 Yuqi Shao1,2†
Yuqi Shao1,2† Saisai Yang1,2†Jiafu Li1
Saisai Yang1,2†Jiafu Li1 Lin Cheng1,2Jiawei Kang1,2Juan Liu1,2Jianhong Ma1,2
Lin Cheng1,2Jiawei Kang1,2Juan Liu1,2Jianhong Ma1,2 Jie Duan1,2*
Jie Duan1,2* Yuanzhen Zhang1,2*
Yuanzhen Zhang1,2*- 1Department of Obstetrics, Zhongnan Hospital of Wuhan University, Wuhan, China
- 2Hubei Clinical Research Center for Prenatal Diagnosis and Birth Health, Wuhan, China
Objective: The article aims to provide genetic counseling to a family with two children who were experiencing growth and developmental delays.
Methods: Clinical information of the proband was collected. Peripheral blood was collected from core family members to identify the initial reason for growth and developmental delays by whole exome sequencing (WES) and Sanger sequencing. To ascertain the consequences of the newly discovered variants, details of the variants detected were analyzed by bioinformatic tools. Furthermore, we performed in vitro experimentation targeting SNX14 gene expression to confirm whether the variants could alter the expression of SNX14.
Results: The proband had prenatal ultrasound findings that included flattened frontal bones, increased interocular distance, widened bilateral cerebral sulci, and shortened long bones, which resulted in subsequent postnatal developmental delays. The older sister also displayed growth developmental delays and poor muscle tone. WES identified compound heterozygous variants of c.712A>T (p.Arg238Ter) and .2744A>T (p.Gln915Leu) in the SNX14 gene in these two children. Both are novel missense variant that originates from the father and mother, respectively. Sanger sequencing confirmed this result. Following the guideline of the American College of Medical Genetics and Genomics (ACMG), the SNX14 c.712A>T (p.Arg238Ter) variant was predicted to be pathogenic (P), while the SNX14 c.2744A>T (p.Gln915Leu) variant was predicted to be a variant of uncertain significance (VUS). The structural analysis revealed that the c.2744A>T (p.Gln915Leu) variant may impact the stability of the SNX14 protein. In vitro experiments demonstrated that both variants reduced SNX14 expression.
Conclusion: The SNX14 gene c.712A>T (p.Arg238Ter) and c.2744A>T (p.Gln915Leu) were identified as the genetic causes of growth and developmental delay in two affected children. This conclusion was based on the clinical presentations of the children, structural analysis of the mutant protein, and in vitro experimental validation. This discovery expands the range of SNX14 gene variants and provides a foundation for genetic counseling and guidance for future pregnancies in the affected children’s families.
Introduction
Cerebellar dysfunction may result in ataxia, a neurological disorder that causes a lack of coordinated muscle movement. Ataxia can be classified as acquired or hereditary (Schmahmann, 2004). Hereditary cerebellar ataxia is a genetically heterogeneous group of neurodegenerative diseases primarily characterized by progressive cerebellar ataxia. It accounts for 10%–15% of inherited neurological disorders and has varying prevalence rates among different regions and ethnicities (Klockgether and Paulson, 2011). Autosomal recessive cerebellar ataxias (ARCAs) are early-onset ataxias that are caused by progressive damage to the cerebellum or afferent nerve-related disorders and typically manifest before the age of 20 years. The prevalence of ARCAs is estimated to be three in one hundred thousand. ARCAs patients exhibit multisystemic phenotypes and often present with irregular gait, imbalance, dysphagia, movement disorders, and other motor-related features. These symptoms show extensive inter-and intra-familial variants (Ruano et al., 2014; Pan et al., 2021). The discovery of an increasing number of genes associated with ARCAs has been facilitated by the continuous advancement of molecular technologies. However, approximately 50% of ARCAs patients remain undiagnosed at the molecular level due to the emergence of new gene variants, as well as variants in regulatory regions, epigenetic regulation, gene dosage, or digenic inheritance patterns (Pfeffer et al., 2015; Minnerop et al., 2017). For affected families, it is particularly important to test for the exact causative variants of single-gene disorders and to understand the cause of the disease, which can provide an accurate diagnosis, personalized treatment and an effective follow-up strategy.
Recent studies reported that autosomal recessive spinocerebellar ataxia 20 (SCAR20) is caused by homozygous or compound heterozygous variants in SNX14. SCAR20 is characterized by early-onset cerebellar atrophy, intellectual developmental disorder, ataxia, autism, coarsened facial features, frequent hearing loss, and skeletal abnormalities. This study provided genetic counseling to a family of two clinically affected children who presented with ataxia. After genetic counseling, we carried out whole exome sequencing (WES). Structural analysis and in vitro experimental validation of variant loci were conducted to elucidate the genetic etiology, and the family was ultimately diagnosed with a SNX14 compound heterozygous variant causing SCAR20.
Materials and methods
Subjects
Blood samples were obtained from two patients and their parents who were recruited for prenatal diagnosis at Zhongnan Hospital of Wuhan University. The study received approval from the Ethics Committee of Wuhan University Zhongnan Hospital and obtained written informed consent from the family.
