 Yasser Al-Sarraj1,2,3*
Yasser Al-Sarraj1,2,3* Rowaida Z. Taha2
Rowaida Z. Taha2 Eman Al-Dous1,2Dina Ahram2,4Somayyeh Abbasi2Eman Abuazab2
Eman Al-Dous1,2Dina Ahram2,4Somayyeh Abbasi2Eman Abuazab2 Hibah Shaath2Wesal Habbab2Khaoula Errafii2
Hibah Shaath2Wesal Habbab2Khaoula Errafii2 Yosra Bejaoui2
Yosra Bejaoui2 Maryam AlMotawa2Namat Khattab2Yasmin Abu Aqel2Karim E. Shalaby2Amina Al-Ansari2
Maryam AlMotawa2Namat Khattab2Yasmin Abu Aqel2Karim E. Shalaby2Amina Al-Ansari2 Marios Kambouris2,5Adel Abouzohri2
Marios Kambouris2,5Adel Abouzohri2 Iman Ghazal2Mohammed Tolfat6
Iman Ghazal2Mohammed Tolfat6 Fouad Alshaban1,2
Fouad Alshaban1,2 Hatem El-Shanti2,7
Hatem El-Shanti2,7 Omar M. E. Albagha1,2*
Omar M. E. Albagha1,2*- 1College of Health and Life Sciences, Hamad Bin Khalifa University, Doha, Qatar
- 2Qatar Biomedical Research Institute (QBRI), Hamad Bin Khalifa University, Doha, Qatar
- 3Qatar Genome Program, Qatar Foundation Research, Development and Innovation, Qatar Foundation, Doha, Qatar
- 4Quest Diagnostics Nichols Institute, San Juan Capistrano, CA, United States
- 5Pathology & Laboratory Medicine Department, Genetics Division, Sidra Medicine, Doha, Qatar
- 6The Shafallah Center for Children with Special Needs, Doha, Qatar
- 7Department of Pediatrics, Carver College of Medicine, University of Iowa, Iowa City, IA, United States
Introduction: Autism spectrum disorder (ASD) is characterized by aberrations in social interaction and communication associated with repetitive behaviors and interests, with strong clinical heterogeneity. Genetic factors play an important role in ASD, but about 75% of ASD cases have an undetermined genetic risk.
Methods: We extensively investigated an ASD cohort made of 102 families from the Middle Eastern population of Qatar. First, we investigated the copy number variations (CNV) contribution using genome-wide SNP arrays. Next, we employed Next Generation Sequencing (NGS) to identify de novo or inherited variants contributing to the ASD etiology and its associated comorbid conditions in families with complete trios (affected child and the parents).
Results: Our analysis revealed 16 CNV regions located in genomic regions implicated in ASD. The analysis of the 88 ASD cases identified 41 genes in 39 ASD subjects with de novo (n = 24) or inherited variants (n = 22). We identified three novel de novo variants in new candidate genes for ASD (DTX4, ARMC6, and B3GNT3). Also, we have identified 15 de novo variants in genes that were previously implicated in ASD or related neurodevelopmental disorders (PHF21A, WASF1, TCF20, DEAF1, MED13, CREBBP, KDM6B, SMURF1, ADNP, CACNA1G, MYT1L, KIF13B, GRIA2, CHM, and KCNK9). Additionally, we defined eight novel recessive variants (RYR2, DNAH3, TSPYL2, UPF3B KDM5C, LYST, and WNK3), four of which were X-linked.
Conclusion: Despite the ASD multifactorial etiology that hinders ASD genetic risk discovery, the number of identified novel or known putative ASD genetic variants was appreciable. Nevertheless, this study represents the first comprehensive characterization of ASD genetic risk in Qatar's Middle Eastern population.
1 Introduction
Autism spectrum disorder (ASD) is a neurodevelopmental disorder distinguished by irregular social interaction and communication associated with repetitive behaviors and interests (Marini et al., 2020). ASD can be associated with different comorbidities, such as epilepsy, intellectual disability (ID), and attention deficit hyperactivity disorder (ADHD) (Freitag, 2007; Hyman et al., 2020). The worldwide prevalence of ASD is estimated to be approximately 1%, affecting males about four times more frequently than females (Levy et al., 2009; Hyman et al., 2020). Although there are ASD studies published from the Middle East (Amr, 2012; Alallawi et al., 2020; Yousef et al., 2021), the number is significantly disproportionate to the magnitude of the problem. Recently, a cross-sectional study of Qatari children aged 6–11 years surveyed between 2015 and 2018 revealed an ASD prevalence of 1.14% (95% CI: 0.89–1.46) (Alshaban et al., 2019). The Diagnostic and Statistical Manual of Mental Disorders (DSM-V) identifies ASD to include all social communication/interaction impairments and a minimum of two criteria in the restricted and repetitive behaviors (American Psychiatric Association, 2013).
ASD is one of the most heritable neuropsychiatric disorders, where the estimated recurrence rate is 5%–36% (Ozonoff et al., 2011; Werling and Geschwind, 2015). The high contribution of genetic factors to the etiology of ASD is suggested by a concordance rate of 60%–92% among monozygotic twins compared to 0%–10% among dizygotic twins (Bailey et al., 1995). Nevertheless, the recurrence rates among siblings born after two children affected by ASD per family revealed higher recurrence in males (47.5%) than in females (21.1%) and in siblings of females (44.3%) versus siblings of male probands (30.4%) (Werling and Geschwind, 2015).
ASD is clinically and genetically heterogeneous with several known monogenic disorders presenting with ASD symptoms, such as Fragile X syndrome (FMR1), Tuberous Sclerosis (TSC1, TSC2), and Rett syndrome (MECP2), contributing 1%–5% to the ASD etiology (Betancur and Coleman, 2013). Moreover, high throughput genetic testing identified genetic defects in ∼25% of the ASD cases (Yoo, 2015). These include genes involved in the synaptic formation, remodeling, and maintenance (NRX1, CNTN4, DCLK2, CNT- NAP2, TRIM32, ASTN2, CTNTN5, SYN1), neurotransmission (SYNGAP1, GABRG1, CHRNA7), and DNA methylation and chromatin remodeling (MBD5) (Lo-Castro and Curatolo, 2014). Pathogenic variants were primarily recurrent in TSC1, TSC2, NF1, UBE3A, and MECP2 (Betancur, 2011). The vast majority of known ASD genes have a high incidence of de novo pathogenic variants. The accumulating number of distinct rare genetic causes of ASD (Durand et al., 2007; Morrow et al., 2008; Glessner et al., 2009; Wang et al., 2009) suggests a complex and heterogenous genetic architecture, similar to that of intellectual impairment and epilepsy (Jiang et al., 2004).
The genome-wide microarray studies in ASD patients have reported rare microdeletions or microduplications, collectively called copy number variation (CNV), that contribute to the increased risk of ASD and its associated comorbidities (Marshall et al., 2008; Sanders et al., 2011). Several studies of de novo events confirmed multiple CNVs in a considerable fraction (3%–20%) of ASD cases (Christian et al., 2008; Marshall et al., 2008; Pinto et al., 2010; Sanders et al., 2012). It has now been demonstrated that CNVs explained 3%–20% of the ASD genetic etiology over the past 5 years (Roberts et al., 2014; Zarrei et al., 2019; Yap et al., 2021). Our group described 3 CNV regions that were located in genomic regions implicated in ASD. We reported a patient with distal trisomy 10q syndrome presenting with a few previously undescribed physical features, as well as ASD (Al-Sarraj et al., 2014). Also, we described a case report of a male patient affected by ASD with a mosaic trisomy of the pericentromeric region of chromosome 8 and maternal uniparental disomy of the same chromosome (Ahram et al., 2016). Besides, we reported an ASD female patient with intellectual disability (ID) and epilepsy presenting with recurrent microdeletion [del(15) q24.1-q24.2]) (Ahram et al., 2017).
The genome-wide association studies (GWAS) of ASD conducted over the past 15 years were primarily performed on European populations (Ma et al., 2009; Wang et al., 2009; Weiss et al., 2009; Anney et al., 2010; Anney et al., 2012; Connolly et al., 2013; Autism Spectrum Disorders Working Group of The Genomics Consortium, 2017; Grove et al., 2019), and a few were on the Chinese and Korean populations (Cho et al., 2011; Kuo et al., 2015; Xia et al., 2020). Our group reported on GWAS of ASD in the Middle Eastern population of Qatar using a family-based approach (Al-Sarraj et al., 2021). Results showed that common single nucleotide polymorphisms (SNP) are associated with ASD. Although the identified loci did not reach genome-wide significance, many of the top associated SNPs are located within or near genes that have been implicated in ASD or related neurodevelopmental disorders (Al-Sarraj et al., 2021).
There is a rapid rise in studies utilizing genomic sequencing to identify ASD genetic risk factors (Chapman et al., 2015; Yuen et al., 2015; Satterstrom et al., 2020; Kim et al., 2021). Most of these studies investigated and identified rare de novo variants in multiple genes, such as SCN2A, CHD8, ADNP, SHANK3, PTEN, DEAF1, and ANKRD11 (Yuen et al., 2015; Turner et al., 2017; Satterstrom et al., 2020). Lately, there has been a striking increase in the studies aimed at identifying ASD susceptibility genes in multiple populations using genomic sequencing (Besenbacher et al., 2015; Ye et al., 2015; Chang et al., 2019; Tran et al., 2020; Kim et al., 2021). Nevertheless, improved genomic techniques and analyses have expedited the detection of genes involved in ASD (Shen et al., 2014).
