 Shilin Zhang1Jing Li1Yanhua Zhao1Yujun Tang1Hao Li1Tianzeng Song2Tianwu An3Jiuqiang Guan3Xiaowei Li4
Shilin Zhang1Jing Li1Yanhua Zhao1Yujun Tang1Hao Li1Tianzeng Song2Tianwu An3Jiuqiang Guan3Xiaowei Li4 Ming Zhang1,5,6*
Ming Zhang1,5,6*- 1College of Animal Science and Technology, Sichuan Agricultural University, Chengdu, China
- 2Institute of Animal Science, Tibet Academy of Agricultural and Animal Husbandry Science, Lhasa, China
- 3Sichuan Academy of Grassland Science, Chengdu, China
- 4Breeding Fram of Longri, Agriculture and Rural Bureau of Aba Prefecture in Sichuan, Hongyuan, China
- 5Key Laboratory of Livestock and Poultry Multi-Omics, Ministry of Agriculture and Rural Affairs, College of Animal Science and Technology, Sichuan Agricultural University, Chengdu, China
- 6Farm Animal Genetic Resources Exploration and Innovation Key Laboratory of Sichuan Province, College of Animal Science and Technology, Sichuan Agricultural University, Chengdu, China
The Sichuan-Yunnan region is the main production area of yaks in southwestern China, with rich genetic resources of Yaks. Nevertheless, there have been limited study on the genetic characteristics of the entire yak populations in Tibet and southwestern China. In this study, we performed whole-genome resequencing to identify genetic variation information in a total of 198 individuals from six yak breeds (populations) in Sichuan (Muli yak, Jinchuan yak, Changtai yak, Maiwa yak), Yunnan (Zhongdian yak), and Tibet (Tibetan yak). The aim was to investigate the whole-genome genetic diversity, population genetic structure, and genome selection signatures. We observed that all six populations exhibit abundant genetic diversity. Except for Tibetan yaks, which showed low nucleotide diversity (0.00104), the remaining yak populations generally displayed high nucleotide diversity (0.00129–0.00153). Population genetic structure analysis revealed that, among the six yak populations, Muli yak exhibited greater differentiation from other yak populations and formed a distinct cluster independently. The Maiwa yak population displayed a complex genetic structure and exhibited gene exchange with Jinchuan and Changtai yaks. Positive selection signals were detected in candidate genes associated with growth (GNB4, HMGA2, TRPS1, and LTBP1), reproduction (PI4KB, DYNC1I1, and GRIP1), immunity (CD200 and IL1RAP), lactation (SNX13 and CPM), hypoxia adaptation (NDUFB6, PRKN, and MRPS9), hair (KRT24, KRT25, and KRT26), meat quality (SUCLG2), digestion and absorption (CLDN1), and pigment deposition (OCA2) using the integrated Pi and FST methods. This study provides significant insights into understanding the whole-genome genetic characteristics of yak populations in Tibet and southwestern China.
Introduction
Yaks are a unique livestock species in the plateau region, displaying high-altitude adaptation to extreme and harsh environments, including cold and hypoxia (Wiener et al., 2003; Aguiar et al., 2018; Qi et al., 2019). They hold significant importance in local animal husbandry, providing meat, milk, wool, labor, fuel, and other essential resources to the local herdsmen (Wiener et al., 2003). Additionally, yaks serve as a valuable gene bank for genetic studies (Qiu et al., 2012).
The exploration of population genetic structure of yak has become a prominent field in domestic animal. Studies have been conducted on the genetic diversity of yaks using microsatellites and mitochondrial DNA (Qiu et al., 2015; Wang et al., 2021; Hameed et al., 2022; Ma et al., 2022). Moreover, the assembly and publication of the first yak reference genome in 2012, followed by subsequent improvements, have resulted in a highly complete and accurate yak reference genome at the chromosome level (Qiu et al., 2012; Ji et al., 2021; Zhang et al., 2021). This achievement has laid a robust foundation for investigating genetic variation at the whole-genome level in yaks, leading to the widespread use of whole-genome resequencing technology. This technology has enabled analyses of genetic variation in yaks, covering aspects such as their origin, domestication (Medugorac et al., 2017; Chai et al., 2020; Meng et al., 2022), genetic diversity and structure (Wang et al., 2014; Zhang et al., 2016; Lan et al., 2018; Wang et al., 2019; Li et al., 2022), high-altitude adaptation (Qiu et al., 2012; Qiu et al., 2015; Guang Xin et al., 2019b; Lan et al., 2021; Gao et al., 2022) and selection pressures (Lan et al., 2018; Xie et al., 2018; Guang Xin et al., 2019a; Li et al., 2022).
Southwestern China, specifically Sichuan Province’s Garze Tibetan Autonomous Prefecture, Aba Tibetan and Qiang Autonomous Prefecture, and Yunnan Province’s Diqing Tibetan Autonomous Prefecture, serves as the primary production area for yaks (Lan et al., 2018). In this region, several yak breeds, including Jiulong yak, Maiwa yak, Muli yak, Jinchuan yak, Changtai yak, and Zhongdian yak, have been officially recognized as excellent indigenous breeds by the government (Lan et al., 2021). Each of these yak breeds possesses unique characteristics and outstanding traits. For example, the Jinchuan yak stands out with approximately 52% of individuals having 15 thoracic vertebrae and 15 pairs of ribs, one more thoracic vertebra and one more pair of ribs than other yak breeds (Lan et al., 2018). Additionally, multi-ribbed Jinchuan yaks demonstrate higher meat and milk production, as well as superior reproductive performance compared to other breeds (Mipam et al., 2012). Maiwa yaks, on the other hand, produce milk with a protein content 40%–60% higher than that of native bovine milk (Chen et al., 2021). Protecting, developing, and utilizing the genetic resources of yak populations in this region holds great significance for the advancement of China’s yak industry. However, comprehensive studies on genetic variation detection, and selection signal analysis at the whole-genome level for multiple yak breeds in this region remain unreported.
To address the genetic gap, this study detected genetic variation, elucidate the genetic diversity, explore population genetic structure and genome-wide selection using six yak populations including four from Sichuan (Maiwa yak, Muli yak, Jinchuan yak, and Changtai yak), one from Yunnan (Zhongdian yak), and a collection of Tibetan yak populations. Our findings provide a scientific and theoretical basis for the protection, development, and genetic improvement of yak genetic resources in Tibet and southwestern China.
Materials and methods
Sample collection and whole-genome resequencing
In this study, we collected a total of 69 blood samples from Maiwa yak (MW) (The altitude of each sampling location ranges from 3400 m to 3600 m), 20 from Zhongdian yak (ZD), and 59 from Tibetan yak (XZ). Additionally, we collected 18 tissue samples each from Muli yak (ML) and Changtai yak (CT), and 14 tissue samples from Jinchuan yak (JC). The Tibetan yak population includes 16 Niangya yaks (NY), 11 Chawula yaks (CWL), 6 Sibu yaks (SB), 17 Dangxiong yaks (DX), 3 Naqu yaks (NQ), and 6 Pali yaks (PL) (Supplementary Figure S1; Supplementary Table S1). Genomic DNA was extracted using the TIANamp Genomic DNA Kit (Tiangen Biotech Co., Ltd., Beijing, China). 59 qualified Tibetan yak genomic DNA samples were sent to Biomarker Technologies (Beijing, China) for paired-end sequencing on the Illumina HiSeq 2500 platform. The remaining yak genomic DNA samples were sent to BGI Genomics (Shenzhen, China) for paired-end sequencing on the T7 platform, with each read length of 150 bp.