DNA extraction
Peripheral blood of the family was collected, and the whole genome DNA was extracted with a commercial kit (MyGenostics, China). DNA concentration was measured using Qubit 2.0 fluorometer (Thermo Fisher Scientific, USA).
Whole exome sequencing (WES) analysis and bioinformatic prediction
The WES strategy was used to identify genetic variation. For DNA fragment preparation and hybridization capture, the Nextera DNA Flex Pre-enrichment Library Prep (Illumina, San Diego, USA), IDT’s Illumina Nextera DNA Unique Dual Indexes SetA, and Exome Panels-Enrichment Oligos Only (Illumina, San Diego, USA) were employed. The size of DNA fragments was detected using the Qsep100TM automatic nucleic acid protein analyzer. The DNA fragments underwent sequencing using the NovaSeq6000 platform (Illumina, USA) with 151bp double-end sequencing. The raw files were assessed in Fastq format and any data with less than 90% Q30 were excluded.
The sequencing readings were compared to the human reference genome (hg19/GRCh37) using the Burrows-Wheeler Aligner. PCR repeat fragments were removed using Picard V1.57. Variants were identified using Berry Genomics’ Verita Trekker® Variants Detection System and GATK ANNOVAR and Enliven® were then used to annotate and interpret the variants (Wang et al., 2010). The study analyzed patterns of single-gene inheritance, including autosomal dominant, autosomal recessive, and X-linked recessive. Additionally, the study examined mitochondrial DNA variation and possible imprinted gene variants. The harmful prediction of variants discovered using three different types of software, PolyPhen-2, SIFT, and Mutation Taster, was also evaluated. The variants underwent screening and were compared with the ClinVar database, HGMD, and other human genome databases to determine if they were reported as pathogenic. The pathogenicity of the mutant sites was annotated following the American College of Medical Genetics and Genomics (ACMG) criteria (Yang et al., 2019). Validation was performed using Sanger sequencing.
Conservation analysis of SNX14 protein and re-modeling of mutated site
For conservation analysis, the protein sequences of SNX14 from human (Homo sapiens: NP_001990.2), mouse (Mus musculus: NP_034311.2), cattle (Bos taurus: NP_001265517.1), and zebrafish (Danio rerio: NP_001129262.1) were downloaded from NCBI; further, the sequences were inserted into MEGA11 and aligned using the MUSCLE algorithm (Tamura et al., 2021). For remodeling the structure of the mutated SNX14 protein, the amino acid sequence of SNX14 was first compared in SWISS-MODEL to obtain a template with a high degree of similarity. The predicted 3D structure of the SNX14 protein (AF-Q9Y5W7-F1) in the AlphaFoldDB database was highly fitted. Finally, the p.Gln915Leu variant was remodeled in PyMol based on the template downloaded.
Cell culture
HEK293T cells (Procell, China) were cultured in Dulbecco’s modified Eagle’s medium (Hyclone, USA) with 10% fetal bovine serum (Procell, China) in an atmosphere of 5% CO2 at 37°C.
Plasmids construction and transfection
The coding sequence (CDS) of SNX14 (NM_153816.6) was cloned into the pcDNA3.1(+) vector (Invitrogen, USA) to generate SNX14-WT-HA. Based on SNX14-WT-HA, SNX14-c.712A>T-HA and SNX14-c.2744A>T-HA were generated. The constructs were verified by sequence. Lipofectamine 3000 (Thermo Fisher Scientific, USA) was selected for transfection, details were guided following the manufacturer’s protocol. 48 h after transfection, cells were harvested for the following steps.
RNA extraction and quantitative polymerase chain reaction (qPCR)
Total RNA was extracted using a commercial kit (Aidlab, China), and the purity and amount of RNA were assessed using NanoDrop 2000 (Thermo Fisher Scientific, USA). 1ng RNA was used for reverse transcription with HiScript II mix (Vazyme, China), and qPCR was performed with SYBR mix (Vazyme, China) according to the manufacturer’s protocol. GAPDH was selected as the reference gene. The primer sequences of GAPDH were 5′-CATCATCCCTGCCTCTACTGG-3′ (forward) and 5′-GTGGGTGTCGCTGTTGAAGTC-3′ (reverse), and the primer sequences of SNX14 were 5′-TCCCCGAAACCTTGCTGC-3′ (forward) and 5′-GGCTCGTGTCCAACTGCT-3′ (reverse). The 2−ΔΔCt method was used to calculate relative expression.
Western blot
Total proteins were extracted using RIPA lysis buffer (Beyotime, China) and the concentration was measured according to the instructions of the BCA protein quantification Kit (Beyotime, China). Protein samples were separated by 10% SDS-PAGE (Epizyme, China) and transferred to PVDF membranes (Invitrogen, USA). The membranes were blocked with 5% milk for approximately 1 h at room temperature and then incubated in primary antibodies at 4°C overnight. The primary antibodies used were anti-HA (1:1000, C29F4, CST, USA), anti-β-tubulin (1:2000, 10068-1-AP, Proteintech, China). On the second day, TBST-washed membranes were incubated with horseradish peroxidase (HRP)-conjugated secondary antibodies (1:10000, SA00001-2, Proteintech, China) for 1 h at room temperature. Finally, protein bands were visualized using an ECL kit (Vazyme, China).