In this study, we inspected the contribution of CNVs in 113 patients with ASD, then we investigated the mutational spectrum of ASD patients using whole exome/genome sequencing in the families of 88 ASD cases. This study represents the first comprehensive genetic study of ASD in Qatar’s Middle Eastern population using simplex (trio) and complex families (families with multiple affected individuals).
2 Results
2.1 Characteristics of the study population
The clinical characteristics of the study cohort are shown in Table 1. The study comprised 102 families, of whom 90 were simplex trios, 4 with at least one unaffected sibling, and 8 were multiplex families with more than one affected and/or unaffected sibling. All pedigrees of the study cohort and a summary of the work approach and studied individuals are described in Figure 1. The average age of ASD patients, mother, and father were 8.9, 36.5, and 42.1, respectively (Table 1). In addition, our cohort consisted of 24 ASD individuals who had Epilepsy as a comorbidity.
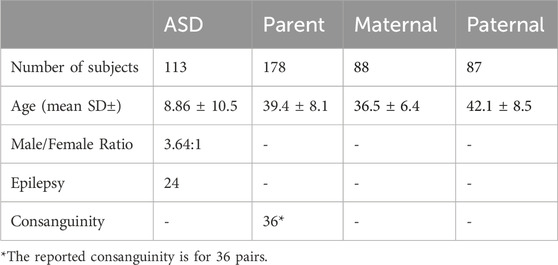
Table 1. Clinical characteristics of the ASD study cohort.
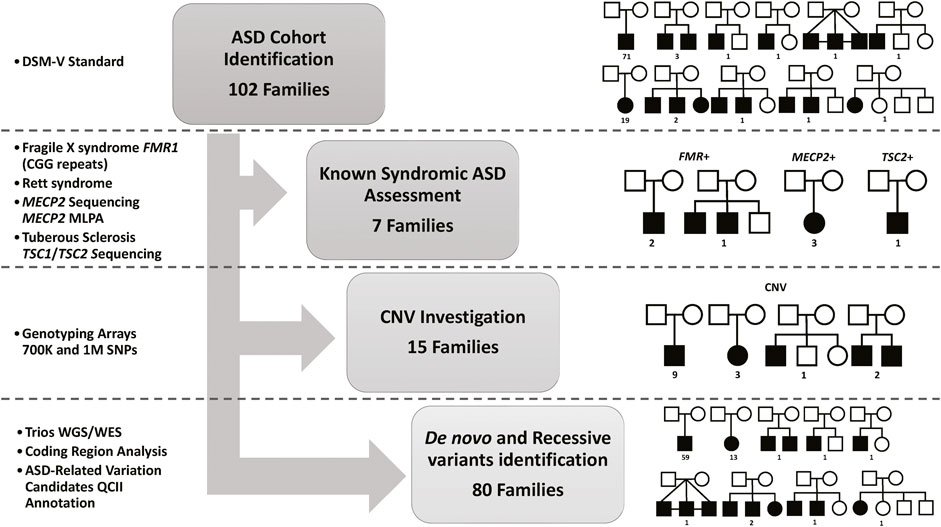
Figure 1. Summary of the Study Cohort and Analysis Approach. DSM-V, the Diagnostic and Statistical Manual of Mental Disorders standards for ASD identification. Fragile X syndrome (Genotyping for CGG expansion in FMR1), Rett syndrome (Sanger Sequencing for MECP2 mutations identification or microdeletion identifications using Multiplex Ligation Dependent Probe Amplification (MLPA)), and Tuberous Sclerosis (Sanger Sequencing for TSC1 or TSC2 mutations identification). Single Nucleotide Polymorphisms (SNP)) markers for Copy Number Variation (CNV) identification. Whole-genome Sequencing (WGS) and Whole-Exome Sequencing (WES) are used for de novo and recessive mutations identification in all trios (both parents and affected children), as well as other affected or healthy siblings for multiplex families. QIAGEN Clinical Insight Interpret (QCII) for variant interpretation and annotation.
2.2 Molecular evaluation of individuals with ASD
We first investigated known syndromic causes of ASD in all identified ASD-affected individuals, namely, fragile X syndrome (caused by CGG expansion in FMR1), Rett syndrome (caused by mutations in MECP2), and Tuberous Sclerosis (caused by mutations in TSC1 or TSC2), as explained in Figure 1. Analysis of the study cohort identified four male subjects with Fragile X syndrome in three families (the repeat length exceeded 200 CGG repeats for all cases), three female subjects with known pathogenic variants in MECP2 (Rett syndrome), and one subject with Tuberous Sclerosis caused by c.5024 C>T/p.Pro1675Leu known pathogenic variant in TSC2. Next, we investigated the contribution of CNVs to ASD etiology in our cohort using SNP genotyping arrays to identify pathogenic CNVs related to ASD, as shown in Figure 1. Analysis of CNVs identified 16 CNVs (7 segmental loss and nine segmental gain) in 16 subjects from 15 families (Supplementary Table S1) that were reported in the DECIPHER database. To illustrate the CNVs identified in our ASD cohort, we compared and aligned examples of the identified CNV regions between the Proband, Mother, and Father for Case 8, Case 11, and Case 16 presented in (Supplementary Figure S1). Detailed information about each detected CNV is presented in Supplementary Table S1). Following the exclusion of classical known ASD disorders and potential CNV regions, our study focused on a cohort of 80 ASD nuclear families. This group consisted of 72 simplex families, each with one affected individual (n = 72 ASD affected), and eight multiplex families. Within the multiplex families, five had additional affected siblings (n = 13 ASD affected), while the remaining three included at least one unaffected sibling (n = 3 ASD affected). This cohort was then subjected to next-generation sequencing (NGS) analysis to identify de novo or novel mutations in genes implicated in the etiology of ASD. These families were evaluated using genome or exome sequencing to identify ASD-related mutations. SNP genotype data analysis identified 28 consanguineous and 52 non-consanguineous families. The male-to-female ratio was 4.2 to 1 (71 males and 17 females), which is consistent with data from previous studies (Yuen et al., 2015; Loomes et al., 2017; Alshaban et al., 2019; Geetha et al., 2019; Samia et al., 2020), and 25% of ASD cases had epilepsy as a comorbidity.
2.3 De novo variant identification in simplex and multiplex ASD families
Next, of the 88 ASD cases, we identified 24 with de novo variants in 22 subjects (Table 2). Two out of the 22 subjects were found to carry two de novo variants. Nevertheless, we identified 24 genes with de novo variants, 19 had established evidence or were predicted to be implicated in ASD, epilepsy, or intellectual disability. For instance, there were nine genes (NLRP5 (Docherty et al., 2015), DRD5 (Nguyen et al., 2014), TCF20 (Vetrini et al., 2019), DEAF1 (Satterstrom et al., 2020), CREBBP (Zheng et al., 2016), KDM6B (Stolerman et al., 2019), ABCA2 (O’ Roak et al., 2012), MYT1L (Blanchet et al., 2017), and CHRNG (Kalınlı et al., 2019) that strongly associated with the Qiagen Clinical Insights (QCI) phenotype-driven ranking scores. Furthermore, we assessed the strength of evidence for our potential candidate identified ASD genes by comparing them with the SFARI database. Candidate genes reported in our patients with no reported evidence of ASD or neurodevelopmental disorders are shown in Supplementary Table S2.