Read mapping and SNP calling
The raw data were filtered through the quality control process conducted by fastp 0.20.0 (Chen et al., 2018). The high-quality clean reads were mapped to the latest yak reference genome (BosGru3.0, GCA_005887515.2) using Burrows–Wheeler Aligner (BWA, v0.7.8-r455) software and SAMtools (Li and Durbin, 2009) to get the original mapping results stored in BAM format. Then, the results were dislodged duplication by Picard (http://broadinstitute.github.io/picard/). After genome mapping we undertook SNP calling for all individuals using SamTools, v1.13, and The “mpileup” command was executed to identify SNPs with the parameters as -m 2 -F 0.002 -d 1000 -u -C 50 (Li et al., 2015). The genotype data were filtered using PLINK 1.9 based on minor allele frequencies, missing genotype rates, and biallelic alleles with parameter: -geno 0.2 --maf 0.05 --biallelic-only. SnpEff (Cingolani et al., 2012) was used for functional annotation of variants. The variation data reported in this paper have been deposited in the Genome Variation Map (GVM) (Li et al., 2021) in National Genomics Data Center, Beijing Institute of Genomics, Chinese Academy of Sciences and China National Center for Bioinformation (CNCB-NGDC Members and Partners, 2022), under accession number GVM000675 (https://bigd.big.ac.cn/gvm/getProjectDetail?Project=GVM000675).
Population genetic diversity analysis
We estimated the genomic nucleotide diversity (Pi) of each yak population using VCFtools (v0.1.16) (Danecek et al., 2011) with the parameters (-window-pi 50,000 -window-pi-step 20,000). VCFtools was also used to calculate the average observed heterozygosity (HO) and expected heterozygosity (HE) as well as the average inbreeding coefficient (F) for six yak populations using the -het parameter. The pattern of linkage disequilibrium (LD) among 198 yaks from six populations can be effectively reflected by calculating the coefficient of determination (r2) between pairwise SNPs using PopLDdecay (Zhang et al., 2019) with default parameter.
Population genetic structure and phylogenetic tree analysis
Linkage sites in the genomic data were removed with parameters (-indep-pair-wise 50 10 0.2) using PLINK (v1.9) software. The genetic structure was analyzed using ADMIXTURE (v1.3.0) and the optimal clustering was also calculated to determine the best K value using ADMIXTURE’s cross-validation procedure. The smartpca module in EIGENSOFT (Patterson et al., 2006) was used to perform principal component analysis (PCA) on six yaks populations. The results were plotted using the ggplot2 package in R for the first principal component (PC1) and the second principal component (PC2). Based on the pairwise distance matrix among samples, a neighbor-joining (NJ) tree was constructed by MEGA (v11.0.11). The original tree file was visualized and beautified using the ggtree package in R. VCFtools was used to calculate the fixation index (FST) between six yak populations.
Genome-wide selective sweep and functional enrichment analysis
Based on the results of FST, we used the Muli yak as the control group and the Maiwa yak as the selection group to perform selection sweep based on two analysis methods, nucleotide diversity (Pi), and the genome-wide distribution of pairwise fixation index (FST). Pi and FST was calculated with 50 kb sliding windows and 20 kb steps between adjacent windows using the VCFtools. The windows with the top 5% FST and -log10 Pi values were considered as candidate region under strong selection and the candidate genes in the regions were identified. The intersection of two analysis results was taken as the final selected genes and a Venn diagram was drawn using the online website jvenn (http://jvenn.toulouse.inra.fr/app/index.html). The functions of the selected genes were further explored by conducting the Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analyses using KOBAS 3.0 (http://kobas.cbi.pku.edu.cn/anno_iden.php).
Results
Genome resequencing and genetic variation
Sequencing of 59 Tibetan yaks yielded 967.591 Gb of raw data and 953.379 Gb of filtered clean data. An additional 139 yaks were sequenced, producing 5274.237 Gb of raw data and 5084.898 Gb of filtered clean data (Supplementary Table S2). After mapping to the yak reference genome (BosGru3.0), the sequencing depth of the 59 individual Tibetan yaks ranged from 3.45 to 4.31×, while the sequencing depth of the remaining 139 yaks ranged from 9.12 to 25.42× (Supplementary Table S3). The mapping rate for all samples was above 96.68%. In total, we obtained 44,262,798 high-quality SNPs. The highest number of SNPs (5,145,209) was detected in Tibetan yaks, followed by Changtai yaks (3,007,140), Jinchuan yaks (2,955,926), Muli yaks (2,951,598), and Maiwa yaks (2,764,492), while the lowest number of SNPs was detected in Zhongdian yaks (2,640,551). Most SNPs were distributed in intergenic and intronic regions, with 63.90% located in intergenic regions and 33.84% in intronic regions. Only 0.61% of SNPs were distributed in exonic regions. Of the SNPs in exonic regions, 39.62% were synonymous and 36.53% were non-synonymous (Table 1).
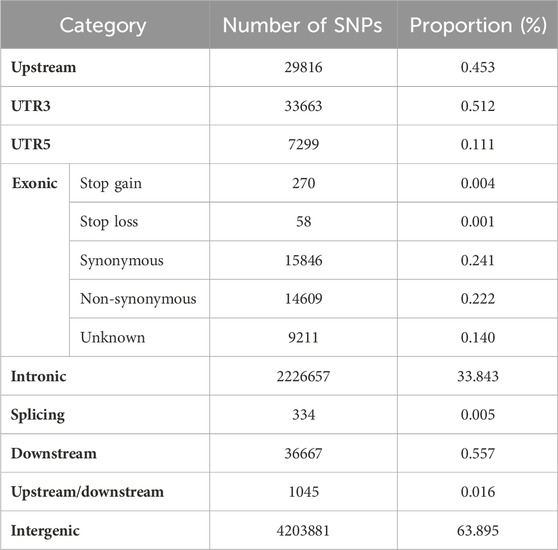
Table 1. SNP functional annotation results.
