targeted exome sequencing of 67 known DSD-associated genes, and the most common mutated genes were AR and CYP17A1. Similar to previous studies, a molecular diagnosis in patients with mild phenotypes such as micropenis or isolated hypospadias was difficult to obtain.
India
In 2019, MR Nagaraja et al. (2019) investigated the monogenic causes and prevalence of 46, XY DSDs and reported that pathogenic genes causative of AIS, steroid 5 alpha-reductase type 2 deficiency (SRD5A2), and gonadal hypoplasia were most common in India.
Summary of the Asian cohorts
Patients with severe phenotypes had a higher molecular diagnosis rate, and patients with DSDs with concurrent hypospadias, micropenis, and cryptorchidism had the highest detection rate (58%) (Wang et al., 2018). Similar results were obtained in a molecular study by Zhang et al. (2019) of 130 Han Chinese individuals with varying degrees of hypospadias, with 25 patients receiving a molecular diagnosis. Of the 25 patients who received a molecular diagnosis, patients with hypospadias combined with micropenis had the highest detection rate (68%), followed by patients with combined micropenis and cryptorchidism (28%) and patients with combined cryptorchidism (4%). Patients with isolated hypospadias in this study did not receive a molecular diagnosis.
We found that the rate of positive diagnosis correlated with the severity of the clinical presentation and possibly with the mode of testing and the number of individuals. However, Vuthy E applied next-generation DNA sequencing panel technology to 297 boys with isolated, mainly mild forms of hypospadias (mostly mild hypospadias, no micropenis, and no undescended testis) and obtained surprising results (Ea et al., 2021). Genes involved in the process of steroid synthesis had a higher detection rate in this cohort, including HSD17B3 (27.78%, 10/36) and SRD5A2 (11.11%, 4/36). The genes involved in the process of gonadal development detected were POR (16.67%, 6/36), NR5A1 (11.11%, 4/36), and STAR (8.33%, 3/36).
The diagnostic rate in this study increased with increasing severity of hypospadias, which is believed to be true for the immediate range (from anterior hypospadias (4.9%) to penoscrotal hypospadias (14.3%)). In this cohort, 5.5% (16/293) of patients with isolated hypospadias were definitively diagnosed (Ea et al., 2021).
The common etiology of 46, XY DSD patients was ultimately attributed to impaired steroid synthesis and action or problems in the gonadal decision differentiation process. Asian cohort studies of patients with 46, XY DSDs have shown AR, NR5A1, and SRD5A2 to be the most commonly mutated genes, which is consistent with a common DSD etiology.
Although some cohorts excluded patients with CAIS, AR was still the most common gene detected (Wang et al., 2018). The results of these cohort studies suggest AR is a common causative gene for DSDs. The AR gene is located on the X chromosome and encodes the androgen receptor, also known as the dihydrotestosterone receptor. CAIS is caused by complete androgen insensitivity due to AR inactivation. Even after excluding CAIS patients, the probability of an AR mutation was still the highest. Hundreds of AR mutations, mainly located in the ligand-binding domain, have been reported in CAIS. The majority of AR mutations originated from the mother (approximately 66.67%), while the remaining mutations (approximately 33.33%) arose from somatic and de novo mutations (Hiort et al., 1998).
However, in these articles, the demographics of the patients in the cohorts are not clear, and we cannot guarantee that all patients are residents of the city where the hospital is located, except for the cohort of Zhang Wangyu et al., who mentioned that all patients were Han Chinese. Therefore, it is not possible to determine the variability in the regional prevalence of XY DSD patients in China.
Europe
The United Kingdom and Ukraine
A total of 73 patients with 46, XY DSDs were included in the UK study, and the diagnostic rate of 46, XY DSDs by a panel (including 30 genes) was 34.2% (25/73). The most common mutated gene in this study was AR (9.59%, 7/73), followed by HSD17B3 (6.85%, 5/73) and SRD5A2 (5.48%, 4/73) (Hughes et al., 2019). The Ukrainian study excluded patients with sex chromosomal abnormalities and patients with CAH and included 71 patients with 46, XY DSDs. Using WES, 46.2% of patients were diagnosed. The most common pathogenic genes detected in this study were AR (15.49%, 11/71) and NR5A1 (7.04%, 5/71) (Globa et al., 2022).