Statistical analysis
Data were calculated as ratios relative to the corresponding negative controls, presented as means ± SD, and were appropriately analyzed by unpaired t-test with GraphPad Prism (Version 9, California). p-values <0.05 were considered statistically significant.
Results
Clinical findings
The proband was diagnosed with a series of abnormalities during the prenatal period. The mother underwent amniocentesis, which revealed no significant chromosomal or gene copy number variants. The mid-term ultrasound examination indicated that the proband femur and humerus lengths were 2 standard deviations (SD) below the normal values, along with a slightly flattened frontal bone and increased interocular distance. A month later, magnetic resonance imaging (MRI) revealed bilateral enlargement of the extracerebral space in the fetal brain. A late-term systematic ultrasound examination showed that the measurements of the fetal long bones ranged from −1.7 to −4.1SD, suggesting possible fetal cartilage dysplasia (Figure 1A). Tracheal intubation was given to aid respiratory treatment, and continuous gastrointestinal decompression and intravenous nutritional support therapy were administered. This was followed by anti-infective, bleeding prevention, myocardial nutrition, and other symptomatic supportive therapies. A follow-up MRI revealed mild asymmetry in the brain parenchyma, widened brain gyri, and slight enlargement of the subarachnoid space in the right frontal, parietal, and temporal lobes, with a slight leftward shift of midline structures (Figure 1B). The infant was delivered by cesarean section at term pregnancy, after birth, the proband showed some severe symptoms such as dyspnea and three concave signs, with arterial oxygen saturation of 89%. At the age of 3, the proband exhibited poor language skills, only understanding simple commands, and was unable to walk independently.
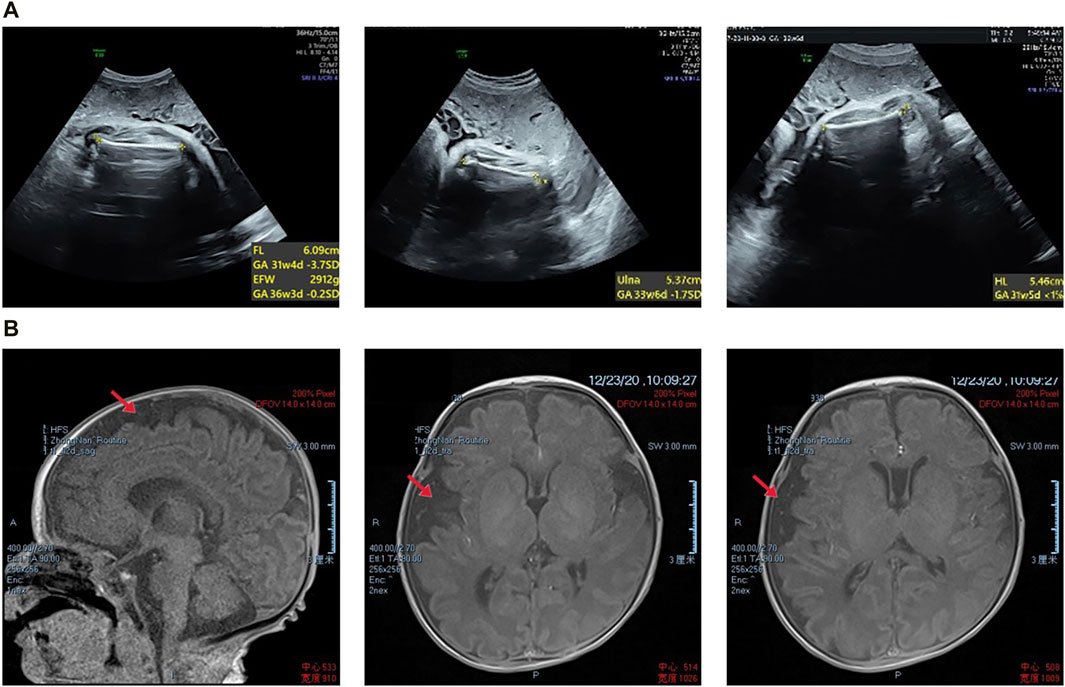
Figure 1. (A) Prenatal transabdominal ultrasound images were taken of the fetus at 31+4 weeks’ gestation, which showed short limbs. (B) The proband’s magnetic resonance imaging results revealed slight asymmetry in the bilateral brain parenchyma, wider gyri, and a slight widening of the subarachnoid space in the right frontal, parietal, and temporal lobes. Additionally, there was a slight leftward tilting of the midline structure.
During the consultation, it was discovered that the proband had an older sister who experienced overall growth delay, with a large head circumference at birth and the ability to walk with assistance at the age of 8. Both affected children exhibited broad foreheads, wide nasal bridges, long eustachian tubes, thick vermilion of the lower lip, and a small and narrow chin (Table 1).