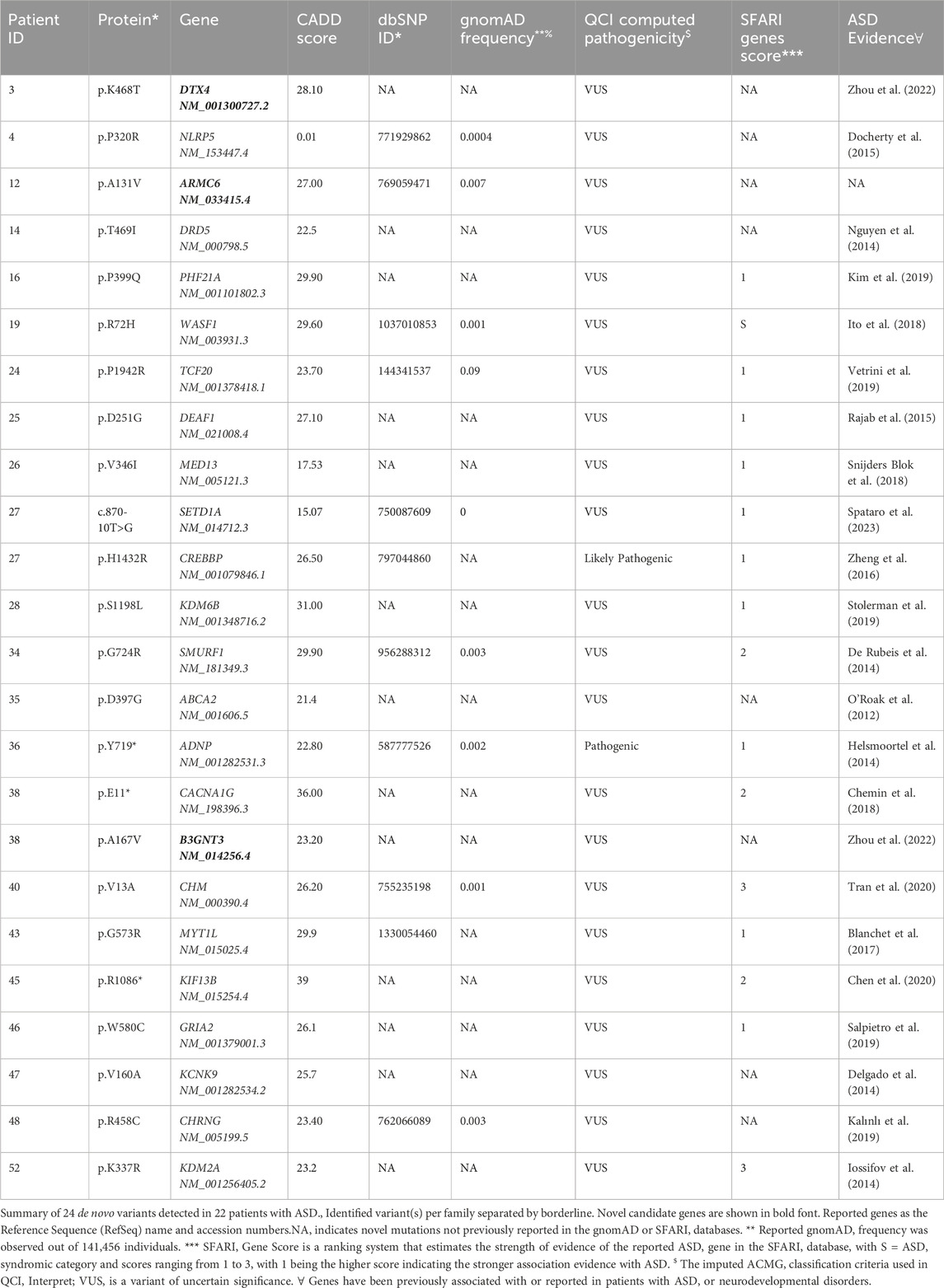
Table 2. De novo variants in ASD patients detected in genes with substantial evidence of involvement with ASD.
In addition, to validate the de novo variants, stringent filtration criteria were implemented on the trio members (Proband, Mother, and Father) to eliminate variants with poor base and mapped quality or variants with low coverage and allele depth. Moreover, de novo variants were only considered if they had high-quality sequencing data from the proband, mother, and father (Supplementary Table S3). The two de novo variants, which were detected in the same ASD subject, and three additional randomly selected variants were tested for validation using Sanger sequencing (Supplementary Figures S2–S4).
Validated de novo variants showed the presence of the variant in the proband but not the parents. Sanger sequencing results confirmed all the five tested variants (Case19 with WASF1 variant (c.215G>A/p.R72H) Figure 2A; case 36 with ADNP variant (c.2157C>A/p.Y719*) Figure 2B; and case34 with SMURF1 variant (c.2170G>A/p.G724R) Supplementary Figure S2). Also, we confirmed the de novo variants in case 38, one in CACNA1G (c.31G>T/p.E11*) and a second in B3GNT3 (c.500C>T/p.A167V) Supplementary Figures S3, S4.
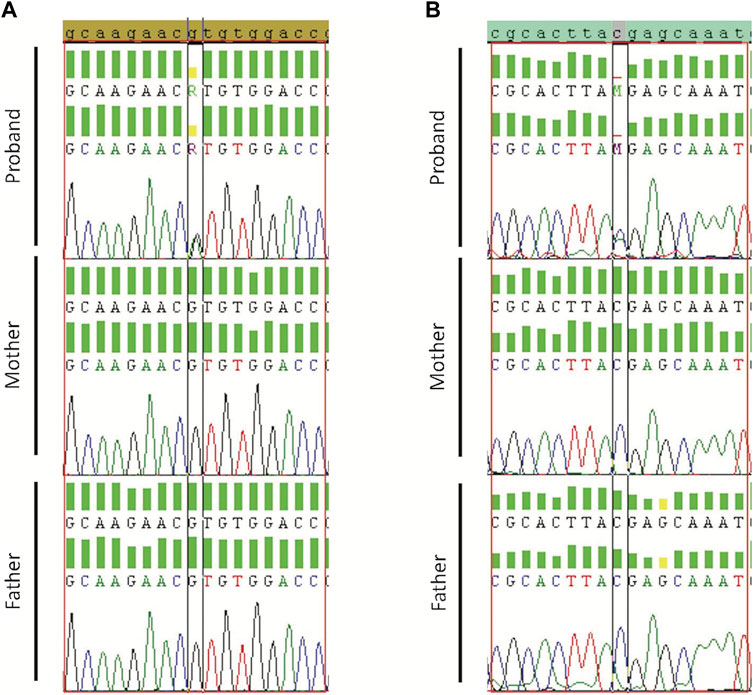
Figure 2. Sanger sequencing results for Proband, Mother, and Father. (A) Validation of WASF1 de novo variant for case19 with WASF1 variant (c.215G>A/p.R72H). (B) Validation of ADNP for case 36 with ADNP variant (c.2157C>A/p.Y719*). Borders indicate de novo alteration (in the child) and wild-type allele (in parents).
2.4 Recessive variants in simplex and multiplex families with ASD
In parallel, ASD-affected individuals from consanguineous parents were processed for recessive and X-linked variant identification (Yu et al., 2013; Cukier et al., 2014). Hence, we identified 21 recessive variants (16 autosomal and 4 X-linked) in 19 ASD subjects (Table 3). Most detected variants were single nucleotide variants (SNVs; n = 17), but we also identified 2 Indels. Interestingly, about 16% of these variants were located in genes implicated in ASD, as revealed by the QIAGEN knowledge base shreds of evidence. Also, our analysis revealed five recessive variants described in genes with strong phenotype-driven ranking scores within the QCI. These include VPS13B (Yu et al., 2013), CADPS2 (Grabowski et al., 2017), LYST (Manoli et al., 2010), TECTA (Toma et al., 2014), and C12orf57 (Akizu et al., 2013).
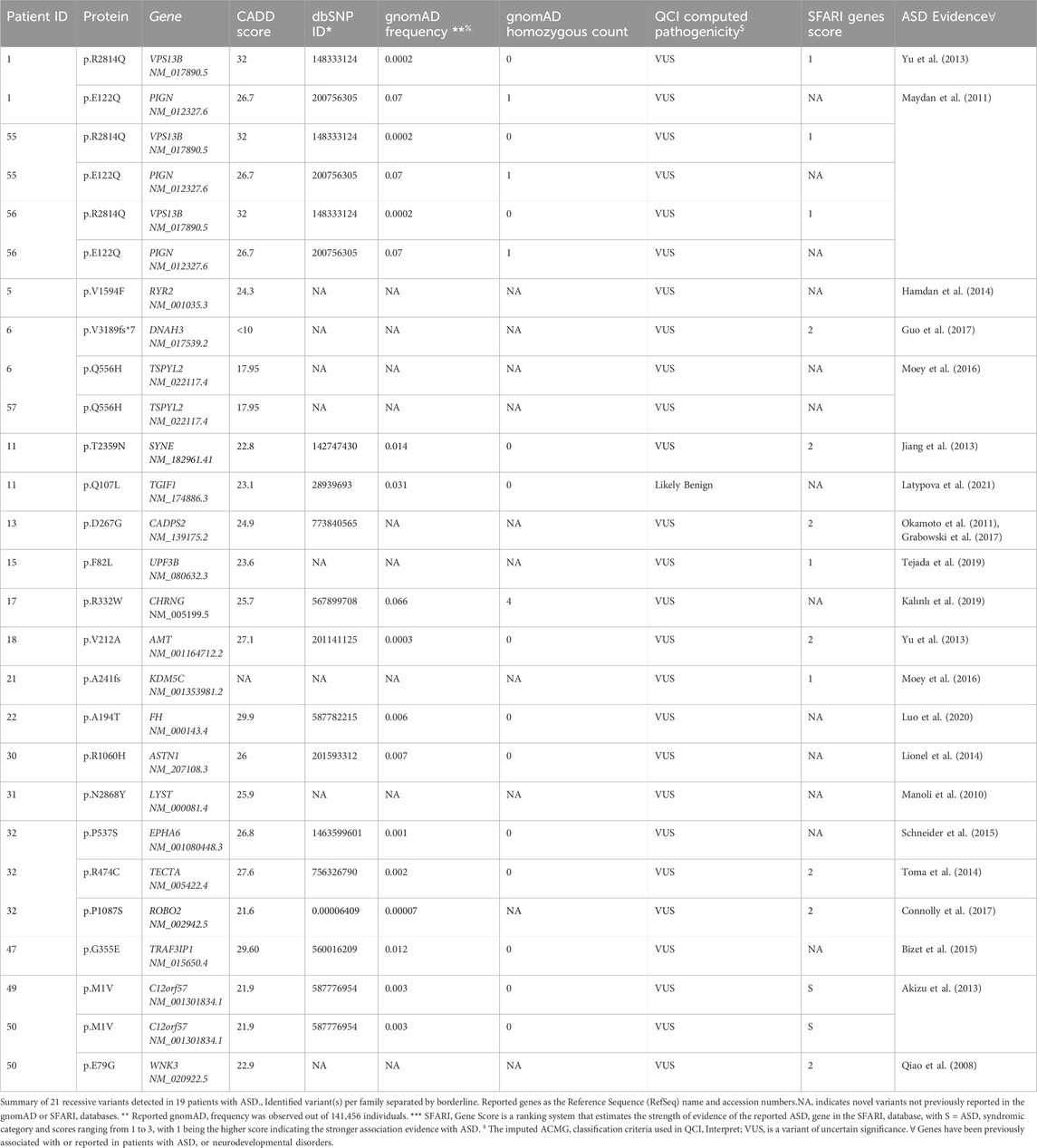
Table 3. Recessive variants in ASD patients detected in genes with substantial evidence of involvement in ASD.