Population genomic diversity
Pi among the six breeds of yaks ranged from 0.00104 to 0.00153 (Figure 1A). Changtai yak exhibited the highest Pi at 0.00153, while Tibetan yak had the lowest at 0.00104. Apart from Tibetan yak, which showed relatively low Pi, the other yak populations generally exhibited high Pi, ranging from 0.00129 to 0.00153, (Supplementary Table S4). Except for Tibetan yak (0.05707), the F of the other five yak breeds was relatively low (−0.07969 to 0.01778) (Supplementary Table S4, Figure 1B). Muli yak had the lowest average inbreeding coefficient at −0.07969. The LD analysis showed that Muli yak had the highest average LD (r2), followed by Jinchuan yak, Changtai yak, Zhongdian yak, and Maiwa yak. In contrast, Tibetan yaks had the lowest average LD (r2) (Figure 1C). Results for heterozygosity indicated that the differences between observed heterozygosity (Ho) and expected heterozygosity (He) among the six breeds of yaks were small (Figure 1D). Muli yak had the highest Ho and He, at 0.36546 and 0.33848, respectively, while Tibetan yak had the lowest Ho and He, at 0.27363 and 0.29020, respectively (Supplementary Table S4). This suggests that Tibetan yak had the fastest LD decay rate among these six breeds, while Muli yak had the slowest decay rate.
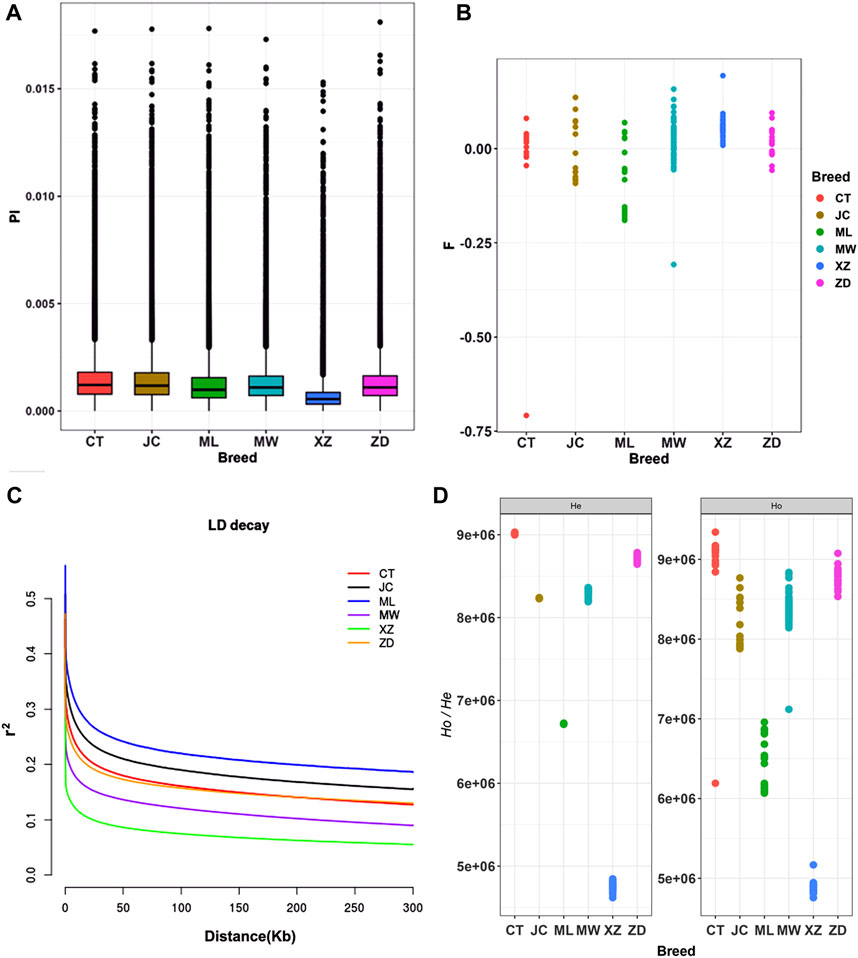
Figure 1. Analysis of genomic diversity, Pi, F and LD for these yak breeds/populations in southwestern China. (A) Box plots of nucleotide diversity (Pi). (B) the average inbreeding coefficient (F). (C) Decay of linkage disequilibrium (LD). (D) The observed heterozygosity (Ho) and expected heterozygosity (He) CT: Changtai yak, JC: Jinchuan yak, ML:Muli yak, MW: Maiwa yak, XZ: Tibet yak, ZD: Zhongdian yak.
Population genetic structure
To examine the population genetic structure and relationships among six different yak breeds, we conducted a series of genome-wide analyses, including population genetic structure, PCA, and phylogenetic analysis. The results of the ancestral component analysis showed that the K value was equal to 3 and the lowest value of the cross-validation (CV) error was reached, proving that the optimal population grouping was when the ancestor population was 3 (Figure 2A). Moreover, when K = 2, the six yak breeds were grouped into two ancestral populations, distinguishing Muli yak from the other yak breeds. When K = 3, a new ancestral population emerged, further distinguishing Muli yak, Tibetan yak, and the other yak breeds. Most Muli yak samples have a relatively homogeneous ancestral population, while Tibetan yak showed a small number of mixed third ancestry. The majority of the other yak breeds showed a composition of three ancestries. When K = 4, the overall ancestry composition of each breed remained consistent with the results at K = 3 (Figure 2B). However, there was an uneven distribution phenomenon in the ancestry composition ratio of each sample of Maiwa yak.
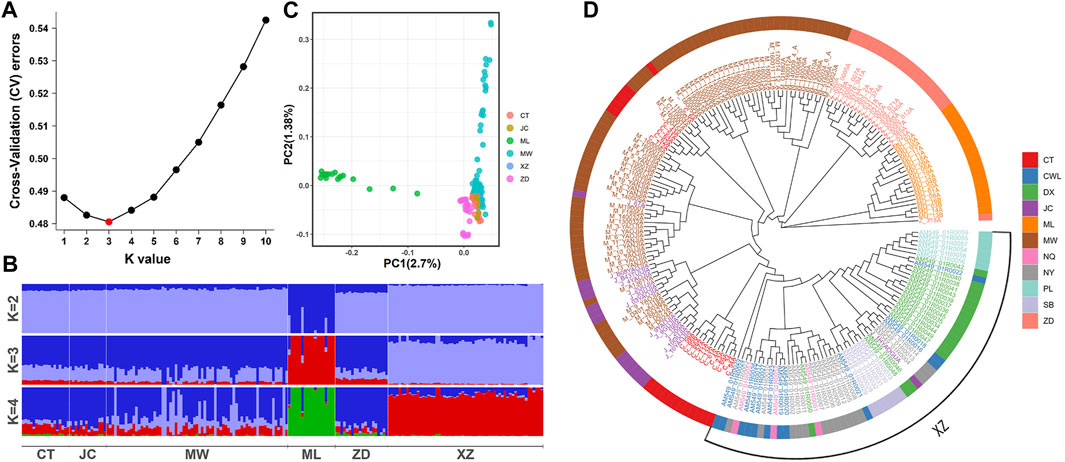
Figure 2. Population genetic structure of six yak breeds/population. (A) Cross-validation (CV) errors. (B) Analysis of ancestor components. (C) Principal components analysis. (D) Phylogenetic tree construction by neighbor-joining method. CT: Changtai yak. JC: Jinchuan yak. ML:Muli yak. MW: Maiwa yak. ZD: Zhongdian yak. XZ: Tibet yak, and it includes CWL, NQ, DX, NY, SB and PL poputation.