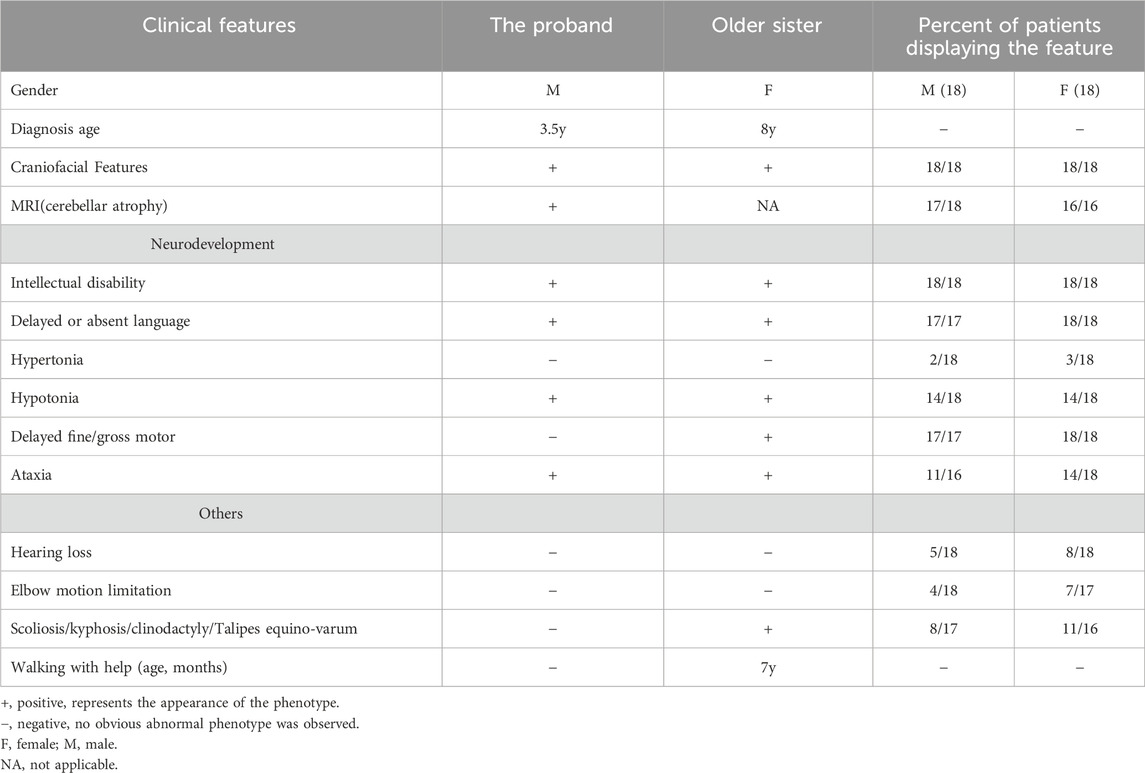
Table 1. Clinical features of patients in this study and the literature. (See Supplementary Table S1 for detailed clinical information).
Variants analysis
Based on OMIM and HGMD, this study successfully identified the proband and his older sister carrying a compound heterozygous variants of the SNX14 gene consisting of c.712A>T (p.Arg238Ter) and c.2744A>T (p.Gln915Leu) by WES, which were respectively originated from the father and mother with normal phenotypes (Figures 2A, B). These variants were confirmed by Sanger sequencing. These missense variants were not present in the HGMD, the single nucleotide polymorphism (SNP) database (dbSNP), the 1000 Genomes Project (TGP) database, or the ClinVar database (Table 2). Furthermore, to our knowledge, these variants have not been described in the universal mutation database SNX14 or reported in any published literature. Based on the ACMG guidelines, the c.712A>T (p.Arg238Ter) variant of SNX14 was predicted to be pathogenic (P) (PVS1 + PM2_Supporting + PP3), and the c.2744A>T (p.Gln915Leu) variant of SNX14 was predicted to be a variant of uncertain significance (VUS) (PM2_Supporting + PP3).