On the other hand, all reported recessive variants were assessed for transmission in trio analysis, and only those showing transmission were considered. The identified variants were then annotated using several available databases, including Genome Aggregation Database (gnomAD) (Karczewski et al., 2020), Exome Aggregation Consortium (ExAC) on non-psychiatric disease samples (Walsh et al., 2017), and Greater Middle Eastern (GME) database (Scott et al., 2016) to identify potential novel findings. Additionally, we evaluated the strength of evidence for our potential ASD candidate genes by comparing them to the SFARI database. Genetic variations in candidate genes with no substantial association or evidence of ASD or neurodevelopmental disorders are shown in Supplementary Table S4. Finally, we assessed if any affected child carries both de novo and recessive variants in simplex and multiplex families with ASD. Of 88 ASD patients, we identified two affected individuals with de novo and inherited variants, as described in (Table 4).
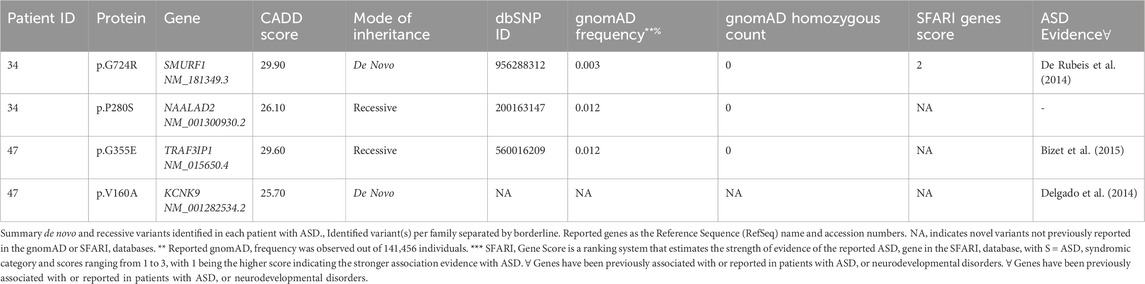
Table 4. De novo and recessive variants in ASD patients detected in genes with substantial evidence of involvement in ASD.
3 Methods
3.1 Study subjects
Three hundred and twenty-nine individuals from 91 simplex and 11 multiplex families were recruited for this study. Study subjects were recruited from the Shafallah Center for Children with Special Needs (Doha, Qatar). ASD diagnosis was ascertained by clinical geneticists according to the Diagnostic and Statistical Manual of Mental Disorders (DSM-V) standards, and the clinical phenotype was delineated in affected individuals, including ASD-associated comorbidities, congenital anomalies, dysmorphic features, the Autism Diagnostic Interview-Revised (ADI-R) and/or Autism Diagnostic Observation Schedule (ADOS), and Intelligence Quotient (IQ) measure when possible. A signed informed consent was obtained from the participants or their Legally Authorized Representatives. The study was approved by the Qatar Biomedical Research Institute’s Institutional Review Board (IRB).
3.2 DNA extraction
About 3 mL of whole blood samples was drawn, in EDTA tubes, from all available family members of the ASD proband. DNA was extracted with the Gentra-Puregene Blood kit from [Qiagen, Valencia, CA], according to the manufacturer’s specifications and protocols. Total DNA amount and purity were measured by NanoDrop 2,200 Spectrophotometers from (ThermoScientific) and Qubit dsDNA BR or HS assay kit and Qubit 2.0 system from [Invitrogen] according to the manufacturers’ guidelines.
3.3 Molecular assessment of known syndromic ASD
Fragile-X syndrome molecular testing was conducted by direct PCR of extracted DNA using primers for FMR1 and fluorescent capillary electrophoresis of the amplified CGG-repeat to determine their number. Alleles above 120 CGGs are challenging to detect using this technique (Chen et al., 2010). If no FMR1 allele is detected in males or a single normal allele is detected in females, the testing continued with a Southern blot analysis. FMR1 was exposed to digestion with two endonuclease enzymes, one methylation-sensitive, followed by Southern hybridization and analysis of the produced fragment (Sofocleous et al., 2009).
The Rett syndrome molecular testing (MECP2) was performed by PCR amplification of extracted DNA followed by Sanger resequencing analysis of the entire coding region of MECP2 and splice sites. Further testing by Multiplex Ligation dependent Probe Amplification (MLPA) (Erlandson et al., 2003) for deletions and duplications was performed for individuals who tested negative via sequencing.
The Tuberous Sclerosis Complex molecular diagnostic testing was performed by PCR of extracted DNA followed by Sanger resequencing using primers for the entire coding region of TSC and TSC2 and splice sites (Milunsky et al., 2009).
3.4 SNP genotyping
SNP genotyping was conducted for all ASD probands and their family members using Illumina’s Infinium Bead Chip Human1M-Duo-v3 (1.119 million SNPs) or HumanOmniExpress-12v1-1 (719,000 SNPs) (Illumina, San Diego, CA) by following the manufacturer’s protocol. SNP genotype data were first analyzed using Genome Studio software followed by PLINK v1.9 (Chang et al., 2015) to perform quality control measures and assess consanguinity. Illumina GenomeStudio-2.0 plug-in was used for the copy number variations analysis by applying two independent algorithms, cnvPartition-v3.2.0 (Tsuang et al., 2010) and QuantiSNP-v2 (Colella et al., 2007).
3.5 Whole-exome and whole-genome sequencing
Exome sequencing library preparation was performed for 298 individuals using the SureSelect Human All Exon v6. The SureSelectXT target size is around 60 Mega Base (MB) which cover the genomic coding and UTRs regions and the splice sites boundaries (site of exon-intron boundaries). Exomes were sequenced on the HiSeq4000 utilizing the HiSeq3000/4000 SBS Kit 300 Cycles reagents. Whole Genome sequencing was performed for 63 samples (21 trios) by BGI genomics (n = 27), or CompleteGenomics (n = 36) (Drmanac et al., 2010).
3.6 Validation by Sanger sequencing
For identified de novo variant validation, primers were designed using the web-based tool Primer3web (https://primer3.ut.ee/). PCR-amplified targets were then subjected to Sanger sequencing using an automated sequencer (Applied Biosystems ABI-3700).
3.7 Data analysis approach for exome/genome sequencing
Data quality control assessment of the generated Fastq files was performed using FastQC (v0.11.5) to estimate quality metrics and evaluate the quality of the raw-sequenced reads. Analysis of sequence data generated from the Hiseq4000 system was done through in-house and optimized scripts using Applied Genome Analysis Tool Kit (GATK-4.0.11) best practices pipeline for short variants (SNPs and Indels). Sequencing reads data were aligned to human genome reference (hg19). PLINK v1.91 (Purcell et al., 2007) was employed to evaluate the quality of the generated variants for all samples (variants call rate, excess heterozygous, and gender mismatch). To assess consanguinity among the recruited families, we computed the inbreeding coefficient for parents using PLINK v1.9, where we determined a subset of independent autosomal SNPs (around 58K pruned SNPs, using a window size of 200 SNPs and LD threshold of r2 = 0.05).
Variants with Mendelian violation were estimated then eliminated by the GATK-4.011 FindMendelianViolation tool to exclude samples with evidence of contamination during the wet lab preparation.
For the genome sequencing performed at BGI and CompleteGenomics data, a list of variants was received for each genome in a standard variant call format (vcf) file. Nevertheless, the WGS data were subjected to the same QC and downstream analysis as the WES data.
The variant interpretation and annotation were processed by QIAGEN Clinical Insight Interpret (QCII) version 9.0 to identify significant variants in genes involved in neurological development as follows described below.
3.8 De novo variant identification
The first set of analyses involved the identification of de novo variants. The QCII-analysis setup and filtrations were initiated to keep all variants with passed upstream pipeline filtering, genotype quality ≥30, and read depth ≥10 in cases and controls. Also, variants outside the top 1% of most exonically variable genes in healthy public genomes (1,000 genomes) were included. Next, de novo variants were excluded for variants with an allele frequency ≥0.1% in the 1,000 genomes project, the NHLBI ESP exomes (All), ExAC, and the gnomAD databases. Variants were then filtered to keep only variants in exons and up to 20 bases into the intron with allele fraction of at least 35 in cases or controls. In this step, variants were annotated for their functional effect and disease-association (clinically relevant) using the QCII platform. The following keywords were used for phenotype: Autism, Autism spectrum disorder, epilepsy, intellectual disability, and mental retardation or combinations of these terms.
