 Chenling Shen1,2,3†
Chenling Shen1,2,3† Yilin Shen1,2†
Yilin Shen1,2† Weiyi Huang1,2†
Weiyi Huang1,2† Andi Zhang
Andi Zhang Tianyuan Zou1,2,3
Tianyuan Zou1,2,3 Dongye Guo1,2Hao Wang1,2
Dongye Guo1,2Hao Wang1,2 Jichang Wu1,2
Jichang Wu1,2 Haixia Hu1,2*
Haixia Hu1,2* Mingliang Xiang1,2*
Mingliang Xiang1,2* Bin Ye1,2*
Bin Ye1,2*- 1Department of Otolaryngology and Head and Neck Surgery, Ruijin Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai, China
- 2Department of Audiology and Speech-Language Pathology, College of Health Science and Technology, Shanghai Jiao Tong University School of Medicine, Shanghai, China
- 3Department of Otolaryngology and Head and Neck Surgery, Shanghai Children’s Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai, China
Introduction: Primary ciliary dyskinesia (PCD) is a rare heterogeneous disease caused by abnormalities in motile cilia. In this case report, we first analyzed the clinical and genetic data of a proband who was suspected of having PCD on the basis of her clinical and radiological findings.
Methods: Whole-exome sequencing was performed, and a variant in the RSPH4A gene was identified in the proband. Sanger sequencing was used for validation of RSPH4A variants in the proband, her sister, her daughter and her parents. Finally, the phenotypic features of the patient were analyzed, and the current literature was reviewed to better understand the gene variants in PCD related to hearing loss and the clinical manifestations of the RSPH4A variant in PCD.
Results: The chief clinical symptoms of this proband included gradual mixed hearing loss, otitis media, anosmia, sinusitis, recurrent cough and infertility. Her DNA sequencing revealed a novel homozygous T to C transition at position 1321 within exon 3 of RSPH4A according to genetic testing results. This variant had never been reported before. The homozygous variant resulted in an amino acid substitution of tryptophan by arginine at position 441 (p.Trp441Arg). The same variant was also found in the proband’s sister, and a heterozygous pathogenic variant was identified among immediate family members, including the proband’s daughter and parents.
Discussion: A literature review showed that 16 pathogenic variants in RSPH4A have been reported. Hearing loss had only been observed in patients with the RSPH4A (c.921+3_6delAAGT) splice site mutation, and the specific type of hearing loss was not described.
Introduction
Primary ciliary dyskinesia (PCD) is a rare genetic disease related to abnormal ciliary structure or function. It is usually inherited in an autosomal recessive pattern, with few X-linked recessive forms. The prevalence of PCD is estimated to be between 1:2000 and 1:40000. In children with recurrent respiratory infection, the incidence is as high as 5% (Chapelin et al., 1997). PCD is mainly caused by defects in the ciliary outer dynein arms, radial spokes (RSs) or central pair (CP), which lead to severely impaired mucociliary clearance (Edelbusch et al., 2017). The main clinical manifestations of PCD include neonatal respiratory distress, bronchiectasis, recurrent upper or lower respiratory infections, and infertility in adulthood (Goutaki et al., 2016). Kartagener’s syndrome is a subgroup of PCD, presenting with a triad of bronchiectasis, sinusitis, as well as situs inversus. It accounts for approximately half of patients with PCD (Kennedy et al., 2007). In pediatric patients with PCD, respiratory and otologic symptoms are common. The slow progression of PCD may affect the growth and development of children. Since detection methods require a certain level of skill, the diagnosis of PCD is usually delayed. In Europe, the median age at diagnosis of PCD is 5.3 years (Kuehni et al., 2010). In China, the median patient age at diagnosis is 7.0 years (Guan et al., 2021). Therefore, it is necessary for Chinese doctors, especially respiratory physicians, pediatricians, and ENT doctors, to improve their awareness of this disease. Eighty-one percent of patients with PCD have recurrent otitis media, including otitis media with effusion (OME) and acute or chronic otitis media, at an age of 2–3 years (Sommer et al., 2011). Recurrent ear infections and otitis media with effusion can lead to temporary or permanent hearing loss, which may lead to delayed speech development during childhood (Pruliere-Escabasse et al., 2010). More than 90% of children with hearing loss have ever been diagnosed of otitis media for at least one time due to the abnormality of mucociliary clearance in the middle ears and Eustachian tubes (Kreicher et al., 2018). In adult patients with PCD, the median age at diagnosis is 23.5 years (Shah et al., 2016). Adults have a higher prevalence of chronic wet cough, sinusitis, diffuse panbronchiolitis, Pseudomonas aeruginosa infection, and radiological bronchiectasis and show worse lung function (Peng et al., 2022). Therefore, if a patient’s chief symptoms are unexplained ENT manifestations, including chronic rhinitis, recurrent or chronic rhinosinusitis and recurrent otitis media, it is important for specialists to be aware of PCD. The diagnosis of PCD mainly relies on typical clinical manifestations combined with biopsy for ultrastructural examination with electron microscopy, nasal nitric oxide testing, gene detection, high-speed video microscopy and immunofluorescence (Goutaki and Shoemark, 2022). Treatments of PCD are largely referred to the international guidelines of cystic fibrosis, which focus on improving the ability of mucus clearance and anti-bacterial treatments of airway infections. Moreover, latest precision medicine therapies, such as mRNA transcription and gene therapy, have also been explored as possible treatment options. The identification of specific genotypes in PCD and their underlying mechanisms is the first step to achieving personalized medicine. The ultimate goal is to restore ciliary function (Paff et al., 2021).
More than fifty genes have been found to be associated with PCD, including DNAH11, DNAH5, DNAI1, CCDC39, and CCC40 genes. The most frequently mutated genes reported in North America are DNAH5 and DNAI1 genes, followed by DNAH11, CCDC39, and CCDC40 genes. Pathogenic variants in these five genes account for more than 50% of PCD cases (OConnor et al., 2021). In China, the highest incidence of variants occurs in DNAH11 gene, followed by DNAH5, CCDC39, DNAH1, and CCNO genes (Guan et al., 2021).