Turkey
The study in Turkey, which was reported in 2016, excluded patients with mutations in the AR and SRD5A2 genes identified by Sanger sequencing, and a total of 20 patients with 46, XY DSDs were enrolled for panel sequencing. The overall diagnostic rate was 45% (9/20) (Özen et al., 2017). An HSD17B3 gene mutation was detected in six patients. The remaining patients harbored mutations in the LHCGR, WT1, and SRY genes. The rate of HSD17B3 mutations in this study may be high because the AR and SRD5A2 mutations were excluded. However, in this cohort, 14 patients had consanguineous parents, and the effect of consanguineous marriage cannot be ruled out (Özen et al., 2017) as the reason for the high rate of HSD17B3 mutations. The policies on consanguineous marriages vary from country to country, and there is no study cohort with a larger sample of consanguineous marriages worldwide.
Africa
Algeria
The Algerian cohort included 122 Algerian patients with 46, XY DSDs analyzed using WES (patients with suspected or confirmed CAH and sex chromosome abnormalities were excluded), and the overall diagnosis rate was 50.82% (62/122); the diagnosis rate of 46, XY nonsyndromic DSD patients was 42.2% (35/83), and that of 46, XY syndromic DSD patients was 69.2% (27/39). The AR, HSD17B3, NR5A1, and SRD5A2 genes were the most frequently mutated genes in this cohort (Zidoune et al., 2022).
North America
The United States of America
Previous studies using WES to diagnose 46, XY DSD identified genetic causes in 35% (14/40) of patients and six variants of uncertain significance (VUSs) (15%) (Arboleda et al., 2013). A related study using panel sequencing (including 64 genes) in the United States included 40 XY patients for whom a genetic diagnosis could not be obtained. No commonly mutated genes, such as AR, NR5A1, or SRA52, were detected. WES was also performed on these patients. The MAP3K1 variant was identified in four patients (Baxter et al., 2015a).
South America
Brazil
Patients with syndromic DSDs, dysmorphic features, developmental delay and/or intellectual disability, and the presence of more than two malformations in addition to genital anomalies were excluded from the São Paulo Hospital, Brazil, study (Gomes et al., 2022). A total of 209 patients were retrospectively included, all of whom had a 46, XY karyotype. Patients were divided according to time, using 2015 as the cutoff; patients before 2015 (168/209) were analyzed using panel testing, and patients after 2015 (41/209) were analyzed using massively parallel signature sequencing (MPSS). The molecular diagnosis rate of patients in the panel group was 59.5% (100/168). Targeted massively parallel sequencing (TMPS) was performed in 59 patients without a diagnosis and in 41 patients after 2015. Ten patients without a molecular diagnosis underwent WES. The most common mutated genes were, in order, AR (9.57%, 20/209), SRD5A2 (7.18%, 15/209), and NR5A1 (6.70%, 14/209). Five patients in this cohort were found to have polygenic mutations. Three of these patients had an unknown clinical etiology, and three coexisting mutations were found: WWOX and DHX37, NR5A1 and DHX37, and MAP3K1 and GATA4 (Gomes et al., 2022). Two patients with gonadal dysgenesis had missense mutations in two genes, FGFR2 and MAP3K1 (Table 2).
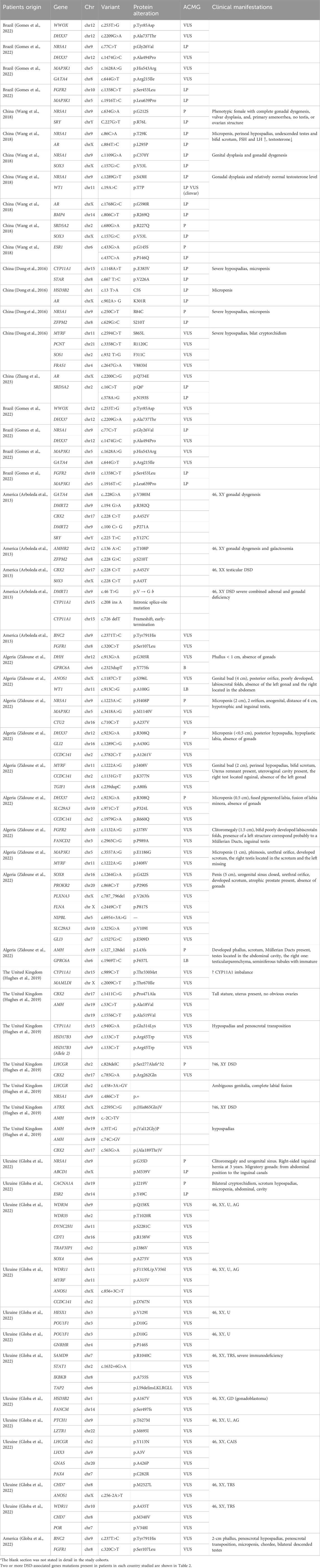
Table 2. Patient has mutations in two or more genes.