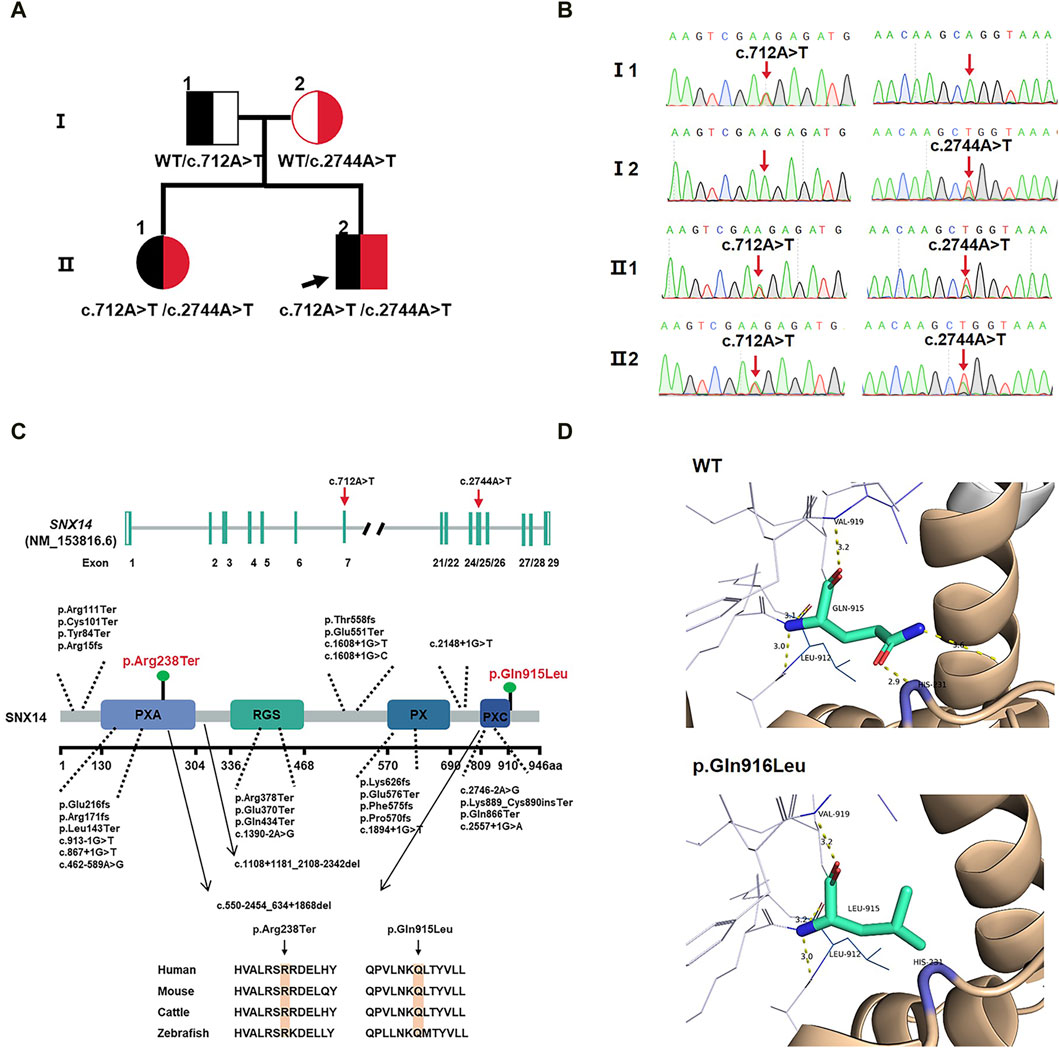
Figure 2. (A) Family pedigree. (B) Sanger sequencing in the proband and II:1 showing compound variants c.712A>T(p.Arg238Ter) and c.2744A>T(p.Gln915Leu) in SNX14, heterozygous in mother and father. (C) SNX14 exons location of variants indicated. The structural model of the SNX14 protein and the position of the two variants identified in this study are shown in blue. Phylogenetic comparison of protein encoded by SNX14 across species. (D) Homology modeling of wild-type and mutant SNX14 c.2744A>T(p.Gln915Leu) variants. The remodeled structure revealed that the variant from Gln to Leu at amino acid 915 broke two hydrogen bonds, which could affect the stability of the SNX14 protein.
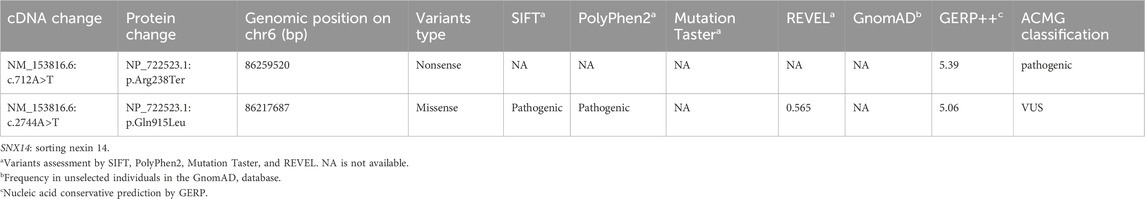
Table 2. SNX14 variants observed in this family.
Protein modeling
The two mutated sites of the SNX14 protein were highly conserved in four species (Figure 2C). We constructed the 3D structure of the p.Gln915Leu variant (Figure 2D). The remodeled structure revealed that the variant from Gln to Leu at amino acid 915 broke two hydrogen bonds, which could affect the stability of the SNX14 protein.
The expression of SNX14
We constructed C-termination HA-tagged plasmids, as shown in Figure 3, c.712A>T (p.Arg238Ter) variant led to early termination of protein translation with no band in the related lane, meanwhile, we found that the mRNA expression of SNX14 was decreased with the mutation of c.712A>T. We surprisingly found that c.2744A>T (p.Gln915Leu) led to the deletion of SNX14 protein expression which was also observed at the mRNA level. Taken together, SNX14 c.712A>T (p.Arg238Ter) and c.2744A>T (p.Gln915Leu) were reclassified as P (PVS1 + PM2_Supporting + PP1 + PP3 + PP4) and VUS (PM2_Supporting + PP1 + PP3 + PP4) by ACMG guidelines (Supplementary Table S2).