According to the PCA results (Figure 2C), we observed that the Muli yak population formed a distinct and separate cluster from the other yak populations. On the other hand, most Maiwa yaks overlapped with the Jinchuan yaks, Changtai yaks, Tibetan yaks, and Zhongdian yaks, forming a clustered group together.
Based on genome-wide data, the NJ tree revealed that Muli yaks and Zhongdian yaks were grouped into two distinct branches, with Maiwa yaks forming a separate branch (Figure 2D). Some individuals from the Changtai yak and Jinchuan yak populations clustered together within the Maiwa yak branch, indicating close genetic relatedness. Similarly, some individuals from the Changtai yak and Tibetan yak populations formed another branch within the Maiwa yak group, suggesting a close genetic relationship between them. Notably, one individual from the Zhongdian yak population exhibited significant hybridization and formed a separated branch from the main population to form its own branch. Furthermore, one individual from the Jinchuan yak population clustered with the Tibetan yak population. Within the Tibetan yak population, the Pali yaks displayed a relatively pure bloodline, reflecting their distant geographic distribution from other Tibetan yaks.
The average FST of Muli yak was found to be the highest at 0.0898 (Table 2), indicating a moderate degree of genetic differentiation between Muli yak and other yak populations. On the other hand, the average fixation index of other yak populations was relatively low, with Maiwa yak having the lowest average differentiation index at 0.0345.
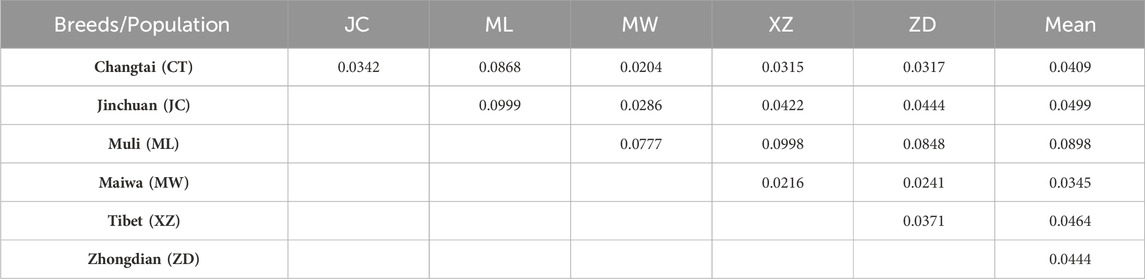
Table 2. FST values between pairwise populations.
Genome-wide selective sweeps and functional enrichment analysis of candidate genes
According to the results of genetic differentiation between populations, we observed that the average FST between Muli and Maiwa yak was the highest, indicating a moderate level of differentiation. Consequently, we designated Muli yak as the control group and Maiwa yak as the selection group for the selection sweep analysis using two methods, Pi and FST. Subsequently, we identied the top 5% of scanning results as candidate regions (Figures 3A, B). As a result, a total of 1935 genes (Pi) and 2344 genes (FST) were identified, with 187 genes overlapping between the two analyses (Figure 3C). These candidate genes were subject to strong selection and mainly participated in biological processes such as growth (GNB4, HMGA2, TRPS1, and LTBP1), reproduction (PI4KB, DYNC1I1, and GRIP1), immunity (CD200 and IL1RAP), neural development (ST3GAL3, KIAA0319L, and SFPQ), lipid metabolism and accumulation (CYP27A1, AGMO, and KBTBD2), lactation (SNX13 and CPM), bone development (ZNF687 and PPP2R2B), hypoxia adaptation (NDUFB6, PRKN, and MRPS9), hair development (KRT24, KRT25, and KRT26), meat quality (SUCLG2), digestion and absorption (CLDN1), and pigment deposition (OCA2).
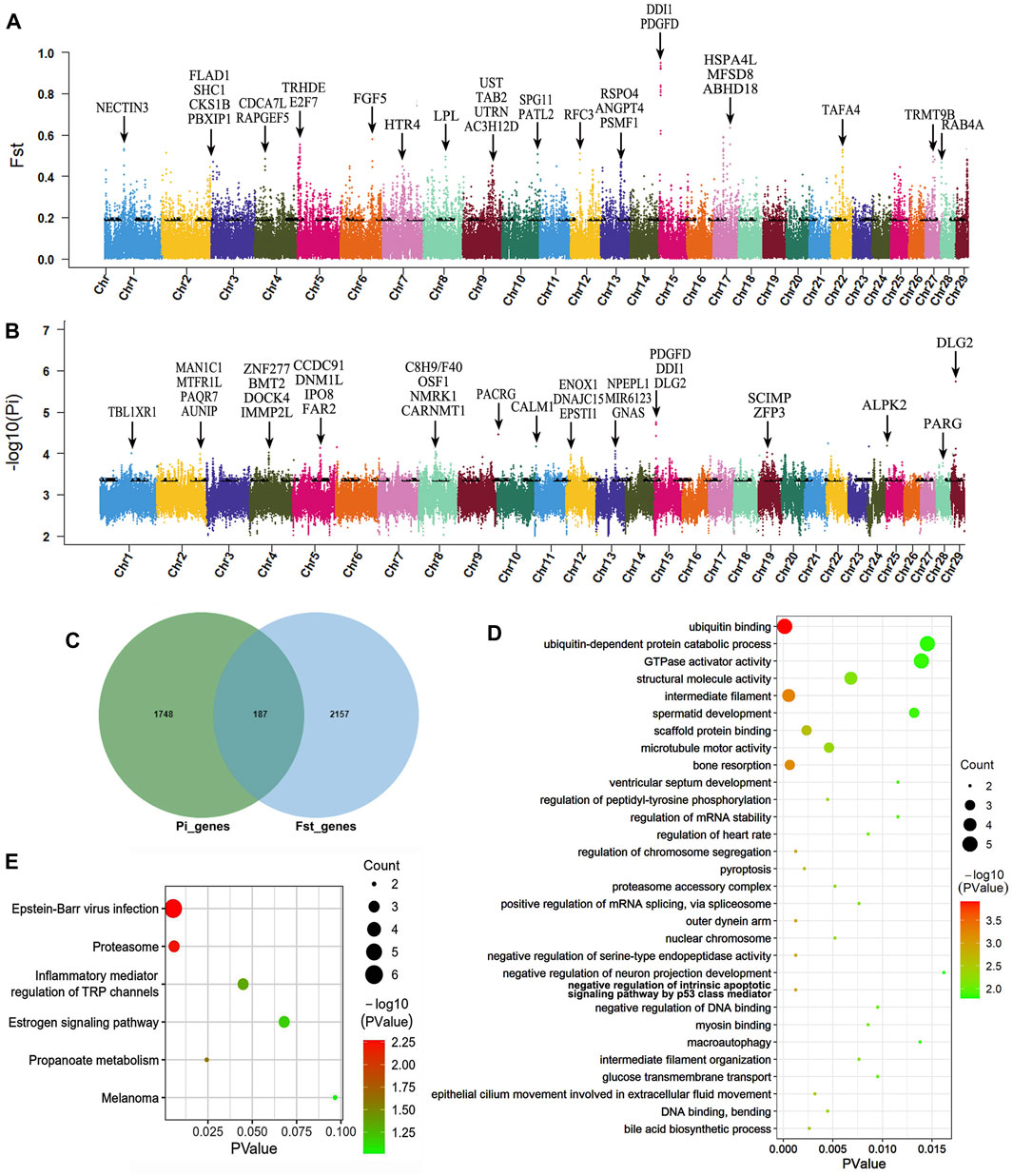
Figure 3. Genome-wide selective sweeps and functional analysis of Maiwa yak. Manhattan map based on FST (A) and Pi (B) selected detection methods. (C) Venn diagram based on two selected detection methods. (D) Top-30 GO items. (E) KEGG enrichment analysis of candidate genes for strong selection. Black arrow indicates candidate genes for strong selection in Figures 3A, B.