Oceania
Australia
In 2016, MPS (including 1,031 genes) was used in Australia to conduct a large cohort study of DSDs. A total of 278 patients with 46, XY DSDs and 48 patients with 46, XX DSDs were enrolled after excluding patients with CAH and sex chromosome abnormalities. The cohort covered 12 countries, and the researchers divided patients into Asia, Australia/New Zealand, and Europe. This study showed that the proportion of patients with DSD variants was similar in different regions, and the diagnosis rate varied (from 33% in Asia [58 of 174 patients] to 45% in Australia/New Zealand [41 of 90 patients]) in different regions. In the 46, XY DSD cohort, genetic diagnosis was possible in 40% of patients with gonadal dysplasia and 60% of patients with androgen synthesis and dysplasia. A total of 43% of 46, XY DSD patients received a possible genetic diagnosis. Variations in the AR (17.22%, 26/151) gene were the most common, followed by NR5A1 (10.60%, 16/151) and SR5A2 (8.61%, 13/151) (Eggers et al., 2016).
Summary of worldwide 46, XY DSD cohorts
In the above-collected studies, the molecular diagnostic rate for patients with 46, XY DSD ranged from a low of 24.3% to a high of 64.3%, and the most common mutated genes were AR, NR5A1, and SRD5A2. We found that patients with androgen synthesis disorders accounted for the greatest percentage of all patients. These three genes, which encode key enzymes for the androgen receptor, steroid nuclear receptor, and dihydrotestosterone, are involved in the process of androgen synthesis and action (Gulía et al., 2018; Dhurat et al., 2020; Morohashi et al., 2020). The gene encoding the androgen receptor (AR), located on the X chromosome, causes AIS (Gulía et al., 2018). NR5A1 is a transcription factor of the steroidogenic nuclear receptor superfamily that regulates the transcription of many reproductive, steroidogenesis, and male sexual differentiation genes (Morohashi et al., 2020). Steroid 5-alpha-reductase catalyzes the conversion of testosterone to the more potent dihydrotestosterone, the lack of which often leads to the development of micropenis (Dhurat et al., 2020).
The results of the individual cohorts were influenced by the inclusion criteria. There are no mutations with a clear geographic or ethnic predominance. Patients with multiple mutations tended to have more severe clinical manifestations. Conversely, patients with milder manifestations tended to have difficulty obtaining a molecular diagnosis. The diagnostic rates for these cohort studies may be correlated with the number of patients and the incidence of disease, as well as coincidence. The number of genes included in gene panels is limited, and WES can only detect coding regions. Therefore, WGS might gradually become the main modality used to detect such diseases, which can expand the genetic spectrum of human disease-causing genes. There are still many genes for which the association with DSDs is unclear, and some patients also lack a definitive molecular diagnosis. The basic information on 55 patients with multiple mutations is listed in Table 2; in total, 38 patients had 2 DSD-related mutations, 10 had 3 DSD-related mutations, and 7 had 4 or more DSD-related mutations. For 46, XY DSDs, there is no clear genotype–phenotype correlation, and the phenotypic variations may be due to other genomic variants or environmental influences on regulatory pathways required for gonad formation and/or function. Therefore, in patients with multiple mutations, it is necessary to determine whether the simultaneous mutation of these genes causes the disease or whether only one of the mutations is associated with the disease.
The results of polygenic testing suggest the diversity of disease etiologies and the complexity of their manifestations and, at the same time, provide direction for diagnosis. Patients with oligogenic mutations do not develop more severe phenotypes. The severity of the manifestation is more likely to be the result of the action of the mutated gene and the altered protein.
Updated monogenic forms of 46, XY DSDs
A monogenic phenotype results from the action of a pair of genes or alleles. The gene variants described here are considered monogenic causes of DSDs (i.e., variants in these genes trigger DSDs, and DSDs would not occur without these variants). The etiology of 46, XY DSDs is often categorized as androgen synthesis or action disorders, AMH synthesis or action disorders, and 46, XY GD.