Here, we report a PCD patient with a novel RSPH4A pathogenic variant in a Chinese family. Studies have shown that RSPH4A localizes to the radial spoke heads of the microtubules in cilia. Variants in RSPH4A mainly destroy the ciliary ultrastructure and lead to the absence of radial spoke heads, which causes dysfunction of mucus clearance (Zhao et al., 2021). A range of motility defects, including reduced ciliary beat frequency, dyskinesia and rotational rather than planar beating patterns, have been reported in PCD patients with RSPH4A variants through nasal biopsies (Frommer et al., 2015). In China, the incidence of variants in RSPH4A gene reported in children with PCD is 1.33% (Guan et al., 2021). To date, a total of five adult PCD patients with RSPH4A variants in China have been reported by Chinese researchers (Bian et al., 2021; Wang et al., 2022; Feng et al., 2023).
Materials and methods
Subjects
Here we described the clinical feature of a young woman with primary ciliary dyskinesia. A three-generation family tree was drawn according to available information obtained from hospital records as well as interview of her immediate family members. The pathogenic variant was also verified on patient’s sibling, parents and daughter. This study was approved by the research ethics committee in Ruijin Hospital. Written informed consents were obtained from all participants.
DNA purification and genetic testing
We performed whole-exome sequencing to detect the presence of any variant. Genomic DNA was extracted from peripheral blood lymphocytes using standard protocols according to previous references (Hu et al., 2022). The sequencing was performed by capturing high-throughput chip technology. The platforms for whole-exome sequencing (NanoWES Human Exome, Berry Genomics Corporation, Beijing, China) were performed on a Illumina NovaSeq 6000 (Illumina, San Diego, United States). Sanger sequencing was used to verify the variants in patients and her family members.
In silico analyses
Single-nucleotide variants (SNVs) and short insertion and deletions (InDels) discovery were performed according to the American College of Medical Genetics and Genomics (ACMG) guidelines (Richards et al., 2015). High-frequency variants (minor allele frequency>0.01) were ruled out combined with using HPO (https://hpo.jax.org/app/), OMIM (https://www.ncbi.nlm.nih.gov/omim/) and GHR (https://ghr.nlm.nih.gov/genetics) database. SNVs and InDels were filtered as follows: 1) Noncoding and intronic variants were excluded; 2) Synonymous missense variants were filtered; 3) Homozygous or compound heterozygous variants were retained. No pathogenic SNV or InDel variant which had been previously reported related to the phenotype of our case was detected. However, one candidate missense variant in RSPH4A was found. A further detailed genetic study of 78 genes (Supplementary Table S1) was carried out by panel sequencing of causal and risk genes known to be associated with ciliary dyskinesia reported by the American College of Medical Genetics and Genomics (ACMG) (Miller et al., 2022). The functional effects and the pathogenicity of the variant were predicted and analyzed using Verita Trekker® variant detection system and Enliven® variant annotation interpretation system independently developed by Berry Genes (Berry Genomics Corporation, Beijing, China) (Hu et al., 2022).
Literature review
A literature review was firstly done using Pubmed for all previously reported articles published between 1975 and 2023 which were related to hearing loss and PCD. A combination of relevant medical subject heading terms along with keywords such as: “cilia,” “ciliary,” “dyskinesia,” “immotile,” “motility,” “hearing,” “hearing loss,” “impairment,” and “deafness” were used (see details of the search strategy in Supplementary Table S2). The reference lists of the relevant articles were also reviewed. Full text articles and conference abstracts of the identified citations were retrieved and reviewed to determine their eligibility for inclusion. In addition, the keyword “RSPH4A” was searched using LOVD3 (Leiden Open Variation Database) (www.lovd.nl/RSPH4A) and PubMed to summarize all pathogenic variants in RSPH4A and related clinical manifestations reported so far.
Results
Clinical findings
The proband is a 31-year-old female patient with gradual hearing loss in the past 3 years accompanied by ear dullness (Figure 1). She also complained of hyposmia and recurrent sinusitis since childhood. Nasal steroids were used to control nasal symptoms (Figure 2A). She had a prolonged wet cough after suffering from COVID-19 and was diagnosed with bronchiectasis by chest computed tomography (Figure 2B). In addition, the patient reported infertility after marriage. Salpingography showed no obvious obstruction in the bilateral fallopian tubes, but they were not as patent as normal (Figure 2C). She finally had a daughter assisted by reproductive technology after fertility consultation. When asking the proband about her family history, she reported that her sister had similar symptoms. The proband’s parents and daughter did not show any symptoms at the time. The parents were not close relatives. No family history of primary ciliary dyskinesia had ever been identified.
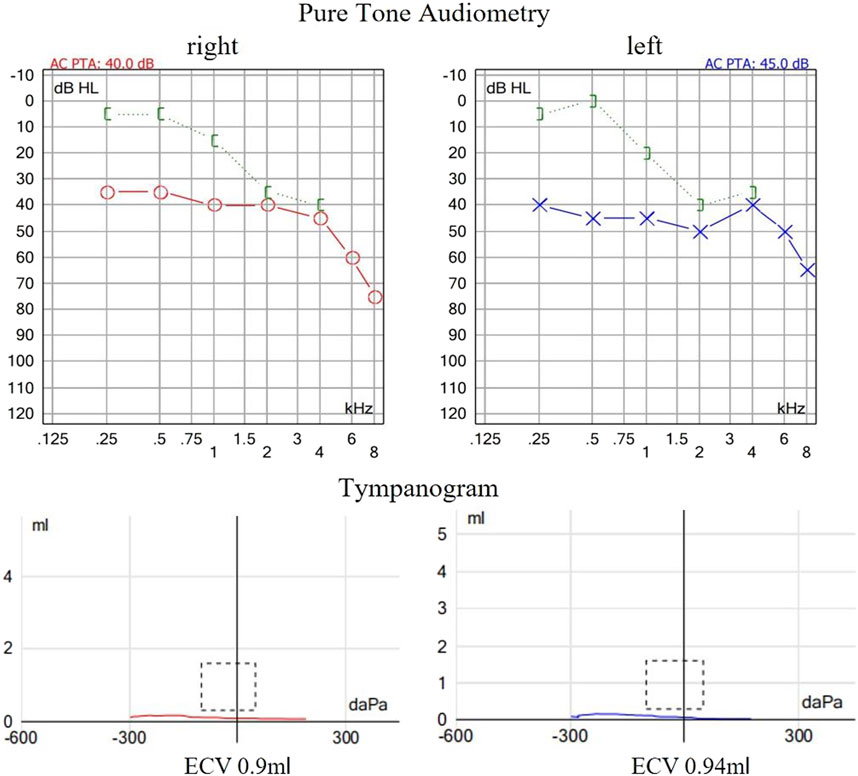
Figure 1. Auditory examination of the proband. The pure tone audiometry revealed bilateral conductive hearing loss at low frequencies and sensorineural hearing loss at high frequencies. Acoustic immittance showed flat tympanogram which indicated bilateral secretory otitis media.