3.9 Recessive variant identification
Initially, we performed the recessive variant analysis in the same way as the first three steps applied in the QCII de novo analysis described above. However, we altered the genetic analysis (mode of inheritance) to keep homozygous or hemizygous variants in at least one of the case samples at the variant level. Simultaneously, these variants should not occur in any control samples (homozygous or hemizygous). Besides, the identified homozygous or hemizygous should be consistent with the inheritance transmitted by parents. Finally, the variants were annotated as described in the QCII de novo analysis using the same phenotype keywords.
3.10 QIAGEN Clinical Insight Interpret version 9.0.0.20220826 freeze
The variants reported correspond to the hg19 human genome reference sequence. The databases used in the variant annotation process are listed in Supplementary Table S5.
3.11 SFARI genes database 01-16-2024 release
To assess the strength of the evidence, we annotated the identified genes to the SFARI (Simons Foundation Autism Research Initiative) genes database, which is a comprehensive resource that catalogs genes potentially associated with ASD. Genes in the SFARI Gene database are assigned scores with S for syndromic ASD cases, then defined ranging score from 1 to 3, with 1 being the higher score indicating stronger evidence of association with ASD (Abrahams et al., 2013).
4 Discussion
We present a comprehensive study for ASD in the Middle Eastern population of Qatar using WGS/WES of simplex (trio) and multiplex families. We identified 40 candidate genes for ASD in 39 ASD subjects. Many of the identified genes had previously been implicated in ASD or related neurodevelopmental disorders. We identified three novel de novo variants in genes not previously linked to ASD (DTX4, ARMC6, and B3GNT3). All three de novo mutations were predicted to be damaging and rare, with either never reported in gnomAD and dbSNP or having allele frequencies less than 0.001%. We have defined these genes as highly conceivable suggested novel genes not reported in the SFARI database. Still, the literature showed that these genes are expressed in the human brain and indirectly illustrate a link to ASD or neurodevelopmental disorders (NDD). However, it necessitates future functional studies to ascertain their potential role in ASD or NDD pathogenesis.
Moreover, recent studies investigating exome or genome sequencing data aggregation of ASD cases have identified de novo mutations in DTX4 and B3GNT3 (Fu et al., 2022; Zhou et al., 2022). DTX4 (Deltex E3 ubiquitin ligase 4) is mainly expressed in the brain and is denoted to be involved in the Notch signaling pathway and protein ubiquitination. NLRP4 recruits DTX4, and through their interaction, it promotes the degradation of TBK1(Cui et al., 2012). Recent studies highlighted ubiquitin’s role in neurodevelopmental disorders and reported their monogenic forms to provoke lesions in gene coding, including ubiquitin proteins. These studies hypothesize a possible autoinflammation involvement in neurodevelopmental disorders pathogenesis (Ebstein et al., 2021). ARMC6 (armadillo repeat containing 6), the function of this gene’s protein product has not been determined. However, this gene is expressed in the brain. ARMC6 belongs to the Armadillo repeat-proteins (ARMCs) family which are distributed in eukaryotes and play prominent roles in cell-cell adhesion, intracellular signaling, and cytoskeletal regulation (Song et al., 2019). Also, in situ hybridization revealed a high expression of Armcx6 in mouse neuronal tissue in the developing and adult nervous system (López-Doménech et al., 2012). B3GNT3 (UDP-GlcNAc:betaGal beta-1,3-N-acetylglucosaminyltransferase3) gene is involved in the biosynthesis of poly-N-acetyllactosamine chains and the biosynthesis of the backbone structure of dimeric sialyl Lewis a (Zhang et al., 2015). This gene is mainly expressed in the intestine, salivary gland, and stomach. B3GNT3 is a member of GlcNAc transferases and part of glycobiology enzymes. Several studies have implicated glycobiology-related genes in autism spectrum disorders (Dwyer and Esko, 2016). Also, mutations in N acetylglucosaminyltransferase 1 have been associated with inherited forms of ASDs (Yuen et al., 2017).
Nevertheless, we found de novo variants in 15 genes that have been previously associated with or reported in patients with ASD or neurodevelopmental disorders. These include PHF21A (Kim et al., 2019), WASF1 (Ito et al., 2018), TCF20 (Vetrini et al., 2019), DEAF1 (Rajab et al., 2015), MED13 (Snijders Blok et al., 2018), CREBBP (Zheng et al., 2016), KDM6B (Stolerman et al., 2019), SMURF1 (De Rubeis et al., 2014), ADNP (Helsmoortel et al., 2014), CACNA1G (Chemin et al., 2018), MYT1L (Blanchet et al., 2017), KIF13B (Chen et al., 2020), GRIA2 (Salpietro et al., 2019), CHM (Tran et al., 2020), and KCNK9 (Delgado et al., 2014). Our results are consistent with many previous reports. For example, we identified a de novo variant (c.215G>A/p.R72H, CADD score: 29.6) in WASF1 in an ASD subject who had epilepsy comorbidity from non-consanguineous parents. This variant was confirmed by Sanger sequencing (Figure 2).
Notably, a recent study has reported three nonsense WASF1 de novo mutations in five unrelated cases with Intellectual Disability and Seizures (Ito et al., 2018). Also, WASF1 is considered a Rho family GTPases protein member, which is known to be involved in the pathogenesis of ASD (Guo et al., 2020). ADNP is another example of an essential and well-defined gene associated with ASD. Our study revealed a validated de novo variant in ADNP (c.2157C>A/p.Y719*, CADD score: 22.8), a gene in which multiple studies have reported variants in ASD patients (Helsmoortel et al., 2014; Vandeweyer et al., 2014; Rossi et al., 2017; Arnett et al., 2018). A study by Helsmoortel et al. (Helsmoortel et al., 2014) listed ten ASD patients with different clinical presentations, mainly ID and facial dysmorphism due to ADNP mutations. Besides, they estimated that nearly 1 in 10,000 ASD cases could have ADNP mutations. A recent in vivo study using Adnp knock-out mice showed a reduction in dendritic spine density and a change in gene expression due to Adnp deficiency (Hacohen-Kleiman et al., 2018). Our sequencing data revealed a deleterious de novo variant in TCF20. Consistent with our study, pathogenic variants in this gene have been reported in patients with ASD, intellectual impairment, behavioral abnormalities, and epilepsy (Babbs et al., 2014; Vetrini et al., 2019). Notably, about twenty-five inherited or de novo pathogenic variants in TCF20 were recently observed in affected patients with clinical presentations similar to the phenotype observed in Smith–Magenis syndrome (Babbs et al., 2014; Vetrini et al., 2019). The novel de novo variant in DEAF1 is predicted to be damaging and positioned in a highly conserved area of the genome and is not reported in the gnomAD database. A family-based study in a consanguineous Omani family using WES reported a homozygous variant in DEAF1 in three affected siblings with autism and ID (Rajab et al., 2015). Also, recent studies have reported many deleterious de novo and recessive variants in DEAF1 implicated with ASD genetic risk (Chen et al., 2017; Satterstrom et al., 2020).
Our study provides additional support for inherited genetic risk in ASD. We have identified three multiplex families with affected individuals who shared the same variants. One interesting case is a consanguineous family with identical triplets’ children (three monozygotic male siblings) who had ASD with variable clinical presentation, but all suffered from epilepsy. WES identified a homozygous missense variant in the VPS13B (c.8441G>A/p.R2814Q, CADD score: 32.0) in all three children (Table 3). Both parents were heterozygous for this variant, and all three ASD patients were homozygotes. The variant was infrequent in the gnomAD database (MAF = 0.00002, homozygous count = 0) and predicted to be pathogenic by functional prediction tools. Current evidence indicates a vital role for VPS13B in the normal growth and development of neurons. Pathogenic variants in VPS13B have been reported in patients with Cohen syndrome, a rare autosomal recessive disease characterized by intellectual disability, dysmorphism, and microcephaly (Yu et al., 2013). Sequencing results also revealed a homozygous missense variant in the PIGN (c.364G>C/p.E122Q, CADD score: 26.7) (Table 3) in the three ASD patients from this family. This mutation was rare in the gnomAD database (MAF = 0.0007, homozygous count = 1) and predicted to be pathogenic. The PIGN protein is involved in glycosylphosphatidylinositol (GPI)-anchor biosynthesis, and homozygous pathogenic variants in PIGN have been reported in patients with multiple congenital anomalies, including delayed psychomotor development, hypotonia, and seizures (Maydan et al., 2011; Tian et al., 2022). Our data reveal the complex genetic architecture of ASD and identify two homozygous variants in genes involved in neuronal development. Notably, expression of VPS13B and PIGN varies significantly during differentiation of wild-type iPSC to neurons suggesting a role for these two genes in neuronal development (Lizio et al., 2019). The variant in PIGN could explain the observed seizures in the patients, but further functional studies will be required to confirm this finding.