The genes involved in hormone synthesis and action are AR, SRD5A2, HSD17B3, DHCR7, LHCGR, STAR, CYP11A1, CYP17A1, CYB5A, HSD3B2, AMH, AMHR2, AKR1C2, and AKR1C4 (Zhang et al., 2023). In the early stages of research into DSDs, most studies favored factors related to hormone synthesis or action, and AR was considered the most likely gene to cause the disease. In the included cohort studies, AR was the most common causative gene for 46, XY DSDs. However, with the continuous development of sequencing technology and improvements in basic experiments, new genes that have not been previously linked to this disease’, such as CBX2, DHX37, GATA4, DHH, and other genes in the SOX gene family, are being identified.
We have realized the updated monogenic mutations involved in the gonadal decision and differentiation phases leading to GD occurrence, including CBX2, DMRT1, GATA4 and 2FPM2 (FOG2), MYRF, the SOX gene family, DHX37, DHH and HHAT, PPP2R3C, and ZNRF3 (Elzaiat et al., 2022). Figure 1 shows that with the continuous updating of testing technology, more genes related to DSDs have been found to have diagnostic significance.
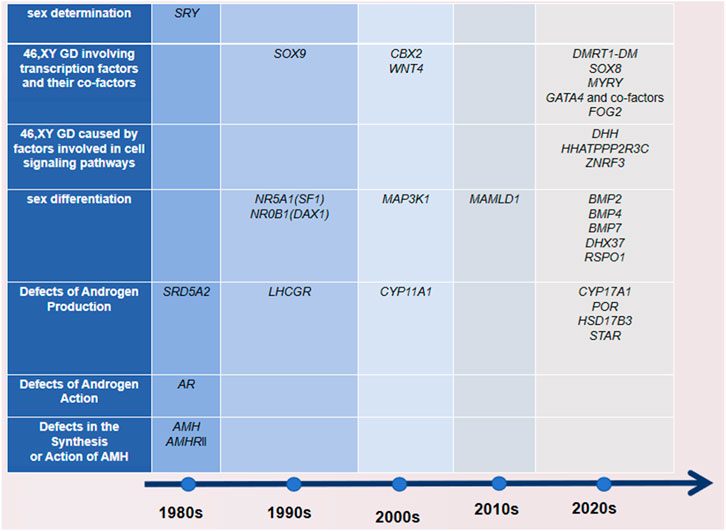
Figure 1. Time of discovery and classification of DSD-related genes. The time standard found here refers to when there are basic experiments, and it is detected in the molecular assay of the patients.
Next, we briefly discuss the genes recently proposed to be responsible for monogenic 46, XY GD in these cohorts. Current data suggest that in the testicular developmental pathway, CBX2 stabilizes the testis pathway by blocking the upregulated expression of genes in the ovarian pathway (Garcia-Moreno et al., 2019). We found a mutation in CBX2 in patients with polygenic mutations (c.228 C > T (p.Ala452Val), c.1411C > G (p.Pro471Ala), c.785G > A (p.Arg262Gln), and c.565G > A (p.Ala189Thr)). The two CBX2 variants found in the United States had the same locus, c.228C > T (p.A452V).
DHX37 is a member of the DEXH family of RNA helicases, and DHX37 mutations have been previously shown to cause 46, XY GD, testicular regression sequence (TRS), or anencephaly. The cause of these testicular developmental disorders has been identified as effects on β-conjugated proteins (Wan et al., 2023). The following mutation sites were found in the molecular assay results from two Algerian polygenic patients: c.2209G>A (p.Ala737Thr), c.1474G>C (p.Ala494Pro), and c.923G>A (p.Arg308Gln).
GATA4 is a zinc finger (ZF) transcription factor characterized by the presence of two conserved type IV ZF structural domains (amino acids 217-241 and 271-295), which interact with NR5A1 to regulate gene expression during testis assay and differentiation (Robert et al., 2006). Similarly, we found GATA4 c.644G>T (p.Arg215Ile) mutations in two patients in Brazil.
Conclusion
AR, NR5A1, and SRD5A2 are the most common mutated genes because of differences in the labeling of the cohorts in different countries, especially China, Brazil, and Australia. The mutation frequency of the AR gene in these cohorts ranged from 4.05% to 55.56%; the highest mutation frequency of NR5A1 was 22.22% and that of SRD5A2 was 11.11%, which were affected by the number of patients in the cohorts.