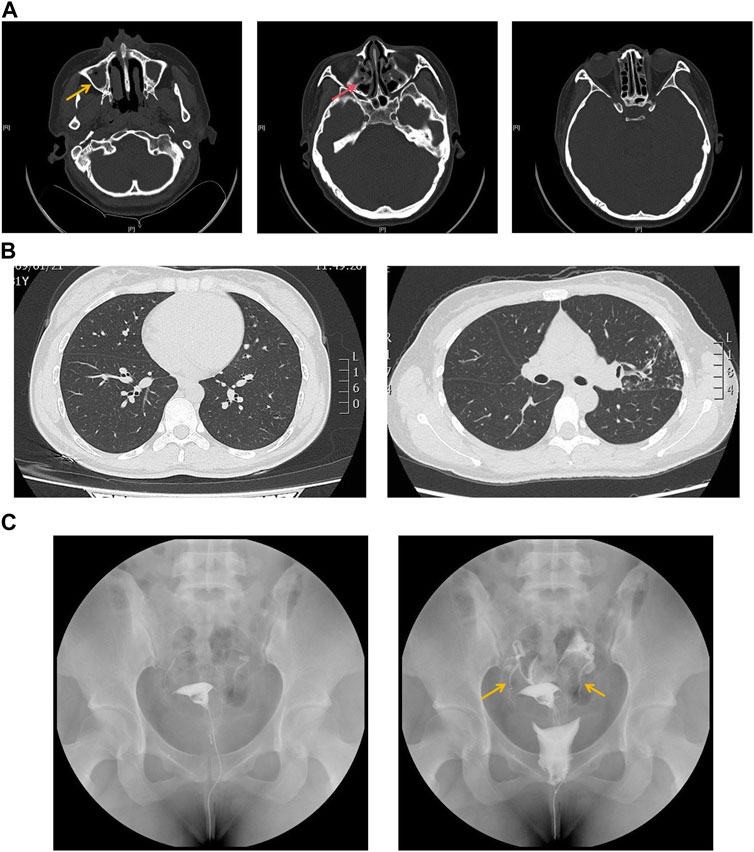
Figure 2. (A) Sinus computed tomography showed inflammation in both maxillary (yellow arrow) and ethmoidal sinuses (red arrow). (B) Chest computed tomography showed bronchiectasis in both sides of lung lobes. (C) Salpingography showed no obstruction in bilateral fallopian tubes but was not patent like as normal (yellow arrow).
Genetic findings and in silico predictions
We sequenced the DNA of patients using Agilent SureSelect Whole Exome capture and Illumina sequencing technology. No pathogenic SNV or InDel variant that had been reported to be related to the phenotype in our patient was detected. However, a homozygous missense variant in exon 3 of RSPH4A was detected. According to ACMG guidelines and recommendations from the ClinGen Sequence Variant Interpretation (SVI) working group (Biesecker et al., 2018), the variant in RSPH4A (NM_001010892.3:exon3:c.1321T>C:p.Trp441Arg) was considered a likely pathogenic variant (PP3+PM2+ PM3+PP1). This variant was further analyzed by REVEL (Rare Exome Variant Ensemble Learner) (Ioannidis et al., 2016), and the results showed that it could cause harmful effects. This variant has not been reported in the 1000 Genomes Project Database, Exome Aggregation Consortium (ExAC) Database or Genome Aggregation Database (gnomAD). According to public databases, RSPH4A (OMIM:612647) variants have been verified to cause primary ciliary dyskinesia. Therefore, this novel variant was considered likely pathogenic combined with the clinical features and family history. Sanger sequencing verified a novel T to C transition at position 1321 within exon 3 of RSPH4A, resulting in an amino acid substitution of tryptophan by arginine at position 441 (Trp441Arg), which led to dysfunction of the RSPH4A protein (Figure 3A).
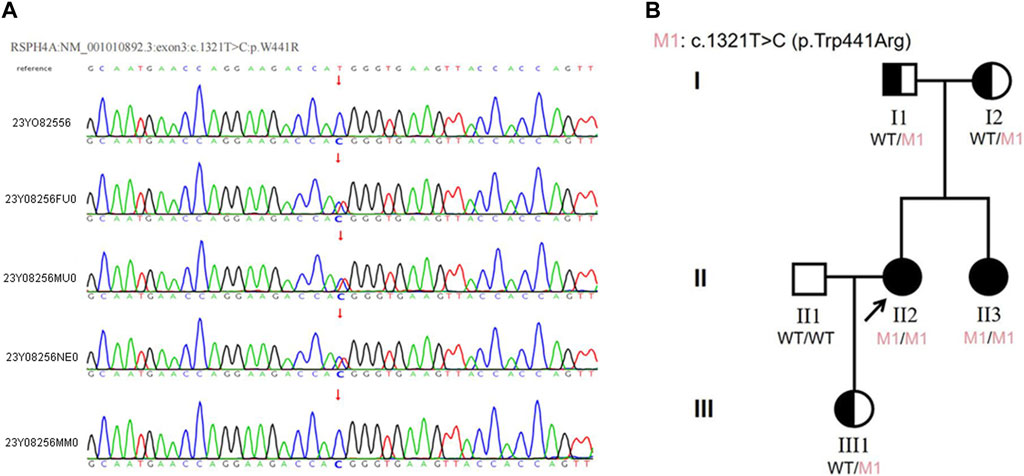
Figure 3. (A)The results of Sanger sequencing in this family. The homozygous variant in the RSPH4A gene was found in both the proband and her sister. The variant was found in the heterozygous state in the proband’s parents and daughter. (23Y08256: proband, 23Y08256FU0: father, 23Y08256MU0: mother, 23Y08256NE0: daughter, 23Y08256MM0: sister). (B) Pedigree of the proband’s family. The RSPH4A variant was found in the proband (II-2). The patient was diagnosed with ciliary dyskinesia at 33 years of age. Arrow indicates proband. One family member-her sister (II-3) had the same clinical symptoms and was also diagnosed with ciliary dyskinesia. Proband and her sister were homozygous for the variant in RSPH4A. Their parents (I1 and I2) and proband’s daughter (III1) all carried the heterozygous variant in RSPH4A.