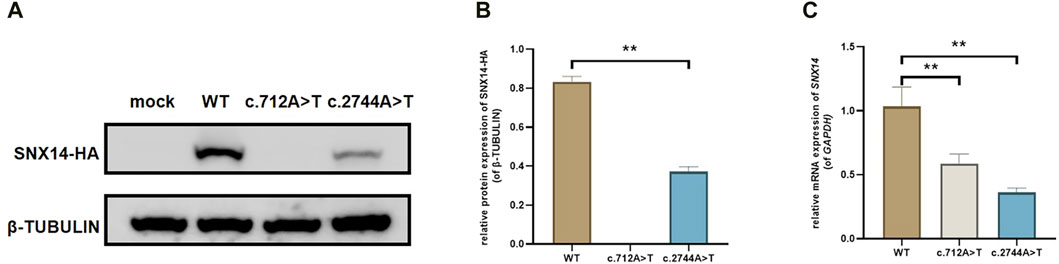
Figure 3. The effect of the variant on SNX14 expression. (A, B) Western blot staining of wild-type and splice site mutant highlighting the relative protein amount. (C) Relative mRNA levels of SNX14 mutant and wild type. The values were the Mean ± SD. *p < 0.05, **p < 0.01, (n = 3).
Discussion
SCAR20 is an early-onset form of ARCAs that was recently reported. It typically occurs in early childhood, although rare cases have been diagnosed during infancy or fetal development (Thomas et al., 2014). The main features of SCAR20 include progressive cerebellar atrophy, ataxia, and severe intellectual disability. Some patients may also exhibit craniofacial abnormalities, such as macrocephaly, a prominent forehead, and thick lips. In addition, patients may exhibit symptoms such as epilepsy, deafness, and skeletal abnormalities (Thomas et al., 2014; Akizu et al., 2015). The primary phenotype of the fetus can only be observed postnatally, and prenatal ultrasound examinations often fail to detect these features. Furthermore, many of the characteristic phenotypes of affected individuals may not be present during the prenatal stage, such as intellectual disability in the absence of gross brain structural anomalies. Currently, there is no description of intrauterine phenotypes associated with SCAR20. This study retrospectively reviewed the prenatal examinations of the proband. The mid-term ultrasound examination showed fetal femur and humerus lengths below -2SD, along with a slightly flattened forehead and increased interocular distance. A month later, a fetal MRI revealed bilateral enlargement of the brain’s extracerebral space. The late-term systematic ultrasound examination revealed that the measurements of the fetal long bones ranged from −1.7SD to −4.1SD. This study contributes to the intrauterine phenotype of SCAR20, highlighting the importance of detailed genetic counseling in cases where prenatal ultrasound suggests fetal limb shortening and the possibility of SCAR20 should be excluded. Both the proband and his older sister in the present study have craniofacial features that are predominantly characterized by broad foreheads, wide nasal bridges, long eustachian tubes, thick vermilion of the lower lip, and a small and narrow chin, and the same facial features were present in all the patients reported (Akizu et al., 2015). In addition, the two affected children in this study and the majority of patients reported in the literature were associated with neurodevelopmental delays such as intellectual disability, delayed or absent language, hypertonia or hypotonia, and ataxia, and MRIs would have shown cerebellar atrophy in the majority of patients, providing imaging support for clinicians (Levchenko et al., 2023). Nevertheless, it should be noted that these clinical signs are progressive and absent in early infancy, making clinical recognition in infants challenging. Thus, there is a greater need to focus on the interrogation of family history and the use of molecular testing techniques. In addition, hearing loss and abnormal skeletal development are also found in patients with SCAR20, but not all patients have these features (Thomas et al., 2014); for example, the proband and his older sister in this study have normal hearing and no significant abnormalities in skeletal development (Table 1; Supplementary Table S1).
Loss-of-function variants in SNX14 cause SCAR20. SNX14 is a member of the sorting nexin (SNX) family, specifically the PXA-RGS-PX-PXC subclass of SNX proteins (Zhang et al., 2021). Research has shown that a higher proportion of patients with early-onset cerebellar atrophy and ataxia carry variants in SNX14 (9.88%) compared to other known pathogenic genes such as GRID2 (2.47%), NPC1 (1.23%), and SETX (1.23%) (Akizu et al., 2015). The majority of these variants are loss-of-function variants occurring primarily in the PX and RGS domains. To date, 30 different pathogenic and potentially pathogenic variants, including nonsense variants, exon deletions, and splice site variants, have been reported in data from 47 patients from 25 families (Maia et al., 2020). Studies on animals have shown that the SNX14 protein is highly expressed in the mouse brain. It gradually increases during neuronal development and maturation and plays a critical role in maintaining normal excitability and synaptic transmission in neurons (Zhang et al., 2021). Knockout mouse models of Snx14 have shown severe motor impairments and cell-autonomous Purkinje cell degeneration, revealing the pathogenic mechanism of cerebellar ataxia (Huang et al., 2014). This study found that the proband and his older sister carried compound heterozygous variants in the SNX14 gene, c.712A>T (p.Arg238Ter) and c.2744A>T (p.Gln915Leu), respectively. These variants were inherited from phenotypically normal parents. According to the ACMG guidelines, the SNX14 c.712A>T (p.Arg238Ter) variant was classified as P (PVS1 + PM2_Supporting + PP3), while the SNX14 c.2744A>T (p.Gln915Leu) variant was classified as VUS (PM2_Supporting + PP3).