Among the known genes associated with nonsyndromic 46, XY gonadal agenesis, variants of SRY, NR5A1, and MAP3K1 are the most common (Reyes et al., 2023). The MAP3K1 mutations detected in the United States, Australia, and South Korea cohorts were completely different. The variants detected in the United States were p. Ly616Arg, p. Rg339Gln, p. Ly616Arg, and p. RO257Leu (Baxter et al., 2015a). In Australia, the MAP3K1 variants were p.Leu189Arg, p.Met312Leu, p.Ala1443Val (Eggers et al., 2016), and in Korea, the main variant was p. E1293K (Kim et al., 2017). For the international molecular diagnosis of DSDs, the limited ethnic or geographic differences we found may result from either the lack of reference to the patient’s ethnic origin in the article or an insufficient number of studies. The results of molecular detection were affected by the inclusion criteria and the detection method.
Multiple mutations have been identified in national studies, although the mutated genes were not commonly thought to be associated with DSDs. However, there is no clear relationship between the number of mutations in a gene and the severity of the disease; rather, the severity of the disease is directly related to the role of the mutated gene in sexual differentiation and development. Mutations in two key genes can lead to more severe manifestations. This also suggests that DSD is not a monogenic disease. This study also provides direction for the molecular diagnosis of DSD patients. Mutations in multiple genes may not only lead to the development of one disease but also to multisystemic lesions. Therefore, it is important to observe and follow patients with multiple abnormal manifestations in multiple systems in the clinic. Obtaining a definitive molecular diagnosis is important for understanding not only the cause of the disease but also the prognosis of the patient, as well as providing targets for gene therapy and even aiding in genetic counseling. The history of the discovery of disease-causing genes shows that an increasing number of disease-causing genes are discovered with the advancement of techniques and technology. Even with the current widespread use of WES, the cause of the disease in nearly half of 46, XY DSD patients still cannot be clearly identified.
The timing of diagnosis is critical for patients with 46, XY DSDs, some of whom may not ever receive a definitive diagnosis. Treatment is a challenge in terms of gender rearing as most of these patients reach puberty and are clarifying their gender identity as well as their sexual orientation, and we need to account for the thoughts of the patients and their families. In the absence of curative treatments, prenatal diagnosis is crucial for DSDs and other rare diseases. Patients with poor gonadal differentiation have an increased risk of germ cell tumors, and gonadoblastoma is the most common in this patient group (Reyes et al., 2023).
The molecular diagnosis of 46, XY DSDs remains a substantial challenge, and with the development and innovation of molecular techniques, the diagnosis of these patients can be greatly improved in the future. With the widespread use of WGS for rare diseases, it may be a better test for the diagnosis of DSDs because the whole genome is sequenced rather than only protein-coding regions.
Author contributions
CJ: Writing–original draft, Writing–review and editing. PH: Writing–original draft, Writing–review and editing. JY: Writing–review and editing. ZX: Writing–review and editing. JH: Writing–review and editing.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This work is funded in part by the research grant from the Application Research Project of Science and Technology Research and Development Plan Joint Fund of Henan Province in China (No. 232103810049) and the Key Project of Science and Technology Research and Development Joint Fund of Henan Province in China (No. 225200810054).
Conflict of interest