Verification of RSPH4A in the patients and family members
The proband reported that her sister had the same clinical manifestations. Sanger sequencing was also used to verify the RSPH4A variant in her family members. The homozygous variant of RSPH4A was also detected in the proband’sister. The variant was found in the heterozygous state in the proband’s parents and daughter (Figure 3A). A family pedigree was drawn (Figure 3B).
Literature review of genes related to hearing loss in PCD
No review has identified genes related to hearing loss in PCD. A literature review was performed by searching the keywords, such as “cilia,” “ciliary,” “dyskinesia,” “immotile,” “motility,” “hearing,” “hearing loss,” “impairment,” and “deafness.” Our searches extracted 108 articles from Pubmed, of which 6 articles met our eligibility criteria (Supplementary Figure S1). Our literature review revealed that the hearing loss was different depending on the gene involved (Table 1). Variants in CCDC114 cause sensorineural hearing loss (SNHL) (Li et al., 2019), while variants in CCNO, CDH3, DNAH5 and OFD1 cause conductive hearing loss (CHL) (Bukowy-Bieryllo et al., 2019; Fan et al., 2019; Henriques et al., 2021; Wang et al., 2021; Yang et al., 2022). In addition, the clinical findings were summarized to conclude the different symptoms, age at onset or diagnosis among PCD patients with CHL and SNHL (Table 2). The first visit to a doctor of PCD patients with CHL was always before 10 years of age, while the time of patients with SNHL was often approximately 10 years of age or later. However, the age at diagnosis was almost after 10 years, demonstrating the lack of awareness of this disease by most clinicans. According to the literature review, CHL is the most common type of hearing loss in PCD due to otitis media, acounting for about 88.9% and only one PCD patient had SNHL. All patients with CHL and SNHL had bronchiectasis and rhinosinusitis. Five patients (62.5%) with CHL also suffered from gastro-oesophageal refux and three patients (32.5%) had situs inversus. The patient with SNHL had the unique symptoms of pulmonary artery stenosis, renal fibrosis and patent ductus arteriosus. These findings implied that specific genotypes of PCD could have different underlying pathogenic mechanisms.
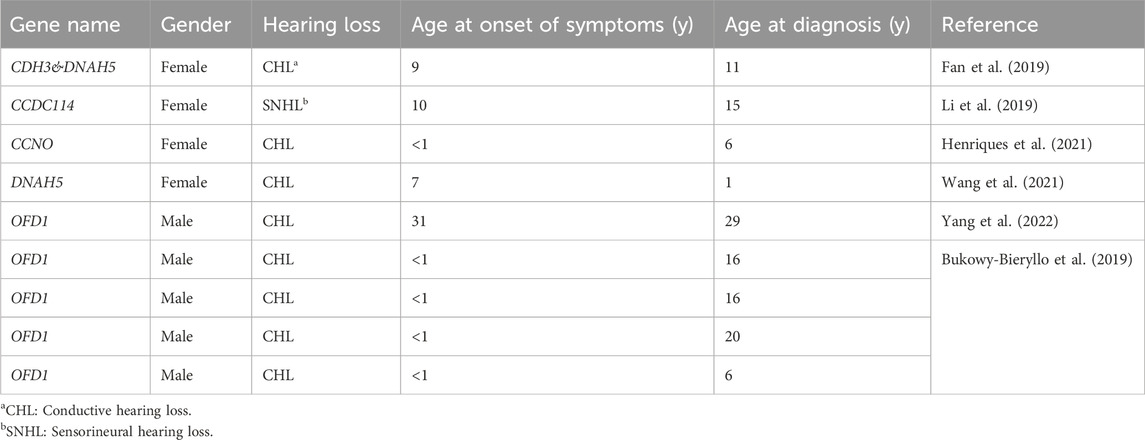
Table 1. Different genes variants and their corresponding type of hearing loss.
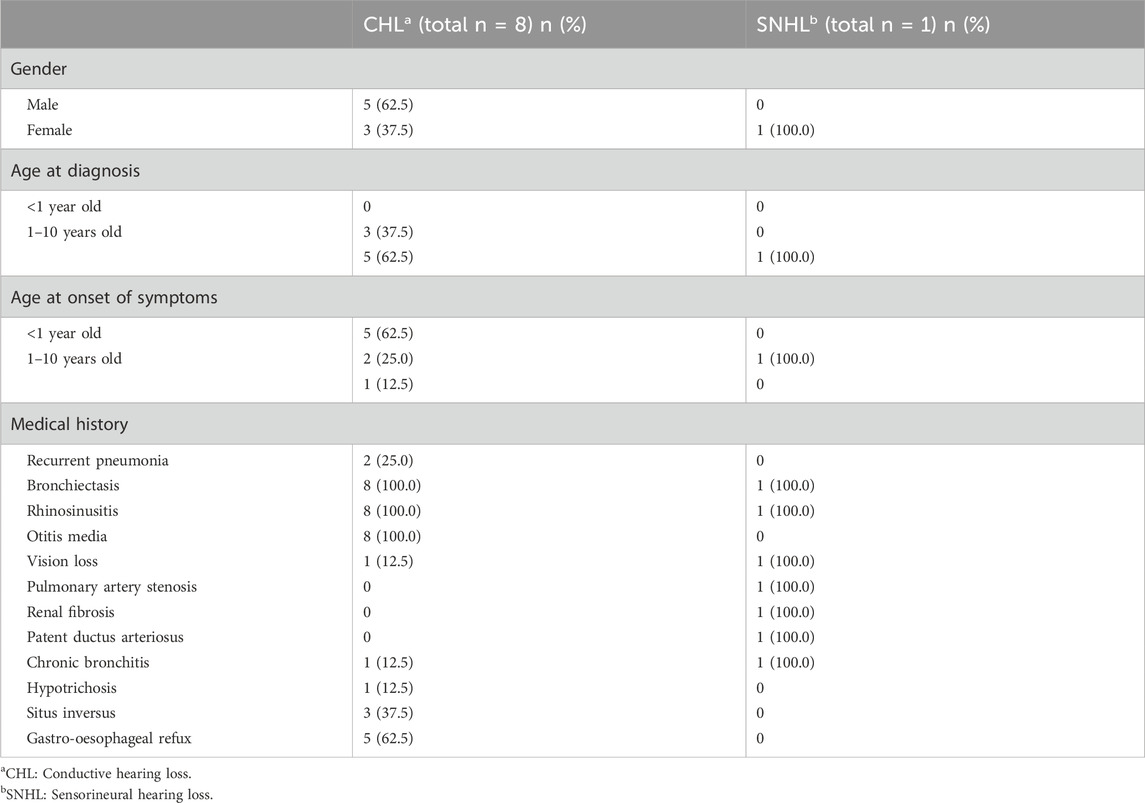
Table 2. Comparison of clinical characteristics in PCD patients with CHL and SNHL.