Functional enrichment analysis using Gene Ontology (GO) was performed on candidate genes, revealing GO terms with a significance level of p < 0.05 (Figure 3D). The analysis showed that The 187 selected genes were predominantly enriched in functions associated with immune regulation, reproductive regulation, regulation of heart rate, cell apoptosis, mitosis, digestive synthesis and metabolism, transmembrane substance transport, epithelial cell proliferation and wound healing, hormone secretion and regulation, bone resorption, heme transport, and ventricular septum development. Furthermore, KEGG pathway enrichment analysis was performed, identifying pathways with a significance level of p < 0.1 (Figure 3E). The identified pathways were linked to virus infection, propanoate metabolism, proteasome, inflammatory mediator regulation of TRP channels, estrogen signaling, and melanoma.
Discussion
Genetic diversity reflects the adaptability of a population to its environment and its evolutionary history. It holds significant value for the protection and further utilization of animal genetic resources. In this study, we observed that the nucleotide diversity of Tibetan yak is low, likely due to their isolated geographical location, leading to limited genetic interchange with other populations. Chai et al. (2020) classeified 91 domestic yaks from 31 populations into three groups based on their phylogenetic relationships.They noted that the nucleotide diversity and genetic diversity of the yak population in the central Tibetan region were both low, aligning with our study results.
The observed heterozygosity (Ho) of Muli yak, Jinchuan yak, and Changtai yak was higher than the expected heterozygosity, and the inbreeding coefficient was generally lower. This suggests that these populations abundant exhibit genetic diversity and a low degree of inbreeding. This may be attributed to the large-scale purebred breeding and hybrid improvement efforts carried out on yaks in Sichuan in recent years. As a result of this work, several yak varieties have been developed for breeding populations and core breeding groups. On the other hand, the observed heterozygosity of Tibetan yak was low, while the inbreeding coefficient was high. This suggests a lack of genetic diversity, possibly due to the traditional feeding practices in the region that have led to significant inbreeding and a decline in reproductive performance. The LD decay analysis results indicated that Muli yak, Jinchuan yak, and Changtai yak have experienced more intense artificial selection in recent years, as evidenced by their slower LD decay rate. as evidenced by their slower LD decay rate. In contrast, Tibetan yak exhibited a faster LD decay compared to other populations, indicating less human intervention. This aligns with previous LD decay analysis results for Jinchuan yak and Tibetan yak (Wang et al., 2020).
In previous studies, Lan et al. (2018) investigated the population genetic structure of Jinchuan yaks at the whole-genome level. The results from phylogenetic tree and PCA analysis both indicated that Jinchuan yaks form an independent branch within the domestic yak population and are distantly related to Zhongdian yak, Tibetan yak, and Qinghai yak. Additionally, the ancestral component analysis results revealed that when K = 3, the Jinchuan yak population was separated from the rest of the domestic yak population. In the present study, the ancestral component analysis, PCA analysis, and FST statistics results of the six yak populations all pointed towards Muli yak exhibiting a relatively independent population genetic structure and distant relationship with other yak populations. Most of the Maiwa yak showed a high degree of hybridization, characterized by the smallest average differentiation index and a complex genetic background. This genetic variation is likely influenced by the diverse geographical environment of the two primary yak breeding regions. Muli yak and Maiwa yak are classified as the “Hengduan Mountains type” and “Qinghai-Tibet Plateau type,” respectively, based on their ecological characteristics. Muli yak predominantly inhabits mountain valleys, leading to grassland fragmentation and restricted gene flow, resulting in isolated breeding populations with. artificial selection. Conversely, Maiwa yak is primarily found in high-altitude terrains with espansive grasslands, where primitive year-round grazing is common, leading to a lower degree of artificial selection. It is speculated that the Maiwa yak population in this study may have originated from two distinct large populations due to the establishment of a core breeding group for Maiwa yak and the implementation of various effective breeding practices. One population may have introduced Jinchuan and Changtai yak bloodlines, while the other population retained a relatively pure genetic lineage without external influences.
The identification of selective signatures in a population is crucial for understanding its evolutionary history and economic traits. A total of 187 candidate positively selected genes were identified in the Maiwa yak population, and their potential effects on relevant important traits were discussed. For example, the CLDN1 gene, a component of endothelial tight junctions, has been demonstrated to decrease intestinal permeability and improve immune adaptability in calves through the upregulation of its expression (Ghaffari et al., 2021). It also plays an effective role in repairing the rumen epithelial barrier of slow-growing yaks (Hu et al., 2019). Thus, the CLDN1 gene may be associated with the stronger gastrointestinal immune function of Maiwa yaks, enabling them to adapt to the challenging high-altitude wetland grassland environment. The GNB4 gene can control cattle growth by influencing the expression of growth-related hormones in the pituitary gland (Lu et al., 2020). The HMGA2 gene, a transcription factor of insulin-like growth factor gene 2 (IGF2), plays a significant role in the growth trait of navel length in beef cattle (Aguiar et al., 2018). Similarly, the TRPS1 gene is significantly associated with the increase in both cattle body weight and eye muscle area (Lee et al., 2011). Therefore, the GNB4, HMGA2, and TRPS1 genes are likely to be closely associated with the growth and development of Maiwa yak.
This study also identified several genes that are specifically associated with reproduction. For instance, PI4KB is responsible for regulating actin aggregation during sperm capacitation (Etkovitz et al., 2007). DYNC1I1 participates in transporting molecules and organelles during oocyte maturation and is the most prevalent transport system in cells (Racedo et al., 2008). GRIP1 has been validated as a marker for detecting cow estrus (Hlaing et al., 2001; Lee et al., 2017). CARPT is a novel regulatory factor for ovarian function during follicular wave emergence in cows and is thought to play a potential role in dominant follicle selection during the follicular wave period in single-ovulating species as an “ovulation quota gatekeeper” (Mihm et al., 2000; Lv et al., 2009; Smith et al., 2010). These genes may have an impact on maintaining a stable reproductive pattern of Maiwa yak in the harsh high-altitude environments.