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Ahmed, S. F., Khwaja, O., and Hughes, I. A. (2000). The role of a clinical score in the assessment of ambiguous genitalia. BJU Int. 85 (1), 120–124. doi:10.1046/j.1464-410x.2000.00354.x
Arboleda, V. A., Lee, H., Sánchez, F. J., Délot, E. C., Sandberg, D. E., Grody, W. W., et al. (2013). Targeted massively parallel sequencing provides comprehensive genetic diagnosis for patients with disorders of sex development. Clin. Genet. 83 (1), 35–43. doi:10.1111/j.1399-0004.2012.01879.x
Baxter, R. M., Arboleda, V. A., Lee, H., Barseghyan, H., Adam, M. P., Fechner, P. Y., et al. (2015a). Exome sequencing for the diagnosis of 46,XY disorders of sex development. J. Clin. Endocrinol. Metab. 100 (2), E333–E344. doi:10.1210/jc.2014-2605
Baxter, R. M., Arboleda, V. A., Lee, H., Barseghyan, H., Adam, M. P., Fechner, P. Y., et al. (2015b). Exome sequencing for the diagnosis of 46,XY disorders of sex development. J. Clin. Endocrinol. Metab. 100 (2), E333–E344. doi:10.1210/jc.2014-2605
Délot, E. C., and Vilain, E. (2021a). Towards improved genetic diagnosis of human differences of sex development. Nat. Rev. Genet. 22 (9), 588–602. doi:10.1038/s41576-021-00365-5
Délot, E. C., and Vilain, E. (2021b). Towards improved genetic diagnosis of human differences of sex development. Nat. Rev. Genet. 22 (9), 588–602. doi:10.1038/s41576-021-00365-5
Dhurat, R., Sharma, A., Rudnicka, L., Kroumpouzos, G., Kassir, M., Galadari, H., et al. (2020). 5-Alpha reductase inhibitors in androgenetic alopecia: shifting paradigms, current concepts, comparative efficacy, and safety. Dermatol Ther. 33 (3), e13379. doi:10.1111/dth.13379
Dong, Y., Yi, Y., Yao, H., Yang, Z., Hu, H., Liu, J., et al. (2016). Targeted next-generation sequencing identification of mutations in patients with disorders of sex development. BMC Med. Genet. 17, 23. doi:10.1186/s12881-016-0286-2
Ea, V., Bergougnoux, A., Philibert, P., Servant-Fauconnet, N., Faure, A., Breaud, J., et al. (2021). How far should we explore hypospadias? Next-Generation sequencing applied to a large cohort of hypospadiac patients. Eur. Urol. 79 (4), 507–515. doi:10.1016/j.eururo.2020.12.036
Eggers, S., Sadedin, S., van den Bergen, J. A., Robevska, G., Ohnesorg, T., Hewitt, J., et al. (2016). Disorders of sex development: insights from targeted gene sequencing of a large international patient cohort. Genome Biol. 17 (1), 243. doi:10.1186/s13059-016-1105-y
Elzaiat, M., McElreavey, K., and Bashamboo, A. (2022). Genetics of 46,XY gonadal dysgenesis. Best. Pract. Res. Clin. Endocrinol. Metab. 36 (1), 101633. doi:10.1016/j.beem.2022.101633
Fukami, M., Wada, Y., Okada, M., Kato, F., Katsumata, N., Baba, T., et al. (2008). Mastermind-like domain-containing 1 (MAMLD1 or CXorf6) transactivates the Hes3 promoter, augments testosterone production, and contains the SF1 target sequence. J. Biol. Chem. 283 (9), 5525–5532. doi:10.1074/jbc.M703289200
Garcia-Moreno, S. A., Lin, Y. T., Futtner, C. R., Salamone, I. M., Capel, B., and Maatouk, D. M. (2019). CBX2 is required to stabilize the testis pathway by repressing Wnt signaling. PLoS Genet. 15 (5), e1007895. doi:10.1371/journal.pgen.1007895
Garnai, S. J., Brinkmeier, M. L., Emery, B., Aleman, T. S., Pyle, L. C., Veleva-Rotse, B., et al. (2019). Variants in myelin regulatory factor (MYRF) cause autosomal dominant and syndromic nanophthalmos in humans and retinal degeneration in mice. PLoS Genet. 15 (5), e1008130. doi:10.1371/journal.pgen.1008130
Globa, E., Zelinska, N., Shcherbak, Y., Bignon-Topalovic, J., Bashamboo, A., and MсElreavey, K. (2022). Disorders of sex development in a large Ukrainian cohort: clinical diversity and genetic findings. Front. Endocrinol. (Lausanne) 13, 810782. doi:10.3389/fendo.2022.810782
Gomes, N. L., Batista, R. L., Nishi, M. Y., Lerário, A. M., Silva, T. E., de Moraes Narcizo, A., et al. (2022). Contribution of clinical and genetic approaches for diagnosing 209 index cases with 46,XY differences of sex development. J. Clin. Endocrinol. Metab. 107 (5), e1797–e1806. doi:10.1210/clinem/dgac064