Literature review of RSPH4A variants in PCD and their clinical symptoms
RSPH4A variants have rarely been reported in China. Up to January 2023, a total of 85 cases with 18 pathogenic variants in RSPH4A had been reported in the Leiden Open Variation Database (www.lovd.nl/RSPH4A). A literature review was performed through the Litvar and PubMed databases and altogether 10 articles from 1875-2023 were selected which described the clinical manifestations of patients with altogether 18 pathogenic variants in RSPH4A gene in details (Supplementary Table S3). We then summarized the clinical features of patients with different varients in RSPH4A for comparison (Table 3). The most common features of RSPH4A variants in PCD patients were bronchiectasis, rhinosinusitis, otitis media, followed by infertility, recurrent pneumonia and chronic bronchitis. Hearing loss was only reported in one patient of RSPH4A (exon1:c.460C>T:p.Gln154) without describing the specific type (Castleman et al., 2009).
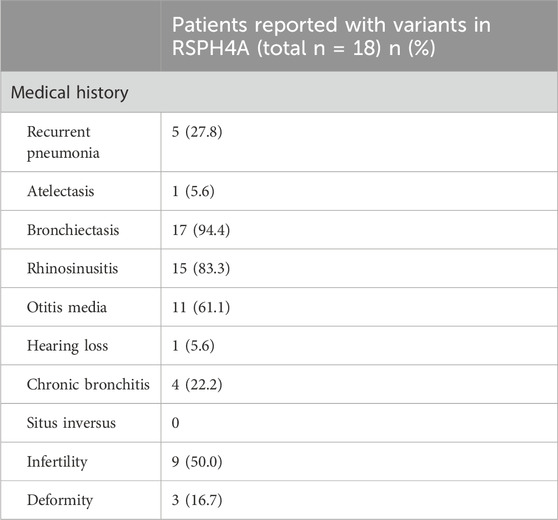
Table 3. Comparison of clinical characteristics of PCD patients with RSPH4A variants.
Discussion
In this study, we reported a patient with gradual hearing loss, otitis media, anosmia, sinusitis, recurrent cough and infertility. She was diagnosed with PCD after performing whole-exome sequencing. A novel homozygous in RSPH4A (NM_001010892.3:exon3:c.1321T>C:p.Trp441Arg) was found. This genetic variant has not been reported before in the literature on individuals with RSPH4A-related disease. Therefore, doctors should be aware of this disease, especially when young patients experience mixed hearing loss. PCD is usually inherited in an autosomal recessive pattern. The pathological basis of PCD is abnormal ciliary movement, which leads to dysfunction of mucociliary clearance in the middle ear and Eustachian tubes. Therefore, PCD patients may have auditory manifestations at an early age. Hearing loss in children will lead to speech or language delay. Therefore, it is necessary to regularly monitor hearing in children with PCD (Shapiro et al., 2016). Symptoms are often alleviated with age, although some patients continue to require hearing aids in adulthood (Lucas et al., 2020). Kreicher et al., 2018 found that CHL was the most common type of hearing loss in PCD patients, although 30% of children had some sensorineural component to their hearing loss. With the treatment of CHL caused by otitis media, hearing thresholds in PCD children can often be improved following conservative management. Tympanogram scores in different age groups also showed a trend toward improvement with age. Most cases resolve by the age of 12 (Wolter et al., 2012). Ventilation tube insertion in children with PCD is controversial because postoperative prolonged otorrhea and persistent perforation of the tympanic membrane are common complications. Hearing aids are recommended in patients with moderate and severe hearing loss (Andersen et al., 2016). In addition to the most common manifestation of secretory otitis media, sensorineural hearing loss can also be found in some PCD patients. We speculate that this may be closely related to abnormal ciliary function in the cochlear hair cells, although to date, no researcher has reported pathological structural and functional changes in hair cells of PCD patients or animal models. PCD is usually caused by motile cilia dysfunction due to variants in genes that encode axonemal motor proteins that are responsible for ciliary beat regulation. However, latest studies have shown that these proteins also play important roles in sensory function (Piatti et al., 2017). Mutation of different proteins may cause corresponding clinical features. From the above literature review, we conclude that pathogenic variants in the CCDC114 gene may cause SNHL, while variants in CCNO, CDH3, DNAH5, and OFD1 genes may cause CHL, suggesting that different types of hearing loss may be closely related to the specific location and function of different proteins. Sometimes manifestations of motor and sensor ciliopathies can overlap.
The difference in the clinical manifestations of PCD usually depends on the specific location of the mutated protein in the cilia. The cross section of cilia shows a typical “9 + 2” configuration, with 9 peripheral microtubule doublets surrounding the central microtubule pair. The outer dynein arm and inner dynein arm are attached to the microtubule doublets and provide motor activity. Radial spoke and nexin-dynein regulatory complexes ensure stability (Horani and Ferkol, 2021). PCD patients whose bronchial biopsies showed the absence of central microtubules under transmission electron microscopy had a higher incidence of otologic features and respiratory tract infections (Tamalet et al., 2001). For example, compared with patients with variants in DNAH5 gene or other outer dynein arm defects, those with variants in CCDC39 or CCDC40 gene have worse lung function and growth indices (Davis et al., 2015; Davis et al., 2019). Patients with variants in CCNO and MCIDAS genes that cause a reduced number of cilia also exhibit more severe pulmonary lesions (Boon et al., 2014). Individuals with variants in genes that encode the ciliary outer dynein arm or radial spoke, including DNAH11, DNAH9 and RSPH1 genes, tend to have less severe respiratory or otologic symptoms (Knowles et al., 2014; Fassad et al., 2018; Shoemark et al., 2021). Therefore, exploring the association between the genotype and phenotype of PCD patients is helpful for the long-term management of disease and accurate prediction of prognosis.