The challenging high-altitude environment and extensive productionpractices have resulted in the strong selection of genes associated with immunity and adaptation to hypoxia in the genome of Maiwa yak. For example, the reduction of the CD200 gene has a significantly affects on the quantity of pulmonary epithelial cells in radiation conditions (Beach et al., 2023). This finding might elucidate the relevant mechanism that enables the Maiwa yak to adapt to high-altitude with intense radiation and maintain lung function without harm. The IL1RAP gene has been identified as a central gene encoding the interleukin-1 receptor accessory protein, which plays a significant role in the immune system (Dos Santos Silva et al., 2019). Furthermore, three genes, NDUFB6, PRKN, and MRPS9, are closely linked to mitochondrial structure and normal function (Buggiotti et al., 2021; Fang et al., 2022; Zhang et al., 2022). Their influence on respiratory and energy metabolism efficiency may be be associated with Maiwa yak’s adaptation to high-altitude hypoxic environments.
In addition, this study identified lactation-related genes SNX13 and CPM among the selected genes of Maiwa yak. The SNX13 gene is highly correlated with milk production traits in cows, while the CPM gene has a significant effect on the fatty acid content in cow milk (Rincón et al., 2009; Shi et al., 2020). The SUCLG2 gene has been identified as a candidate biomarker for beef tenderness in cattle (Li and Li, 2021).
Three keratin family genes, KRT24, KRT25, and KRT26, have been identified. The KRT24 gene play a crucial role in hair growth and follicle development in yaks (Bao et al., 2022). The long and dense wool of Maiwa yak helps them retain heat and withstand cold weather at high altitudes. It is worth noting that a pigment deposition-related gene, Additionally, an OCA2 gene related to pigment deposition has been identified, potentially influencing black pigment deposition in Maiwa yak’s wool (Grønskov et al., 2007; Caduff et al., 2017; Ballan et al., 2022).
Conclusion
In conclusion, this study systematically investigates the genetic diversity, population genetic structure, kinship relationships, genetic differentiation, and gene selection of several yak breeds (populations) in Tibet and southwestern China at the whole-genome level. Overall, all six populations exhibit rich genetic diversity. Muli yak shows higher differentiation from other yak populations and forms an independent cluster. The Maiwa yak population demonstrates a complex genetic structure. The candidate genes identified for Maiwa yak are associated with various functions such as growth, reproduction, immunity, lactation, hypoxia adaptation, hair development, meat quality, digestion and absorption, and pigment deposition. These findings provide a scientific and theoretical foundation for the exploration and conservation of yak genetic resources in this region, as well as for the selection and breeding of yak breeds.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://bigd.big.ac.cn/gvm/getProjectDetail?Project=GVM000675, GVM000675.
Ethics statement
The animal studies were approved by the Animal Welfare and Ethics Committee of Sichuan Agricultural University. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent was obtained from the owners for the participation of their animals in this study.
Author contributions
SZ: Data curation, Formal Analysis, Methodology, Writing–original draft, Software. JL: Data curation, Methodology, Investigation, Writing–original draft. YZ: Writing–original draft, Data curation, Resources. YT: Data curation, Methodology, Writing–review and editing. HL: Data curation, Methodology, Writing–review and editing. TS: Data curation, Methodology, Resources, Writing–review and editing. TA: Data curation, Resources, Writing–review and editing. JG: Data curation, Resources, Writing–review and editing. XL: Data curation, Resources, Writing–review and editing. MZ: Conceptualization, Funding acquisition, Supervision, Writing–review and editing, Project administration.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This research was supported by National Key Research and Development Projects of China (2021YFD1600201).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2024.1382128/full#supplementary-material
References
Aguiar, T. S., Torrecilha, R. B. P., Milanesi, M., Utsunomiya, A. T. H., Trigo, B. B., Tijjani, A., et al. (2018). Association of copy number variation at intron 3 of HMGA2 with navel length in Bos indicus. Front. Genet. 9, 627. doi:10.3389/fgene.2018.00627
Ballan, M., Bovo, S., Schiavo, G., Schiavitto, M., Negrini, R., and Fontanesi, L. (2022). Genomic diversity and signatures of selection in meat and fancy rabbit breeds based on high-density marker data. Genet. Sel. Evol. 54 (1), 3. doi:10.1186/s12711-022-00696-9
Bao, Q., Zhang, X., Bao, P., Liang, C., Guo, X., Yin, M., et al. (2022). Genome-wide identification, characterization, and expression analysis of keratin genes (KRTs) family in yak (Bos grunniens). Gene 818, 146247. doi:10.1016/j.gene.2022.146247
Beach, T. A., Finkelstein, J. N., and Chang, P. Y. (2023). Epithelial responses in radiation-induced lung injury (RILI) allow chronic inflammation and fibrogenesis. Radiat. Res. doi:10.1667/rade-22-00130.1
Buggiotti, L., Cheng, Z., Salavati, M., and Wathes, C. D.Genotype plus Environment Consortium (2021). Comparison of the transcriptome in circulating leukocytes in early lactation between primiparous and multiparous cows provides evidence for age-related changes. BMC Genomics 22 (1), 693. doi:10.1186/s12864-021-07977-5
Caduff, M., Bauer, A., Jagannathan, V., and Leeb, T. (2017). OCA2 splice site variant in German Spitz dogs with oculocutaneous albinism. PLoS One 12 (10), e0185944. doi:10.1371/journal.pone.0185944
Chai, Z. X., Xin, J. W., Zhang, C. F., Dawayangla, , Luosang, , Zhang, Q., et al. (2020). Whole-genome resequencing provides insights into the evolution and divergence of the native domestic yaks of the Qinghai-Tibet Plateau. BMC Evol. Biol. 20 (1), 137. doi:10.1186/s12862-020-01702-8
Chen, S., Zhou, Y., Chen, Y., and Gu, J. (2018). fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34 (17), i884–i890. doi:10.1093/bioinformatics/bty560