Gulía, C., Baldassarra, S., Zangari, A., Briganti, V., Gigli, S., Gaffi, M., et al. (2018). Androgen insensitivity syndrome. Eur. Rev. Med. Pharmacol. Sci. 22 (12), 3873–3887. doi:10.26355/eurrev_201806_15272
Hiort, O., Sinnecker, G. H., Holterhus, P. M., Nitsche, E. M., and Kruse, K., Inherited and de novo androgen receptor gene mutations: investigation of single-case families. J. Pediatr., 1998;132(6):939–943. PMID: 9627582.doi:10.1016/s0022-3476(98)70387-7
Huang, C. C., and Yao, H. H. (2010). Diverse functions of Hedgehog signaling in formation and physiology of steroidogenic organs. Mol. Reprod. Dev. 77 (6), 489–496. doi:10.1002/mrd.21174
Hughes, I. A., Houk, C., Ahmed, S. F., and Lee, P. A., (2006). Consensus statement on management of intersex disorders. Arch. Dis. Child. 91 (7), 554–563. doi:10.1136/adc.2006.098319
Hughes, L. A., McKay-Bounford, K., Webb, E. A., Dasani, P., Clokie, S., Chandran, H., et al. (2019). Next generation sequencing (NGS) to improve the diagnosis and management of patients with disorders of sex development (DSD). Endocr. Connect. 8 (2), 100–110. doi:10.1530/EC-18-0376
Kim, J. H., Kang, E., Heo, S. H., Kim, G. H., Jang, J. H., Cho, E. H., et al. (2017). Diagnostic yield of targeted gene panel sequencing to identify the genetic etiology of disorders of sex development. Mol. Cell Endocrinol. 444, 19–25. doi:10.1016/j.mce.2017.01.037
King, T. F., and Conway, G. S. (2014). Swyer syndrome. Curr. Opin. Endocrinol. Diabetes Obes. 21 (6), 504–510. doi:10.1097/MED.0000000000000113
Lee, P. A., Houk, C. P., Ahmed, S. F., and Hughes, I. A. (2006). Consensus statement on management of intersex disorders. International Consensus Conference on Intersex. Pediatrics 118, e488–e500. doi:10.1542/peds.2006-0738
Matson, C. K., Murphy, M. W., Sarver, A. L., Griswold, M. D., Bardwell, V. J., and Zarkower, D. (2011). DMRT1 prevents female reprogramming in the postnatal mammalian testis. Nature 476 (7358), 101–104. doi:10.1038/nature10239
McElreavey, K., and Bashamboo, A. (2023). Monogenic forms of DSD: an update. Horm. Res. Paediatr. 96 (2), 144–168. doi:10.1159/000521381
Morohashi, K. I., Inoue, M., and Baba, T. (2020). Coordination of multiple cellular processes by nr5a1/nr5a1. Endocrinol. Metab. Seoul. 35 (4), 756–764. doi:10.3803/EnM.2020.402
Nagaraja, M. R., Gubbala, S. P., Delphine Silvia, C. R. W., and Amanchy, R., (2019). Molecular diagnostics of disorders of sexual development: an Indian survey and systems biology perspective. Syst. Biol. Reprod. Med. 65 (2), 105–120. doi:10.1080/19396368.2018.1549619
Özen, S., Onay, H., Atik, T., Solmaz, A. E., Özkınay, F., Gökşen, D., et al. (2017). Rapid molecular genetic diagnosis with next-generation sequencing in 46,XY disorders of sex development cases: efficiency and cost assessment. Horm. Res. Paediatr. 87 (2), 81–87. doi:10.1159/000452995
Reyes, A. P., León, N. Y., Frost, E. R., and Harley, V. R. (2023). Genetic control of typical and atypical sex development. Nat. Rev. Urol. 20 (7), 434–451. doi:10.1038/s41585-023-00754-x
Robert, N. M., Miyamoto, Y., Taniguchi, H., and Viger, R. S. (2006). LRH-1/NR5A2 cooperates with GATA factors to regulate inhibin alpha-subunit promoter activity. Mol. Cell Endocrinol. 257 (258), 65–74. doi:10.1016/j.mce.2006.06.011
Sekido, R. S. R. Y. (2010). SRY: a transcriptional activator of mammalian testis determination. Int. J. Biochem. Cell Biol. 42 (3), 417–420. doi:10.1016/j.biocel.2009.12.005
Speiser, P. W., Arlt, W., Auchus, R. J., Baskin, L. S., Conway, G. S., Merke, D. P., et al. (2018). Congenital adrenal hyperplasia due to steroid 21-hydroxylase deficiency: an endocrine society clinical practice guideline. J. Clin. Endocrinol.Metab. 103, 4043–4088. doi:10.1210/jc.2018-01865