The missense variant c.712A>T (p.Arg238Ter) is located in the PXA structural domain of the SNX14 protein. This variant resulted in the premature termination of protein translation (Bryant et al., 2018). In vitro experiments showed decreased expression of SNX14 protein and mRNA levels. SNX14 is an ER-anchored integral membrane protein, and the PXA structural domain is responsible for SNX14 binding to organelle membranes. It is hypothesized that the c.712A>T variant might affect cellular lipid transport, and excess lipids in neuronal cells disrupt the balance of lipid metabolism, which may affect mitochondrial function. On the other hand, the c.2744A>T (p.Gln915Leu) variant is located in none of the important structural domains of the SNX14 protein. The structure of this mutation was remodeled, and it was found that the mutation altered the hydrogen bonding between the amino groups, which may reduce the stability of SNX14. In vitro experiments demonstrated that the expression of SNX14 protein and mRNA was downregulated. Both variants decreased SNX14 expression, and insufficient levels of SNX14 in vivo could result in SCAR20 which may be the result of neuronal lipotoxicity and mitochondrial dysfunction. However, SNX14 c.2744A>T (p.Gln915Leu) remains classified as VUS (PM2_Supporting+PP1+PP3+PP4) following ACMG guidelines. The present study has identified two variants in SNX14, c.712A>T (p.Arg238Ter) and c.2744A>T (p.Gln915Leu), as the genetic cause of SCAR20 in the family. However, as the c.2744A>T is a VUS variant, it is necessary to further study the role of this variant in regulating protein structure and function by in vivo experiments. This will not only expand the spectrum of SNX14 gene variants and the prenatal clinical phenotype of SCAR20 but also guide prenatal diagnosis and clinical genetic consultation.
Currently, ARCAs classification is based on pathogenic genes, pathogenesis, and associated signaling pathways, which include mitochondrial metabolism-related, DNA repair/genome homeostasis-related, and lipid metabolism-related. This classification is useful for treating these diseases. If a treatment is effective for a certain disease, it is expected to have the same effect on other diseases with the same molecular mechanism. However, ARCAs have a multi-systemic phenotype, often with extensive inter- and intra-familial variation. SCAR20 is a disease that is challenging to distinguish from other ARCAs in terms of clinical presentation. The development and advancement of molecular diagnostic techniques have made it possible to definitively diagnose SCAR20 using WES. This is essential for informing parents about their pregnancy options and management, as well as providing more accurate genetic diagnostics of the risk of recurrence in future pregnancies and prior to implantation. Additionally, it allows for more precise genetic counseling for preimplantation genetic diagnosis.
Conclusion
This study reports the identification of the c.712A>T (p.Arg238Ter) and c.2744A>T (p.Gln915Leu) variants in a Chinese family for the first time. Bioinformatics and in vitro experiments have demonstrated that these variants impact the structure and expression of the SNX14 protein, resulting in SCAR20. This expands the spectrum of variants in the SNX14 gene. SCAR20 is rare during the fetal period and childhood and can be difficult to distinguish from other ARCAs. The presentation during the fetal period may be atypical, and the etiology of the fetal abnormalities of the limbs, long bones, and/or facial development should be emphasized. Attention should also be given to the cause of fetal short limbs and/or facial developmental abnormalities. It is important to inquire about the family history of the disease and improve genetic testing to clarify the genetic cause. This will provide a theoretical basis for pregnancy management and guidance for future childbearing.
Data availability statement
The datasets presented in this study can be found in online repositories. The data presented in the study are deposited in the GenBank repository, accession numbers are SAMN40659395.
Ethics statement
The studies involving humans were approved by Zhongnan Hospital of Wuhan University. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants’ legal guardians/next of kin. Written informed consent was obtained from the individual(s), and minor(s)’ legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
Author contributions
YS: Data curation, Writing–original draft. SY: Investigation, Writing–original draft. JiL: Data curation, Resources, Writing–review and editing. LC: Writing–review and editing. JK: Data curation, Writing–review and editing. JuL: Conceptualization, Writing–review and editing. JM: Data curation, Writing–review and editing. JD: Resources, Supervision, Writing–review and editing. YZ: Resources, Supervision, Writing–review and editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This article was funded by Medical technology innovation platform support project of Zhongnan Hospital of Wuhan University (PTXM2021015).
Acknowledgments
We wish to thank the family for participation in the study.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2024.1379366/full#supplementary-material
SUPPLEMENTARY FIGURE S1 | Uncut western blot figure.