Chen, Y., Qu, S., Huang, Z., Ren, Y., Wang, L., and Rankin, S. A. (2021). Analysis and comparison of key proteins in Maiwa yak and bovine milk using high-performance liquid chromatography mass spectrometry. J. Dairy Sci. 104 (8), 8661–8672. doi:10.3168/jds.2021-20269
Cingolani, P., Platts, A., Wang le, L., Coon, M., Nguyen, T., Wang, L., et al. (2012). A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly. (Austin) 6 (2), 80–92. doi:10.4161/fly.19695
CNCB-NGDC Members and Partners (2022). Database resources of the national genomics data center, China national center for bioinformation in 2022. Nucleic Acids Res. 50, D27–D38. doi:10.1093/nar/gkab951
Danecek, P., Auton, A., Abecasis, G., Albers, C. A., Banks, E., DePristo, M. A., et al. (2011). The variant call format and VCFtools. Bioinformatics 27 (15), 2156–2158. doi:10.1093/bioinformatics/btr330
Dos Santos Silva, D. B., Fonseca, L. F. S., Pinheiro, D. G., Muniz, M. M. M., Magalhães, A. F. B., Baldi, F., et al. (2019). Prediction of hub genes associated with intramuscular fat content in Nelore cattle. BMC Genomics 20 (1), 520. doi:10.1186/s12864-019-5904-x
Etkovitz, N., Rubinstein, S., Daniel, L., and Breitbart, H. (2007). Role of PI3-kinase and PI4-kinase in actin polymerization during bovine sperm capacitation. Biol. Reprod. 77 (2), 263–273. doi:10.1095/biolreprod.106.056705
Fang, Z., Liu, G., Zhu, M., Wang, S., Jiang, Q., Loor, J. J., et al. (2022). Low abundance of mitophagy markers is associated with reactive oxygen species overproduction in cows with fatty liver and causes reactive oxygen species overproduction and lipid accumulation in calf hepatocytes. J. Dairy Sci. 105 (9), 7829–7841. doi:10.3168/jds.2021-21774
Gao, X., Wang, S., Wang, Y. F., Li, S., Wu, S. X., Yan, R. G., et al. (2022). Long read genome assemblies complemented by single cell RNA-sequencing reveal genetic and cellular mechanisms underlying the adaptive evolution of yak. Nat. Commun. 13 (1), 4887. doi:10.1038/s41467-022-32164-9
Ghaffari, M. H., Sadri, H., Steinhoff-Wagner, J., Hammon, H. M., and Sauerwein, H. (2021). Effects of colostrum feeding on the mRNA abundance of genes related to toll-like receptors, key antimicrobial defense molecules, and tight junctions in the small intestine of neonatal dairy calves. J. Dairy Sci. 104 (9), 10363–10373. doi:10.3168/jds.2021-20386
Grønskov, K., Ek, J., and Brondum-Nielsen, K. (2007). Oculocutaneous albinism. Orphanet J. Rare Dis. 2, 43. doi:10.1186/1750-1172-2-43
Guang Xin, E., Basang, W. D., and Zhu, Y. B. (2019b). Whole-genome analysis identifying candidate genes of altitude adaptive ecological thresholds in yak populations. J. Anim. Breed. Genet. 136 (5), 371–377. doi:10.1111/jbg.12403
Guang Xin, E., Yang, B. G., Basang, W. D., Zhu, Y. B., An, T. W., and Luo, X. L. (2019a). Screening for signatures of selection of Tianzhu white yak using genome-wide re-sequencing. Anim. Genet. 50 (5), 534–538. doi:10.1111/age.12817
Hameed, A., Schlecht, E., Tariq, M., Buerkert, A., Scheper, C., König, S., et al. (2022). Phenotypic and genetic diversity of domestic yak (Bos grunniens) in high-altitude rangelands of Gilgit-Baltistan, Pakistan. J. Anim. Breed. Genet. 139 (6), 723–737. doi:10.1111/jbg.12730
Hlaing, M., Nam, K., Lou, J., Pope, W. F., and Nephew, K. P. (2001). Evidence for expression of estrogen receptor cofactor messenger ribonucleic acid in the ovary and uterus of domesticated animals (sheep, cow and pig). Life Sci. 68 (12), 1427–1438. doi:10.1016/s0024-3205(01)00937-7
Hu, R., Zou, H., Wang, Z., Cao, B., Peng, Q., Jing, X., et al. (2019). Nutritional interventions improved rumen functions and promoted compensatory growth of growth-retarded yaks as revealed by integrated transcripts and microbiome analyses. Front. Microbiol. 10, 318. doi:10.3389/fmicb.2019.00318
Ji, Q. M., Xin, J. W., Chai, Z. X., Zhang, C. F., Dawa, Y., Luo, S., et al. (2021). A chromosome-scale reference genome and genome-wide genetic variations elucidate adaptation in yak. Mol. Ecol. Resour. 21 (1), 201–211. doi:10.1111/1755-0998.13236
Lan, D., Ji, W., Xiong, X., Liang, Q., Yao, W., Mipam, T. D., et al. (2021). Population genome of the newly discovered Jinchuan yak to understand its adaptive evolution in extreme environments and generation mechanism of the multirib trait. Integr. Zool. 16 (5), 685–695. doi:10.1111/1749-4877.12484
Lan, D., Xiong, X., Mipam, T. D., Fu, C., Li, Q., Ai, Y., et al. (2018). Genetic diversity, molecular phylogeny, and selection evidence of jinchuan yak revealed by whole-genome resequencing. G3 (Bethesda) 8 (3), 945–952. doi:10.1534/g3.118.300572
Lee, S. H., van der Werf, J. H., Kim, N. K., Lee, S. H., Gondro, C., Park, E. W., et al. (2011). QTL and gene expression analyses identify genes affecting carcass weight and marbling on BTA14 in Hanwoo (Korean Cattle). Mamm. Genome 22 (9-10), 589–601. doi:10.1007/s00335-011-9331-9
Lee, W. Y., Park, M. H., Kim, K. W., Song, H., Kim, K. B., Lee, C. S., et al. (2017). Identification of lactoferrin and glutamate receptor-interacting protein 1 in bovine cervical mucus: a putative marker for oestrous detection. Reprod. Domest. Anim. 52 (1), 16–23. doi:10.1111/rda.12744
Li, C., Tian, D., Tang, B., Liu, X., Teng, X., Zhao, W., et al. (2021). Genome Variation Map: a worldwide collection of genome variations across multiple species. Nucleic Acids Res. 49, D1186–D1191. doi:10.1093/nar/gkaa1005
Li, G., Luo, J., Wang, F., Xu, D., Ahmed, Z., Chen, S., et al. (2022). Whole-genome resequencing reveals genetic diversity, differentiation, and selection signatures of yak breeds/populations in Qinghai, China. Front. Genet. 13, 1034094. doi:10.3389/fgene.2022.1034094
Li, H., and Durbin, R. (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25 (14), 1754–1760. doi:10.1093/bioinformatics/btp324
Li, K. X., Hong, W., Jiao, H. W., Wang, G. D., Rodriguez, K. A., Buffenstein, R., et al. (2015). Sympatric speciation revealed by genome-wide divergence in the blind mole rat Spalax. PNAS 112 (38), 11905–11910. doi:10.1073/pnas.1514896112
Li, S., and Li, C. (2021). Proteomics discovery of protein biomarkers linked to yak meat tenderness as determined by label-free mass spectrometry. Anim. Sci. J. 92 (1), e13669. doi:10.1111/asj.13669