Tevosian, S. G., Albrecht, K. H., Crispino, J. D., Fujiwara, Y., Eicher, E. M., and Orkin, S. H. (2002). Gonadal differentiation, sex determination and normal Sry expression in mice require direct interaction between transcription partners GATA4 and FOG2. Development 129 (19), 4627–4634. doi:10.1242/dev.129.19.4627
Thyen, U., Lanz, K., Holterhus, P. M., and Hiort, O. (2006). Epidemiology and initial management of ambiguous genitalia at birth in Germany. Horm. Res. 66, 195–203. doi:10.1159/000094782
Wan, Y., Yu, R., Luo, J., Huang, P., Zheng, X., Sun, L., et al. (2023). A novel DEAH-box helicase 37 mutation associated with differences of sex development. Front. Endocrinol. (Lausanne) 14, 1059159. doi:10.3389/fendo.2023.1059159
Wang, H., Zhang, L., Wang, N., Zhu, H., Han, B., Sun, F., et al. (2018). Next-generation sequencing reveals genetic landscape in 46, XY disorders of sexual development patients with variable phenotypes. Hum. Genet. 137 (3), 265–277. doi:10.1007/s00439-018-1879-y
Wisniewski, A. B., Batista, R. L., Costa, E. M. F., Finlayson, C., Sircili, M. H. P., Dénes, F. T., et al. (2019). Management of 46,XY differences/disorders of sex development (DSD) throughout life. Endocr. Rev. 40 (6), 1547–1572. doi:10.1210/er.2019-00049
Xia, J., Wu, J., Chen, C., Zhao, Z., Xie, Y., Bai, Z., et al. (2021). Molecular study and genotype-phenotype in Chinese female patients with 46, XY disorders of sex development. Gynecol. Endocrinol. 37 (10), 934–940. doi:10.1080/09513590.2021.1960307
Xie, Q. G., Luo, P., Xia, K., Li, Z. Q., Xu, Z., Su, C., et al. 46,XY disorders of sex development: the use of NGS for prevalent variants. Hum. Genet., 2022;141(12):1863–1873. Epub 2022 Jun 21. PMID: 35729303.doi:10.1007/s00439-022-02465-6
Yu, B. Q., Liu, Z. X., Gao, Y. J., Wang, X., Mao, J. F., Nie, M., et al. (2021). Prevalence of gene mutations in a Chinese 46,XY disorders of sex development cohort detected by targeted next-generation sequencing. Asian J. Androl. 23 (1), 69–73. doi:10.4103/aja.aja_36_20
Zhang, W., Mao, J., Wang, X., Zhao, Z., Zhang, X., Sun, B., et al. The genetic spectrum of a Chinese series of patients with 46, XY disorders of the sex development. Andrology. 12, 98, 108. 2023. Epub ahead of print. PMID: 37147882.doi:10.1111/andr.13446
Zhang, W., Shi, J., Zhang, C., Jiang, X., Wang, J., Wang, W., et al. (2019). Identification of gene variants in 130 Han Chinese patients with hypospadias by targeted next-generation sequencing. Mol. Genet. Genomic Med. 7 (8), e827. doi:10.1002/mgg3.827
Zheng, G. Y., Chu, G. M., Li, P. P., and He, R. (2023). Phenotype and genetic characteristics in 20 Chinese patients with 46,XY disorders of sex development. J. Endocrinol. Invest. 46 (8), 1613–1622. doi:10.1007/s40618-023-02020-8
Zidoune, H., Ladjouze, A., Chellat-Rezgoune, D., Boukri, A., Dib, S. A., Nouri, N., et al. (2022). Novel genomic variants, atypical phenotypes and evidence of a digenic/oligogenic contribution to disorders/differences of sex development in a large north african cohort. Front. Genet. 13, 900574. doi:10.3389/fgene.2022.900574
Keywords: 46, XY DSD, genetic diagnosis, worldwide cohort, geographic and ethnic differences, diagnostic method, multiple mutations
Citation: Jiali C, Huifang P, Yuqing J, Xiantao Z and Hongwei J (2024) Worldwide cohort study of 46, XY differences/disorders of sex development genetic diagnoses: geographic and ethnic differences in variants. Front. Genet. 15:1387598. doi: 10.3389/fgene.2024.1387598
Received: 26 February 2024; Accepted: 14 May 2024;
Published: 10 June 2024.
Edited by:
Ani Amelia Zainuddin, National University of Malaysia, MalaysiaReviewed by:
Sitara Roy, Harvard Medical School, United StatesTerje Raudsepp, Texas A and M University, United States
Copyright © 2024 Jiali, Huifang, Yuqing, Xiantao and Hongwei. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jiang Hongwei, amlhbmdod0BoYXVzdC5lZHUuY24=; Peng Huifang, cGVuZ2h1aWZhbmdfc2t5QDE2My5jb20=; Zeng Xiantao, emVuZ3hpYW50YW8xMTI4QHdodS5lZHUuY24=
†These authors have contributed equally to this work