RSPH4A localizes to the radial spoke heads and plays a central role in the three types of RS assembly. It is generally found in the trachea, ependymal tissues and oviduct of mice and modulates the planar beating of motile cilia (Yoke et al., 2020). The research team of De Jesus-Rojas first identified a pathogenic variant in RSPH4A gene (c.921 + 3_921+6delAAGT) in Puerto Rico in 2021 and traced it back to the ancestral haplotype. Sixty-nine percent of the patients had hearing loss, but the specific type of hearing loss was not explicitly reported. Bian et al. reported homozygous variants in the RSPH4A gene (c.667delA, p.S223Afs*15) in a patient with neurofibromatosis and confirmed the diagnosis of PCD at the same time. No hearing loss was found in this patient. Wang et al. reported three novel compound RSPH4A variants: 1) c.2T>C, p. (Met1Thr), 2) c.1774_1775del, p. (Leu592Aspfs*5), and 3) c.351dupT, p. (Pro118Serfs*2), which were related to infertility in PCD patients in China. The symptoms were similar to those reported in our case, but no hearing loss was found in these three families. Hearing was also normal in a child with a variant of c.1454G>A (p.Arg485Gln) in RSPH4A gene (Guan et al., 2021). In the above literature review, the main clinical features of PCD caused by RSPH4A variants were summarized, including bronchiectasis, rhinosinusitis, otitis media and infertility. Only one article reported symptom of hearing loss caused by RSPH4A variant, and not all variants in RSPH4A would cause otitis media and hearing loss. One of the most obvious features the patient exhibited in our case was mixed hearing loss, which had never been reported before in RSPH4A-related PCD. Thus, we consider that different RSPH4A variants exert different effects on cilia movement and function.
We believe that it is important for ENT doctors not only to focus on the patient’s otologic or nasal symptoms but also to pay attention to the clinical manifestations in other organs and the medical history. In this case, we simply connected the patient’s hearing loss and secretory otitis media to her long-term sinusitis at first, but mixed hearing loss was found when more attention was given to the auditory examination. Then, her bronchiectasis and infertility and the similar symptoms in her sister made us further suspect a genetic disease. Finally, gene sequencing confirmed the diagnosis of PCD. Untill now, the treatments of PCD still focus on treating the symptoms and no therapy has been shown to be effective in restoring ciliary function. As a result, expanding the genetic spectrum of this disease will not only raise the awareness of PCD by doctors, but also lead to timely intervention and can put emphasis on therapeutic plan. Different variants will also promote the development of personalized gene therapy in the future.
Data availability statement
The datasets for this article are not publicly available due to concerns regarding participant/patient anonymity. Requests to access the datasets should be directed to the corresponding author.
Ethics statement
The studies involving humans were approved by the Ethical Review Board in Ruijin Hospital affiliated to Shanghai Jiaotong University School of Medicine. The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author contributions
CS: Writing–original draft. YS: Writing–original draft. WH: Writing–original draft, Formal Analysis. AZ: Writing–original draft. TZ: Writing–original draft. DG: Writing–original draft. HW: Writing–original draft. JW: Writing–original draft. HH: Writing–review and editing. MX: Writing–review and editing. BY: Writing–review and editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This study was supported by the National Natural Science Foundation of China (Grant Nos 82101212, 82101209, 82301296, and 82301297), Science and Technology Commission of Shanghai Municipality (Grant Nos 23ZR1440200, 21ZR1440200, and SHDC2020CR1044B-003), Shanghai “Rising Stars of Medical Talents” Youth Development Program and Shanghai Municipal Hospital ENT Specialist Alliance.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2024.1364476/full#supplementary-material
References
Andersen, T. N., Alanin, M. C., von Buchwald, C., and Nielsen, L. H. (2016). A longitudinal evaluation of hearing and ventilation tube insertion in patients with primary ciliary dyskinesia. Int. J. Pediatr. Otorhinolaryngol. 89, 164–168. doi:10.1016/j.ijporl.2016.08.011
Bian, C., Zhao, X., Liu, Y., Chen, M., Zheng, S., Tian, X., et al. (2021). Case report of neurofibromatosis type 1 combined with primary ciliary dyskinesia. Front. Med. 15 (6), 933–937. doi:10.1007/s11684-021-0860-7
Biesecker, L. G., and Harrison, S. M.ClinGen Sequence Variant Interpretation Working Group (2018). The ACMG/AMP reputable source criteria for the interpretation of sequence variants. Genet. Med. 20 (12), 1687–1688. doi:10.1038/gim.2018.42
Boon, M., Wallmeier, J., Ma, L., Loges, N. T., Jaspers, M., Olbrich, H., et al. (2014). MCIDAS mutations result in a mucociliary clearance disorder with reduced generation of multiple motile cilia. Nat. Commun. 5, 4418. doi:10.1038/ncomms5418
Bukowy-Bieryllo, Z., Rabiasz, A., Dabrowski, M., Pogorzelski, A., Wojda, A., Dmenska, H., et al. (2019). Truncating mutations in exons 20 and 21 of OFD1 can cause primary ciliary dyskinesia without associated syndromic symptoms. J. Med. Genet. 56 (11), 769–777. doi:10.1136/jmedgenet-2018-105918