Lu, X., Arbab, A. A. I., Zhang, Z., Fan, Y., Han, Z., Gao, Q., et al. (2020). Comparative transcriptomic analysis of the pituitary gland between cattle breeds differing in growth: yunling cattle and leiqiong cattle. Anim. (Basel) 10 (8), 1271. doi:10.3390/ani10081271
Lv, L., Jimenez-Krassel, F., Sen, A., Bettegowda, A., Mondal, M., Folger, J. K., et al. (2009). Evidence supporting a role for cocaine- and amphetamine-regulated transcript (CARTPT) in control of granulosa cell estradiol production associated with dominant follicle selection in cattle. Biol. Reprod. 81 (3), 580–586. doi:10.1095/biolreprod.109.077586
Ma, Z. J., Li, G. Z., Chen, S. M., Han, J. L., and Hanif, Q. (2022). Rich maternal and paternal genetic diversity and divergent lineage composition in wild yak (Bos mutus). Anim. Biotechnol. 33 (6), 1382–1386. doi:10.1080/10495398.2021.1884567
Medugorac, I., Graf, A., Grohs, C., Rothammer, S., Zagdsuren, Y., Gladyr, E., et al. (2017). Whole-genome analysis of introgressive hybridization and characterization of the bovine legacy of Mongolian yaks. Nat. Genet. 49 (3), 470–475. doi:10.1038/ng.3775
Meng, G., Bao, Q., Ma, X., Chu, M., Huang, C., Guo, X., et al. (2022). Analysis of copy number variation in the whole genome of normal-haired and long-haired tianzhu white yaks. Genes (Basel) 13 (12), 2405. doi:10.3390/genes13122405
Mihm, M., Austin, E. J., Good, T. E., Ireland, J. L., Knight, P. G., Roche, J. F., et al. (2000). Identification of potential intrafollicular factors involved in selection of dominant follicles in heifers. Biol. Reprod. 63 (3), 811–819. doi:10.1095/biolreprod63.3.811
Mipam, T. D., Wen, Y., Fu, C., Li, S., Zhao, H., Ai, Y., et al. (2012). Maternal phylogeny of a newly-found yak population in China. Int. J. Mol. Sci. 13 (9), 11455–11470. doi:10.3390/ijms130911455
Patterson, N., Price, A. L., and Reich, D. (2006). Population structure and eigenanalysis. PLoS Genet. 2 (12), e190. doi:10.1371/journal.pgen.0020190
Poplin, R., Ruano-Rubio, V., DePristo, M. A., Fennell, T. J., Carneiro, M. O., Van der Auwera, G. A., et al. (2018). Scaling accurate genetic variant discovery to tens of thousands of samples. bioRxiv, 201178. doi:10.1101/201178
Qi, X., Zhang, Q., He, Y., Yang, L., Zhang, X., Shi, P., et al. (2019). The transcriptomic landscape of yaks reveals molecular pathways for high altitude adaptation. Genome Biol. Evol. 11 (1), 72–85. doi:10.1093/gbe/evy264
Qiu, Q., Wang, L., Wang, K., Yang, Y., Ma, T., Wang, Z., et al. (2015). Yak whole-genome resequencing reveals domestication signatures and prehistoric population expansions. Nat. Commun. 6, 10283. doi:10.1038/ncomms10283
Qiu, Q., Zhang, G., Ma, T., Qian, W., Wang, J., Ye, Z., et al. (2012). The yak genome and adaptation to life at high altitude. Nat. Genet. 44 (8), 946–949. doi:10.1038/ng.2343
Racedo, S. E., Wrenzycki, C., Herrmann, D., Salamone, D., and Niemann, H. (2008). Effects of follicle size and stages of maturation on mRNA expression in bovine in vitro matured oocytes. Mol. Reprod. Dev. 75 (1), 17–25. doi:10.1002/mrd.20770
Rincón, G., Islas-Trejo, A., Casellas, J., Ronin, Y., Soller, M., Lipkin, E., et al. (2009). Fine mapping and association analysis of a quantitative trait locus for milk production traits on Bos taurus autosome 4. J. Dairy Sci. 92 (2), 758–764. doi:10.3168/jds.2008-1395
Shi, L., Liu, L., Lv, X., Ma, Z., Li, C., Li, Y., et al. (2020). Identification of genetic effects and potential causal polymorphisms of CPM gene impacting milk fatty acid traits in Chinese Holstein. Anim. Genet. 51 (4), 491–501. doi:10.1111/age.12936
Smith, G. W., Sen, A., Folger, J. K., and Ireland, J. J. (2010). Putative role of cocaine- and amphetamine-regulated transcript (CARTPT) in dominant follicle selection in cattle. Soc. Reprod. Fertil. Suppl. 67, 105–117. doi:10.5661/rdr-vii-105
Wang, H., Chai, Z., Hu, D., Ji, Q., Xin, J., Zhang, C., et al. (2019). A global analysis of CNVs in diverse yak populations using whole-genome resequencing. BMC Genomics 20 (1), 61. doi:10.1186/s12864-019-5451-5
Wang, K., Hu, Q., Ma, H., Wang, L., Yang, Y., Luo, W., et al. (2014). Genome-wide variation within and between wild and domestic yak. Mol. Ecol. Resour. 14 (4), 794–801. doi:10.1111/1755-0998.12226
Wang, X., Pei, J., Bao, P., Cao, M., Guo, S., Song, R., et al. (2021). Mitogenomic diversity and phylogeny analysis of yak (Bos grunniens). BMC Genomics 22 (1), 325. doi:10.1186/s12864-021-07650-x
Wang, Y., Cai, H., Luo, X., Ai, Y., Jiang, M., and Wen, Y. (2020). Insight into unique somitogenesis of yak (Bos grunniens) with one additional thoracic vertebra. BMC Genomics 21 (1), 201. doi:10.1186/s12864-020-6598-9
Xie, X., Yang, Y., Ren, Q., Ding, X., Bao, P., Yan, B., et al. (2018). Accumulation of deleterious mutations in the domestic yak genome. Anim. Genet. 49 (5), 384–392. doi:10.1111/age.12703
Zhang, C., Dong, S. S., Xu, J. Y., He, W. M., and Yang, T. L. (2019). PopLDdecay: a fast and effective tool for linkage disequilibrium decay analysis based on variant call format files. Bioinformatics 35 (10), 1786–1788. doi:10.1093/bioinformatics/bty875
Zhang, S., Liu, W., Liu, X., Du, X., Zhang, K., Zhang, Y., et al. (2021). Structural variants selected during yak domestication inferred from long-read whole-genome sequencing. Mol. Biol. Evol. 38 (9), 3676–3680. doi:10.1093/molbev/msab134
Zhang, T., Wang, T., Niu, Q., Zheng, X., Li, H., Gao, X., et al. (2022). Comparative transcriptomic analysis reveals region-specific expression patterns in different beef cuts. BMC Genomics 23 (1), 387. doi:10.1186/s12864-022-08527-3
Keywords: Bos grunniens, whole-genome resequencing, genomic diversity, population genetic structure, selection signature
Citation: Zhang S, Li J, Zhao Y, Tang Y, Li H, Song T, An T, Guan J, Li X and Zhang M (2024) Whole-genome resequencing reveals genetic diversity, differentiation, and selection signatures of yak breeds/populations in southwestern China. Front. Genet. 15:1382128. doi: 10.3389/fgene.2024.1382128
Received: 05 February 2024; Accepted: 17 April 2024;
Published: 30 May 2024.
Edited by:
Zhihong Liu, Inner Mongolia Agricultural University, ChinaReviewed by:
Rugang Tian, Inner Mongolia Academy of Agricultural and Animal Husbandry Sciences, ChinaZhixin Chai, Southwest Minzu University, China
Copyright © 2024 Zhang, Li, Zhao, Tang, Li, Song, An, Guan, Li and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ming Zhang, emhhbmdtaW5nQHNpY2F1LmVkdS5jbg==