Also, our exome data presented another extended family with two affected boys (only one of them has seizures at 30 months) and one healthy sister. Interestingly, a homozygous missense variant in C12orf57 (Table 3) was detected in both affected brothers but not in their healthy sister. This variant has been previously reported (c.A1G/p.M1V, CADD score: 21.9) in several consanguineous Saudi and Kuwaiti patients who displayed diverse clinical characteristics such as a profound global development delay, autistic features, and epilepsy (Akizu et al., 2013; Salih et al., 2013; Zahrani et al., 2013). Besides, C12orf57 is associated with Temtamy syndrome comprised of hypotonia, moderate to severe ID with ASD features, corpus callosum hypoplasia, seizures, and microphthalmia (Akizu et al., 2013; Salih et al., 2013; Zahrani et al., 2013; Wang et al., 2019). Notably, our homozygosity mapping analysis showed that C12orf57 falls within a 22 Mb run of homozygosity on chromosome 2 in both affected brothers. WES for the affected ASD patient with epilepsy revealed a deleterious X-linked variant in WNK3 (c.236A>G/p.E79G, CADD score: 22.9) (Table 3) but this variant was not detected in his brother who had ASD without epilepsy. WNK3 was reported to be partially deleted in two brothers with autism, ID, and cleft lip/palate and a maternally inherited microdeletion (Qiao et al., 2008). WNK3 protein was co-localized with Cl2 transporters (NKCC1 and KCC) and regulate the chloride (Cl2) ion transportation that revealed a critical role in the excitability of gamma-Aminobutyric acid (GABA) receptors in neurons (Kahle et al., 2005).
The third multiplex family includes two affected boys from a consanguineous marriage who share a likely pathogenic variant in TSPYL2 (c.1668G>C/p.Q556H) on the X chromosome. TSPYL2 was described in many patients with Xp11.2 microduplications and neurodevelopmental disorders (Moey et al., 2016). Furthermore, one affected individual had an additional rare variant (c.9565delG/p.V3189fs*7) in dynein axonemal heavy chain 3 (DNAH3) (Table 3). Loss of function variants in DNAH3 has been described in large-scale studies of patients with ASD (the Simons Simplex Collection (Krumm et al., 2015) and in a Chinese study (Guo et al., 2017)).
Collectively, the evidence from this study suggests that 75% of multiplex families (6 out of 8) showed at least two siblings sharing the same variants. This is higher than previous studies (that reported about 30% of affected siblings carrying same variants relevant to ASD risk) which is likely due to the prevalent consanguinity. Besides, there were novel genetic findings that have an additional individual risk effect to ASD.
Furthermore, data from one ASD trio showed a likely pathogenic homozygous variant in CADPS2. The identified variant was in a highly conserved genomic area with no recorded allele frequencies in gnomAD or dbSNP databases. This gene is located in the 7q31 region that is has been reported to be deleted in patients with ASD (Okamoto et al., 2011; Grabowski et al., 2017). Studies using animal models showed that Cadps2-knockout mice exhibited a brain impairment and autistic-like behavioral phenotypes (Sadakata et al., 2007). Besides, CADPS2 variant screening in Italian patients with ID and with ASD subjects have shown parental inheritance for deleterious variants with a suggestion of parent-of-origin effect (Bonora et al., 2014).
It is well known that ASD complexities may be subjected to the multiple heterogeneity threshold or dosage (Schaaf et al., 2011; Cristina et al., 2013), where affected subjects may inherit different genetic risks (either novel or de novo mutations) in known genes, as well as chromosomal anomalies (CNV). Together these genetic risks may contribute as additive interacting factors to ASD-threshold (Cook and Scherer, 2008). Significantly, we recognized such multifactorial effects in several families (Table 4). For instance, one simplex family had variants in two candidate genes; a de novo p.G727R in SMURF1 which was validated by Sanger Sequencing (Supplementary Figure S1), and a recessive p.P635L in NAALAD2. SMURF1 encodes a ubiquitin ligase protein that is essential as a regulatory receptor for the SMAD proteins in the bone morphogenetic protein (BMP) pathway (Cui et al., 2011). Also, SMURF1 was found to play a role in controlling cell motility, signaling, and polarity (Zhu et al., 1999). Moreover, SMURF1 was significantly associated with ASD risk as a de novo splice site variant, as recently described (De Rubeis et al., 2014) by the Autism Sequencing Consortium patient in the Hartwell Autism Research and Technology Initiative (iHART) cohort (Ruzzo et al., 2019). Regarding the NAALAD2 gene, the NAALAD2 protein is considered one of the N-acetylated alpha-linked acidic dipeptidase (NAALADase) gene families. NAALADase was proposed to have an associated role with N-acetyl-L-aspartate-L-glutamate (NAAG) in the central nervous systems. But there is no clear link between NAALAD2 and ASD or any neurodevelopmental disorders (Pangalos et al., 1999).
Another example of oligo-heterogeneity was observed in a consanguineous family with a child affected with ASD and early onset epilepsy (9 months). The WES data identified a de novo variant in KCNK9 (c.479T>C/p.V160A, CADD score: 25.7) (Table 4). KCNK9 (potassium channel subfamily K member 9) is a channelopathy protein associated with Birk-Barel syndrome. This gene is imprinted with paternal silencing, and the mutation entirely abolishes the potassium channel activity when acting as a homodimer and when functioning as a heterodimer (Barel et al., 2008). Two recent studies have described patients with developmental delay and central hypotonia with de novo variants in KCNK9 by exome sequencing (Graham et al., 2016; Šedivá et al., 2020). Nevertheless, the child had also a likely pathogenic homozygous variant in TRAF3IP1 (c.1064G>A/p.G355E, CADD score: 29.6). The variant is rare, with reported allele frequencies of 0.01% in gnomAD and 0.02% in the 1,000 genomes. TRAF3IP1 is an autosomal recessive ciliopathy gene associated with Senior-Loken syndrome nine that is represented by early-onset nephronophthisis and pigmentary retinopathy (Bizet et al., 2015). Besides, a study reported five consanguineous families with four homozygous recessive and one compound heterozygous mutations using exome sequencing, where few families had affected subjects with global developmental delay and severe cognitive impairment.(Bizet et al., 2015).
Our study also identified a novel missense mutation in SUOX (in case 33; c.1156G>A/p.V386M, CADD score: 26.7 Supplementary Table S4). This gene encodes an enzyme, Sulfite oxidase, that catalyzes the oxidation of sulfite to sulfate (Irreverre et al., 1967). The deficiency of this enzyme has been associated with hereditary metabolic disorders and lethal neurological complications that occur at an early age (Rupar et al., 1996). Nevertheless, several previously published variants of SUOX were reported in patients with seizures, feeding difficulties, microcephaly, and brain atrophy.(Rupar et al., 1996; Johnson et al., 2002). Further work needs to be done, including the follow-up of the subject with this identified inborn metabolic errors for re-examining and the testing of sulfite oxidase deficiency or other metabolic profiling.
This study highlighted many genes that warrant further investigation. Future studies should aim at functional studies to characterize the identified genes deeply using animal models and human-induced pluripotent stem cells (hiPSC). The functional studies for the identified putative ASD genes using the known animal models in mice (Ergaz et al., 2016), Drosophila (Bellosta and Soldano, 2019), or zebrafish (Meshalkina et al., 2018); will help us understand the etiology, pathogenesis, and treatment of human ASD. Furthermore, taking advantage of patient-derived stem cell research, the researchers are now using the human-induced potent stem cell technology to generate neurons in vitro to model ASDs (Russo et al., 2019; Pensado-López et al., 2020).
We are aware that our project has limitations since our analysis focused on the coding regions for most cases. These constraints were due to the sequencing-platform variabilities. Another limitation of this study is that it was not possible to validate all identified de novo mutations using Sanger sequencing. However, all those picked variants with a minimum depth coverage were validated. Additionally, the combined sequencing depth for most of the remaining mutations was very high.
In summary, in our study, the known syndromic neurodevelopmental disorders indicated that ∼7% (7/102) were similar to the reported contribution of classical ASD etiology (1%–5%). Additionally, we identified ∼15% (15/102) of the ASD families due to possessing potential CNV regions. On the other hand, our study showed that 44.32% (39 out of 88 ASD cases) of studied cases had variants in genes that are relevant to ASD or related neurodevelopmental disorders. This finding is consistent with the power of clinical NGS as a diagnostic tool for neurodevelopmental (Álvarez-Mora et al., 2022). However, this rate is slightly higher than what has been reported previously for ASD (Stefanski et al., 2021) because it was estimated for genes related to ASD from the literature and the SAFARI database. However, some of these genes warrant functional studies to confirm their involvement in ASD. After crossing the reported genes in this study with the SAFARI database and the recent large SPARK study (Zhou et al., 2022), the rate was approximately ∼28% (25/88).
Furthermore, we crossed our findings with the most recent study from the region that shared a similar scope to our project (Abdi et al., 2023). Regarding the patient background, our cohort had a broader representation of the Middle East and North Africa, with ∼89% versus approximately 50% of Arab in Abdi et al. study. There was no overlap in top candidate variants between the two studies which is expected in highly heterogenous disorders such as ASD. Our study has revealed a significant homozygous burden within our ASD cohort, with an estimated rate of 63% (12/19) among consanguineous ASD cases and 12% (2/17) among non-consanguineous cases. Notably, Our findings are in alignment with the homozygous burden observed in consanguineous versus non-consanguineous families in the Abdi et al. Study.
Our data have revealed several genetic factors associated with either known or rare syndromes that have neurodevelopmental impacts only or have multiple body system effects combining with the nervous system (Birk-Barel syndrome (KCNK9), Temtamy syndrome (C12orf57), Rubinstein-Taybi syndrome 1 (CREBBP), Chediak-Higashi syndrome (LYST), Deafness (TECTA), Cohen syndrome (VPS13B), Escobar syndrome (CHRNG), and Arrhythmogenic right ventricular dysplasia 2 (RYR2)). We acknowledge that in our study, the information provided to our research team was limited to ASD-positive cases and the reported consanguinity within families. Although we recognize the importance of comprehensive clinical characterization, the constraints on available clinical data necessitated our reliance on standardized assessment tools and limited information provided by the families. However, the replication of these findings resulted from various applied molecular genetic testing, which showed high confidence in explaining the multifactorial condition of ASD.