References
Akizu, N., Cantagrel, V., Zaki, M. S., Al-Gazali, L., Wang, X., Rosti, R. O., et al. (2015). Biallelic mutations in SNX14 cause a syndromic form of cerebellar atrophy and lysosome-autophagosome dysfunction. Nat. Genet. 47 (5), 528–534. doi:10.1038/ng.3256
Bryant, D., Liu, Y., Datta, S., Hariri, H., Seda, M., Anderson, G., et al. (2018). SNX14 mutations affect endoplasmic reticulum-associated neutral lipid metabolism in autosomal recessive spinocerebellar ataxia 20. Hum. Mol. Genet. 27 (11), 1927–1940. doi:10.1093/hmg/ddy101
Huang, H.-S., Yoon, B.-J., Brooks, S., Bakal, R., Berrios, J., Larsen, R. S., et al. (2014). Snx14 regulates neuronal excitability, promotes synaptic transmission, and is imprinted in the brain of mice. PloS One 9 (5), e98383. doi:10.1371/journal.pone.0098383
Klockgether, T., and Paulson, H. (2011). Milestones in ataxia. Mov. Disord. Official J. Mov. Disord. Soc. 26 (6), 1134–1141. doi:10.1002/mds.23559
Levchenko, O., Filatova, A., Mishina, I., Antonenko, A., and Skoblov, M. (2023). Homozygous deep intronic variant in SNX14 cause autosomal recessive Spinocerebellar ataxia 20: a case report. Front. Genet. 14, 1197681. doi:10.3389/fgene.2023.1197681
Maia, N., Soares, G., Silva, C., Marques, I., Rodrigues, B., Santos, R., et al. (2020). Two compound heterozygous variants in SNX14 cause stereotypies and dystonia in autosomal recessive spinocerebellar ataxia 20. Front. Genet. 11, 1038. doi:10.3389/fgene.2020.01038
Minnerop, M., Kurzwelly, D., Wagner, H., Soehn, A. S., Reichbauer, J., Tao, F., et al. (2017). Hypomorphic mutations in POLR3A are a frequent cause of sporadic and recessive spastic ataxia. Brain a J. Neurology 140 (6), 1561–1578. doi:10.1093/brain/awx095
Pan, Y., Yan, L., Chen, Q., Wei, C., Dai, Y., Tong, X., et al. (2021). Dysfunction of Shh signaling activates autophagy to inhibit trophoblast motility in recurrent miscarriage. Exp. Mol. Med. 53 (1), 52–66. doi:10.1038/s12276-020-00530-6
Pfeffer, G., Pyle, A., Griffin, H., Miller, J., Wilson, V., Turnbull, L., et al. (2015). SPG7 mutations are a common cause of undiagnosed ataxia. Neurology 84 (11), 1174–1176. doi:10.1212/WNL.0000000000001369
Ruano, L., Melo, C., Silva, M. C., and Coutinho, P. (2014). The global epidemiology of hereditary ataxia and spastic paraplegia: a systematic review of prevalence studies. Neuroepidemiology 42 (3), 174–183. doi:10.1159/000358801
Schmahmann, J. D. (2004). Disorders of the cerebellum: ataxia, dysmetria of thought, and the cerebellar cognitive affective syndrome. J. Neuropsychiatry Clin. Neurosci. 16 (3), 367–378. doi:10.1176/jnp.16.3.367
Tamura, K., Stecher, G., and Kumar, S. (2021). MEGA11: molecular evolutionary genetics analysis version 11. Mol. Biol. Evol. 38 (7), 3022–3027. doi:10.1093/molbev/msab120
Thomas, A. C., Williams, H., Setó-Salvia, N., Bacchelli, C., Jenkins, D., O'Sullivan, M., et al. (2014). Mutations in SNX14 cause a distinctive autosomal-recessive cerebellar ataxia and intellectual disability syndrome. Am. J. Hum. Genet. 95 (5), 611–621. doi:10.1016/j.ajhg.2014.10.007
Wang, K., Li, M., and Hakonarson, H. (2010). ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 38 (16), e164. doi:10.1093/nar/gkq603
Yang, K., Shen, M., Yan, Y., Tan, Y., Zhang, J., Wu, J., et al. (2019). Genetic analysis in fetal skeletal dysplasias by trio whole-exome sequencing. BioMed Res. Int. 2019, 2492590. doi:10.1155/2019/2492590
Keywords: whole exome sequencing, growth and developmental delay, SNX14, genetic counseling, prenatal
Citation: Shao Y, Yang S, Li J, Cheng L, Kang J, Liu J, Ma J, Duan J and Zhang Y (2024) Compound heterozygous mutation of the SNX14 gene causes autosomal recessive spinocerebellar ataxia 20. Front. Genet. 15:1379366. doi: 10.3389/fgene.2024.1379366
Received: 06 February 2024; Accepted: 20 March 2024;
Published: 09 April 2024.
Edited by:
Marco Fichera, University of Catania, ItalyReviewed by:
Jorge Diogo Da Silva, University of Minho, PortugalAngelica D’Amore, Boston Children’s Hospital and Harvard Medical School, United States
Copyright © 2024 Shao, Yang, Li, Cheng, Kang, Liu, Ma, Duan and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yuanzhen Zhang, emhhbmd5dWFuemhlbkB3aHUuZWR1LmNu; Jie Duan, amllZHVhbjEzMUBob3RtYWlsLmNvbQ==
†These authors have contributed equally to this work