Castleman, V. H., Romio, L., Chodhari, R., Hirst, R. A., de Castro, S. C. P., Parker, K. A., et al. (2009). Mutations in radial spoke head protein genes RSPH9 and RSPH4A cause primary ciliary dyskinesia with central-microtubular-pair abnormalities. Am. J. Hum. Genet. 84 (2), 197–209. doi:10.1016/j.ajhg.2009.01.011
Chapelin, C., Coste, A., Reinert, P., Boucherat, M., Millepied, M. C., Poron, F., et al. (1997). Incidence of primary ciliary dyskinesia in children with recurrent respiratory diseases. Ann. Otol. Rhinol. Laryngol. 106 (10 Pt 1), 854–858. doi:10.1177/000348949710601008
Davis, S. D., Ferkol, T. W., Rosenfeld, M., Lee, H. S., Dell, S. D., Sagel, S. D., et al. (2015). Clinical features of childhood primary ciliary dyskinesia by genotype and ultrastructural phenotype. Am. J. Respir. Crit. Care Med. 191 (3), 316–324. doi:10.1164/rccm.201409-1672OC
Davis, S. D., Rosenfeld, M., Lee, H. S., Ferkol, T. W., Sagel, S. D., Dell, S. D., et al. (2019). Primary ciliary dyskinesia: longitudinal study of lung disease by ultrastructure defect and genotype. Am. J. Respir. Crit. Care Med. 199 (2), 190–198. doi:10.1164/rccm.201803-0548OC
Edelbusch, C., Cindrić, S., Dougherty, G. W., Loges, N. T., Olbrich, H., Rivlin, J., et al. (2017). Mutation of serine/threonine protein kinase 36 (STK36) causes primary ciliary dyskinesia with a central pair defect. Hum. Mutat. 38 (8), 964–969. doi:10.1002/humu.23261
Fan, K. C., Patel, N. A., Yannuzzi, N. A., Prakhunhungsit, S., Negron, C. I., Basora, E., et al. (2019). A unique case of vision loss in a patient with hypotrichosis and juvenile macular dystrophy and primary ciliary dyskinesia. Am. J. Ophthalmol. Case Rep. 15, 100486. doi:10.1016/j.ajoc.2019.100486
Fassad, M. R., Shoemark, A., Legendre, M., Hirst, R. A., Koll, F., le Borgne, P., et al. (2018). Mutations in outer dynein arm heavy chain DNAH9 cause motile cilia defects and situs inversus. Am. J. Hum. Genet. 103 (6), 984–994. doi:10.1016/j.ajhg.2018.10.016
Feng, M., Yu, X., Yue, Y., Zhong, J., and Wang, L. (2023). Novel mutations in RSPH4A and TTN genes lead to primary ciliary dyskinesia-hereditary myopathy with early respiratory failure overlap syndrome. Genes Dis. 10 (3), 743–745. doi:10.1016/j.gendis.2022.10.013
Frommer, A., Hjeij, R., Loges, N. T., Edelbusch, C., Jahnke, C., Raidt, J., et al. (2015). Immunofluorescence analysis and diagnosis of primary ciliary dyskinesia with radial spoke defects. Am. J. Respir. Cell Mol. Biol. 53 (4), 563–573. doi:10.1165/rcmb.2014-0483OC
Goutaki, M., Meier, A. B., Halbeisen, F. S., Lucas, J. S., Dell, S. D., Maurer, E., et al. (2016). Clinical manifestations in primary ciliary dyskinesia: systematic review and meta-analysis. Eur. Respir. J. 48 (4), 1081–1095. doi:10.1183/13993003.00736-2016
Goutaki, M., and Shoemark, A. (2022). Diagnosis of primary ciliary dyskinesia. Clin. Chest Med. 43 (1), 127–140. doi:10.1016/j.ccm.2021.11.008
Guan, Y., Yang, H., Yao, X., Xu, H., Liu, H., Tang, X., et al. (2021). Clinical and genetic spectrum of children with primary ciliary dyskinesia in China. Chest 159 (5), 1768–1781. doi:10.1016/j.chest.2021.02.006
Henriques, A. R., Constant, C., Descalço, A., Pinto, A., Moura Nunes, J., Sampaio, P., et al. (2021). Primary ciliary dyskinesia due to CCNO mutations-A genotype-phenotype correlation contribution. Pediatr. Pulmonol. 56 (8), 2776–2779. doi:10.1002/ppul.25440
Horani, A., and Ferkol, T. W. (2021). Understanding primary ciliary dyskinesia and other ciliopathies. J. Pediatr. 230, 15–22.e1. doi:10.1016/j.jpeds.2020.11.040
Hu, K., Wan, Y., Lee, F. T., Chen, J., Wang, H., Qu, H., et al. (2022). Functional analysis of an intronic FBN1 pathogenic gene variant in a family with marfan syndrome. Front. Genet. 13, 857095. doi:10.3389/fgene.2022.857095
Ioannidis, N. M., Rothstein, J. H., Pejaver, V., Middha, S., McDonnell, S. K., Baheti, S., et al. (2016). REVEL: an Ensemble method for predicting the pathogenicity of rare missense variants. Am. J. Hum. Genet. 99 (4), 877–885. doi:10.1016/j.ajhg.2016.08.016
Kennedy, M. P., Noone, P. G., Leigh, M. W., Zariwala, M. A., Minnix, S. L., Knowles, M. R., et al. (2007). High-resolution CT of patients with primary ciliary dyskinesia. AJR Am. J. Roentgenol. 188 (5), 1232–1238. doi:10.2214/AJR.06.0965
Knowles, M. R., Ostrowski, L. E., Leigh, M. W., Sears, P. R., Davis, S. D., Wolf, W. E., et al. (2014). Mutations in RSPH1 cause primary ciliary dyskinesia with a unique clinical and ciliary phenotype. Am. J. Respir. Crit. Care Med. 189 (6), 707–717. doi:10.1164/rccm.201311-2047OC
Kreicher, K. L., Schopper, H. K., Naik, A. N., Hatch, J. L., and Meyer, T. A. (2018). Hearing loss in children with primary ciliary dyskinesia. Int. J. Pediatr. Otorhinolaryngol. 104, 161–165. doi:10.1016/j.ijporl.2017.11.005
Kuehni, C. E., Frischer, T., Strippoli, M. P. F., Maurer, E., Bush, A., Nielsen, K. G., et al. (2010). Factors influencing age at diagnosis of primary ciliary dyskinesia in European children. Eur. Respir. J. 36 (6), 1248–1258. doi:10.1183/09031936.00001010
Li, P., He, Y., Cai, G., Xiao, F., Yang, J., Li, Q., et al. (2019). CCDC114 is mutated in patient with a complex phenotype combining primary ciliary dyskinesia, sensorineural deafness, and renal disease. J. Hum. Genet. 64 (1), 39–48. doi:10.1038/s10038-018-0514-z