Finally, despite the ASD multifactorial etiology that complicates the discovery of ASD risk variants, the number of identified novel or known putative ASD genes from our study was appreciable. Moreover, this study represents the first large-scale characterization of the ASD genetic risk in Qatar’s Middle Eastern population.
Data availability statement
The datasets presented in this article are not readily accessible as the data is protected and inaccessible due to privacy regulations that aim to safeguard the privacy and consent of research participants. However, the data that supports the findings of this study is available upon request from the corresponding author [YA-S and OA].
Ethics statement
The study involving humans were approved by Institutional Review Board of Qatar Biomedical Research Institute (Protocol No.2010-002). (As an extension from the IRB of Shafallah). The study was conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participant’s; legal guardians/next of kin. Written informed consent was obtained from the minor(s)’ legal guardian/next of kin for the publication of any potentially identifiable images or data included in this article.
Author contributions
YA-S: Formal Analysis, Investigation, Methodology, Project administration, Validation, Writing–original draft, Writing–review and editing. RT: Data curation, Methodology, Visualization, Writing–review and editing. EA-D: Data curation, Methodology, Writing–review and editing. DA: Data curation, Methodology, Writing–review and editing. SA: Data curation, Methodology, Writing–review and editing. EA: Data curation, Methodology, Writing–review and editing. HS: Data curation, Methodology, Writing–review and editing. WH: Data curation, Methodology, Writing–review and editing. KE: Data curation, Methodology, Writing–review and editing. YB: Data curation, Methodology, Writing–review and editing. MA: Data curation, Methodology, Writing–review and editing. NK: Data curation, Methodology, Writing–review and editing. YA: Data curation, Methodology, Writing–review and editing. KS: Data curation, Methodology, Writing–review and editing. AA-A: Data curation, Methodology, Writing–review and editing. MK: Data curation, Methodology, Writing–review and editing. AA: Methodology, Resources, Writing–review and editing. IG: Data curation, Resources, Writing–review and editing. MT: Data curation, Resources, Writing–review and editing. FA: Data curation, Resources, Writing–review and editing. HE-S: Investigation, Project administration, Supervision, Writing–original draft, Writing–review and editing. OA: Conceptualization, Funding acquisition, Investigation, Project administration, Supervision, Writing–original draft, Writing–review and editing.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This research was funded by start-up grants to OA. from the college of health and life sciences and the Qatar Biomedical Research Institute at Hamad Bin Khalifa University. YA. and EA. are supported by a Ph.D. scholarship from Hamad Bin Khalifa University. The recruitment of patients was initiated by funds from Shafallah Medical Genetics Center and the Shafallah Center for Children with Special Needs. The study was conducted according to the guidelines of the Declaration of Helsinki and approved by the Institutional Review Board of Qatar Biomedical Research Institute (Protocol No.010-002). (As an extension from the IRB of Shafallah).
Acknowledgments
We thank the patients and their families who participated in this study for their infinite assistance and patience.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2024.1363849/full#supplementary-material
Abbreviations
ASD, Autism spectrum disorder; ID, Intellectual Disability; ADHD, Attention Deficit Hyperactivity Disorder; DSM-V, Diagnostic and Statistical Manual of Mental Disorders; CNV, Copy Number Variations; GWAS, Genome-Wide Association Studies; SNP, Single Nucleotide Polymorphisms; ADI-R, The Autism Diagnostic Interview-Revised; ADOS, Autism Diagnostic Observation Schedule; IQ, Intelligence Quotient; IRB, Institutional Review Board; MLPA, Multiplex Ligation dependent Probe Amplification; WES, Whole-Exome Sequencing; WGS, Whole-genome Sequencing; QCII, QIAGEN Clinical Insight Interpret.
References
Abdi, M., Aliyev, E., Trost, B., Kohailan, M., Aamer, W., Syed, N., et al. (2023). Genomic architecture of autism spectrum disorder in Qatar: the BARAKA-Qatar Study. Genome Med. 15, 81. doi:10.1186/s13073-023-01228-w
Abrahams, B. S., Arking, D. E., Campbell, D. B., Mefford, H. C., Morrow, E. M., Weiss, L. A., et al. (2013). SFARI Gene 2.0: a community-driven knowledgebase for the autism spectrum disorders (ASDs). Mol. Autism 4, 36. doi:10.1186/2040-2392-4-36
Ahram, D. F., Al-Sarraj, Y., Taha, R. Z., Elhag, S. F., Al-Shaban, F. A., El-Shanti, H., et al. (2017). A chromosomal microdeletion of 15q in a female patient with epilepsy, ID, and autism spectrum disorder: a case report. Clin. Case Rep. 5, 1013–1017. doi:10.1002/ccr3.945
Ahram, D. F., Stambouli, D., Syrogianni, A., Al-Sarraj, Y., Gerou, S., El-Shanti, H., et al. (2016). Mosaic partial pericentromeric trisomy 8 and maternal uniparental disomy in a male patient with autism spectrum disorder. Clin. Case Rep. 4, 1125–1131. doi:10.1002/ccr3.705
Akizu, N., Shembesh, N. M., Ben-Omran, T., Bastaki, L., Al-Tawari, A., Zaki, M. S., et al. (2013). Whole-exome sequencing identifies mutated C12orf57 in recessive corpus callosum hypoplasia. Am. J. Hum. Genet. 92, 392–400. doi:10.1016/j.ajhg.2013.02.004
Alallawi, B., Hastings, R. P., and Gray, G. (2020). A systematic scoping review of social, educational, and psychological research on individuals with autism spectrum disorder and their family members in Arab countries and cultures. Rev. J. Autism Dev. Disord. 7, 364–382. doi:10.1007/s40489-020-00198-8
Al-Sarraj, Y., Al-Dous, E., Taha, R. Z., Ahram, D., Alshaban, F., Tolfat, M., et al. (2021). Family-based genome-wide association study of autism spectrum disorder in middle eastern families. Genes (Basel) 12, 761. doi:10.3390/genes12050761
Al-Sarraj, Y., Al-Khair, H. A., Taha, R. Z., Khattab, N., El Sayed, Z. H., Elhusein, B., et al. (2014). Distal trisomy 10q syndrome, report of a patient with duplicated q24.31 - qter, autism spectrum disorder and unusual features. Clin. Case Rep. 2, 201–205. doi:10.1002/ccr3.94
Alshaban, F., Aldosari, M., Al-Shammari, H., El-Hag, S., Ghazal, I., Tolefat, M., et al. (2019). Prevalence and correlates of autism spectrum disorder in Qatar: a national study. J. Child. Psychol. Psychiatry 60, 1254–1268. doi:10.1111/jcpp.13066
Álvarez-Mora, M. I., Sánchez, A., Rodríguez-Revenga, L., Corominas, J., Rabionet, R., Puig, S., et al. (2022). Diagnostic yield of next-generation sequencing in 87 families with neurodevelopmental disorders. Orphanet J. Rare Dis. 17, 60. doi:10.1186/s13023-022-02213-z
American Psychiatric Association (2013). Diagnostic and statistical manual of mental disorders. 5th ed. doi:10.1176/appi.books.9780890425596.744053
Amr, M., Bu Ali, W., Hablas, H., Raddad, D., El-Mehesh, F., El-Gilany, A. H., et al. (2012). Sociodemographic factors in Arab children with autism spectrum disorders. Pan Afr. Med. J. 13, 65. doi:10.11604/pamj.2012.13.65.1839
Anney, R., Klei, L., Pinto, D., Almeida, J., Bacchelli, E., Baird, G., et al. (2012). Individual common variants exert weak effects on the risk for autism spectrum disorders. Hum. Mol. Genet. 21, 4781–4792. doi:10.1093/hmg/dds301
Anney, R., Klei, L., Pinto, D., Regan, R., Conroy, J., Magalhaes, T. R., et al. (2010). A genome-wide scan for common alleles affecting risk for autism. Hum. Mol. Genet. 19, 4072–4082. doi:10.1093/hmg/ddq307
Arnett, A. B., Rhoads, C. L., Hoekzema, K., Turner, T. N., Gerdts, J., Wallace, A. S., et al. (2018). The autism spectrum phenotype in ADNP syndrome. Autism Res. 11, 1300–1310. doi:10.1002/aur.1980
Autism Spectrum Disorders Working Group of The Genomics Consortium (2017). Meta-analysis of GWAS of over 16,000 individuals with autism spectrum disorder highlights a novel locus at 10q24.32 and a significant overlap with schizophrenia. Mol. Autism 8, 21. doi:10.1186/s13229-017-0137-9