Lucas, J. S., Davis, S. D., Omran, H., and Shoemark, A. (2020). Primary ciliary dyskinesia in the genomics age. Lancet Respir. Med. 8 (2), 202–216. doi:10.1016/S2213-2600(19)30374-1
Miller, D. T., Lee, K., Abul-Husn, N. S., Amendola, L. M., Brothers, K., Chung, W. K., et al. (2022). ACMG SF v3.1 list for reporting of secondary findings in clinical exome and genome sequencing: a policy statement of the American College of Medical Genetics and Genomics (ACMG). Genet. Med. 24 (7), 1407–1414. doi:10.1016/j.gim.2022.04.006
OConnor, M. G., Horani, A., and Shapiro, A. J. (2021). Progress in diagnosing primary ciliary dyskinesia: the North American perspective. Diagn. (Basel) 11 (7), 1278. doi:10.3390/diagnostics11071278
Paff, T., Omran, H., Nielsen, K. G., and Haarman, E. G. (2021). Current and future treatments in primary ciliary dyskinesia. Int. J. Mol. Sci. 22 (18), 9834. doi:10.3390/ijms22189834
Peng, B., Gao, Y. H., Xie, J. Q., He, X. W., Wang, C. C., Xu, J. F., et al. (2022). Clinical and genetic spectrum of primary ciliary dyskinesia in Chinese patients: a systematic review. Orphanet J. Rare Dis. 17 (1), 283. doi:10.1186/s13023-022-02427-1
Piatti, G., De Santi, M. M., Torretta, S., Pignataro, L., Soi, D., and Ambrosetti, U. (2017). Cilia and ear. Ann. Otol. Rhinol. Laryngol. 126 (4), 322–327. doi:10.1177/0003489417691299
Pruliere-Escabasse, V., Coste, A., Chauvin, P., Fauroux, B., Tamalet, A., Garabedian, E. N., et al. (2010). Otologic features in children with primary ciliary dyskinesia. Arch. Otolaryngol. Head. Neck Surg. 136 (11), 1121–1126. doi:10.1001/archoto.2010.183
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of medical genetics and genomics and the association for molecular pathology. Genet. Med. 17 (5), 405–424. doi:10.1038/gim.2015.30
Shah, A., Shoemark, A., MacNeill, S. J., Bhaludin, B., Rogers, A., Bilton, D., et al. (2016). A longitudinal study characterising a large adult primary ciliary dyskinesia population. Eur. Respir. J. 48 (2), 441–450. doi:10.1183/13993003.00209-2016
Shapiro, A. J., Zariwala, M. A., Ferkol, T., Davis, S. D., Sagel, S. D., Dell, S. D., et al. (2016). Diagnosis, monitoring, and treatment of primary ciliary dyskinesia: PCD foundation consensus recommendations based on state of the art review. Pediatr. Pulmonol. 51 (2), 115–132. doi:10.1002/ppul.23304
Shoemark, A., Rubbo, B., Legendre, M., Fassad, M. R., Haarman, E. G., Best, S., et al. (2021). Topological data analysis reveals genotype-phenotype relationships in primary ciliary dyskinesia. Eur. Respir. J. 58 (2), 2002359. doi:10.1183/13993003.02359-2020
Sommer, J. U., Schäfer, K., Omran, H., Olbrich, H., Wallmeier, J., Blum, A., et al. (2011). ENT manifestations in patients with primary ciliary dyskinesia: prevalence and significance of otorhinolaryngologic co-morbidities. Eur. Arch. Otorhinolaryngol. 268 (3), 383–388. doi:10.1007/s00405-010-1341-9
Tamalet, A., Clement, A., Roudot-Thoraval, F., Desmarquest, P., Roger, G., Boulé, M., et al. (2001). Abnormal central complex is a marker of severity in the presence of partial ciliary defect. Pediatrics 108 (5), E86. doi:10.1542/peds.108.5.e86
Wang, L., Wang, R., Yang, D., Lu, C., Xu, Y., Liu, Y., et al. (2022). Novel RSPH4A variants associated with primary ciliary dyskinesia-related infertility in three Chinese families. Front. Genet. 13, 922287. doi:10.3389/fgene.2022.922287
Wang, L., Zhao, X., Liang, H., Zhang, L., and Li, C. (2021). Novel compound heterozygous mutations of DNAH5 identified in a pediatric patient with Kartagener syndrome: case report and literature review. BMC Pulm. Med. 21 (1), 263. doi:10.1186/s12890-021-01586-4
Wolter, N. E., Dell, S. D., James, A. L., and Campisi, P. (2012). Middle ear ventilation in children with primary ciliary dyskinesia. Int. J. Pediatr. Otorhinolaryngol. 76 (11), 1565–1568. doi:10.1016/j.ijporl.2012.07.011
Yang, B., Lei, C., Yang, D., Lu, C., Xu, Y., Wang, L., et al. (2022). Identification of a novel OFD1 variant in a patient with primary ciliary dyskinesia. Pharmgenomics Pers. Med. 15, 697–704. doi:10.2147/PGPM.S365740
Yoke, H., Ueno, H., Narita, A., Sakai, T., Horiuchi, K., Shingyoji, C., et al. (2020). Rsph4a is essential for the triplet radial spoke head assembly of the mouse motile cilia. PLoS Genet. 16 (3), e1008664. doi:10.1371/journal.pgen.1008664
Keywords: mixed hearing loss, novel mutation, primary ciliary dyskinesia, RSPH4A, whole-exome sequencing
Citation: Shen C, Shen Y, Huang W, Zhang A, Zou T, Guo D, Wang H, Wu J, Hu H, Xiang M and Ye B (2024) A novel homozygous RSPH4A variant in a family with primary ciliary dyskinesia and literature review. Front. Genet. 15:1364476. doi: 10.3389/fgene.2024.1364476
Received: 02 January 2024; Accepted: 10 April 2024;
Published: 16 May 2024.
Edited by:
Claudia Gonzaga-Jauregui, Universidad Nacional Autónoma de México, MexicoReviewed by:
Seyed Abbas Rafat,University of Tabriz, IranXunlun Sheng, Ningxia Hui Autonomous Region People’s Hospital, China
Copyright © 2024 Shen, Shen, Huang, Zhang, Zou, Guo, Wang, Wu, Hu, Xiang and Ye. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bin Ye, eWIxMjIzN0ByamguY29tLmNu; Mingliang Xiang, eG1sMTIxMjhAcmpoLmNvbS5jbg==; Haixia Hu, aGh4MTIxOTVAcmpoLmNvbS5jbg==
†These authors have contributed equally to this work