Babbs, C., Lloyd, D., Pagnamenta, A. T., Twigg, S. R. F., Green, J., McGowan, S. J., et al. (2014). De novo and rare inherited mutations implicate the transcriptional coregulator TCF20/SPBP in autism spectrum disorder. J. Med. Genet. 51, 737–747. doi:10.1136/jmedgenet-2014-102582
Bailey, A., Le Couteur, A., Gottesman, I., Bolton, P., Simonoff, E., Yuzda, E. R. M., et al. (1995). Autism as a strongly genetic disorder: evidence from a British twin study. Psychol. Med. 25, 63–77. doi:10.1017/s0033291700028099
Barel, O., Shalev, S. A., Ofir, R., Cohen, A., Zlotogora, J., Shorer, Z., et al. (2008). Maternally inherited Birk barel mental retardation dysmorphism syndrome caused by a mutation in the genomically imprinted potassium channel KCNK9. Am. J. Hum. Genet. 83, 193–199. doi:10.1016/j.ajhg.2008.07.010
Bellosta, P., and Soldano, A. (2019). Dissecting the genetics of autism spectrum disorders: a Drosophila perspective. Front. Physiol. 10, 987. doi:10.3389/fphys.2019.00987
Besenbacher, S., Liu, S., Izarzugaza, J. M. G., Grove, J., Belling, K., Bork-Jensen, J., et al. (2015). Novel variation and de novo mutation rates in population-wide de novo assembled Danish trios. Nat. Commun. 6, 5969. doi:10.1038/ncomms6969
Betancur, C. (2011). Etiological heterogeneity in autism spectrum disorders: more than 100 genetic and genomic disorders and still counting. Brain Res. 1380, 42–77. doi:10.1016/j.brainres.2010.11.078
Betancur, C., and Coleman, M. (2013). “Etiological heterogeneity in autism spectrum disorders: role of rare variants,” in The neuroscience of autism spectrum disorders. Editors J. D. Buxbaum, and P. R. Hof (Academic Press). doi:10.1016/B978-0-12-391924-3.00008-9
Bizet, A. A., Becker-Heck, A., Ryan, R., Weber, K., Filhol, E., Krug, P., et al. (2015). Mutations in TRAF3IP1/IFT54 reveal a new role for IFT proteins in microtubule stabilization. Nat. Commun. 6, 8666. doi:10.1038/ncomms9666
Blanchet, P., Bebin, M., Bruet, S., Cooper, G. M., Thompson, M. L., Duban-Bedu, B., et al. (2017). MYT1L mutations cause intellectual disability and variable obesity by dysregulating gene expression and development of the neuroendocrine hypothalamus. PLoS Genet. 13, e1006957. doi:10.1371/journal.pgen.1006957
Bonora, E., Graziano, C., Minopoli, F., Bacchelli, E., Magini, P., Diquigiovanni, C., et al. (2014). Maternally inherited genetic variants of CADPS2 are present in Autism Spectrum Disorders and Intellectual Disability patients. EMBO Mol. Med. 6, 795–809. doi:10.1002/emmm.201303235
Chang, C. C., Chow, C. C., Tellier, L. C. A. M., Vattikuti, S., Purcell, S. M., and Lee, J. J. (2015). Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience 4, 7. doi:10.1186/s13742-015-0047-8
Chang, Y. S., Lin, C. Y., Huang, H. Y., Chang, J. G., and Kuo, H. T. (2019). Chromosomal microarray and whole-exome sequence analysis in Taiwanese patients with autism spectrum disorder. Mol. Genet. Genomic Med. 7, e996. doi:10.1002/mgg3.996
Chapman, N. H., Nato, A. Q., Bernier, R., Ankenman, K., Sohi, H., Munson, J., et al. (2015). Whole exome sequencing in extended families with autism spectrum disorder implicates four candidate genes. Hum. Genet. 134, 1055–1068. doi:10.1007/s00439-015-1585-y
Chemin, J., Siquier-Pernet, K., Nicouleau, M., Barcia, G., Ahmad, A., Medina-Cano, D., et al. (2018). De novo mutation screening in childhood-onset cerebellar atrophy identifies gain-of-function mutations in the CACNA1G calcium channel gene. Brain 141, 1998–2013. doi:10.1093/brain/awy145
Chen, L., Hadd, A., Sah, S., Filipovic-Sadic, S., Krosting, J., Sekinger, E., et al. (2010). An information-rich CGG repeat primed PCR that detects the full range of fragile X expanded alleles and minimizes the need for southern blot analysis. J. Mol. Diagnostics 12, 589–600. doi:10.2353/jmoldx.2010.090227
Chen, L., Jensik, P. J., Alaimo, J. T., Walkiewicz, M., Berger, S., Roeder, E., et al. (2017). Functional analysis of novel DEAF1 variants identified through clinical exome sequencing expands DEAF1-associated neurodevelopmental disorder (DAND) phenotype. Hum. Mutat. 38, 1774–1785. doi:10.1002/humu.23339
Chen, S., Wang, J., Cicek, E., Roeder, K., Yu, H., and Devlin, B. (2020). De novo missense variants disrupting protein-protein interactions affect risk for autism through gene co-expression and protein networks in neuronal cell types. Mol. Autism 11, 76. doi:10.1186/s13229-020-00386-7
Cho, S. C., Yoo, H. J., Park, M., Cho, I. H., Kim, B. N., Kim, J. W., et al. (2011). Genome-wide association scan of Korean autism spectrum disorders with language delay: a preliminary study. Psychiatry Investig. 8, 61–66. doi:10.4306/pi.2011.8.1.61
Christian, S. L., Brune, C. W., Sudi, J., Kumar, R. A., Liu, S., Karamohamed, S., et al. (2008). Novel submicroscopic chromosomal abnormalities detected in autism spectrum disorder. Biol. Psychiatry 63, 1111–1117. doi:10.1016/j.biopsych.2008.01.009
Colella, S., Yau, C., Taylor, J. M., Mirza, G., Butler, H., Clouston, P., et al. (2007). QuantiSNP: an objective bayes hidden-markov model to detect and accurately map copy number variation using SNP genotyping data. Nucleic Acids Res. 35, 2013–2025. doi:10.1093/nar/gkm076
Connolly, J. J., Glessner, J. T., and Hakonarson, H. (2013). A genome-wide association study of autism incorporating autism diagnostic interview-revised, autism diagnostic observation Schedule, and social responsiveness scale. Child. Dev. 84, 17–33. doi:10.1111/j.1467-8624.2012.01838.x
Connolly, S., Anney, R., Gallagher, L., and Heron, E. A. (2017). A genome-wide investigation into parent-of-origin effects in autism spectrum disorder identifies previously associated genes including SHANK3. Eur. J. Hum. Genet. 25, 234–239. doi:10.1038/ejhg.2016.153
Cook, E. H., and Scherer, S. W. (2008). Copy-number variations associated with neuropsychiatric conditions. Nature 455, 919–923. doi:10.1038/nature07458
Cristina, A., Luiza, A., and Pereira-Nascimento, P. (2013). “Genetic etiology of autism,” in Recent advances in autism spectrum disorders - volume I. Editor M. Fitzgerald (London, United Kingdom: InTech). doi:10.5772/53106
Cui, J., Li, Y., Zhu, L., Liu, D., Songyang, Z., Wang, H. Y., et al. (2012). NLRP4 negatively regulates type I interferon signaling by targeting the kinase TBK1 for degradation via the ubiquitin ligase DTX4. Nat. Immunol. 13, 387–395. doi:10.1038/ni.2239
Cui, Y., He, S., Xing, C., Lu, K., Wang, J., Xing, G., et al. (2011). SCFFBXL¹⁵ regulates BMP signalling by directing the degradation of HECT-type ubiquitin ligase Smurf1. EMBO J. 30, 2675–2689. doi:10.1038/emboj.2011.155
Cukier, H. N., Dueker, N. D., Slifer, S. H., Lee, J. M., Whitehead, P. L., Lalanne, E., et al. (2014). Exome sequencing of extended families with autism reveals genes shared across neurodevelopmental and neuropsychiatric disorders. Mol. Autism 5, 1. doi:10.1186/2040-2392-5-1
Delgado, M. S., Camprubí, C., Tümer, Z., Martínez, F., Milà, M., and Monk, D. (2014). Screening individuals with intellectual disability, autism and Tourette’s syndrome for KCNK9 mutations and aberrant DNA methylation within the 8q24 imprinted cluster. Am. J. Med. Genet. Part B Neuropsychiatric Genet. 165, 472–478. doi:10.1002/ajmg.b.32250
De Rubeis, S., He, X., Goldberg, A. P., Poultney, C. S., Samocha, K., Ercument Cicek, A., et al. (2014). Supple: synaptic, transcriptional and chromatin genes disrupted in autism. Nature 515, 209–215. doi:10.1038/nature13772
Docherty, L. E., Rezwan, F. I., Poole, R. L., Turner, C. L. S., Kivuva, E., Maher, E. R., et al. (2015). Mutations in NLRP5 are associated with reproductive wastage and multilocus imprinting disorders in humans. Nat. Commun. 6, 8086–8087. doi:10.1038/ncomms9086
Drmanac, R., Sparks, A. B., Callow, M. J., Halpern, A. L., Burns, N. L., Kermani, B. G., et al. (2010). Human genome sequencing using unchained base reads on self-assembling DNA nanoarrays. Science 327, 78–81. doi:10.1126/science.1181498
Durand, C. M., Betancur, C., Boeckers, T. M., Bockmann, J., Chaste, P., Fauchereau, F., et al. (2007). Mutations in the gene encoding the synaptic scaffolding protein SHANK3 are associated with autism spectrum disorders. Nat. Genet. 39, 25–27. doi:10.1038/ng1933
Dwyer, C. A., and Esko, J. D. (2016). Glycan susceptibility factors in autism spectrum disorders. Mol. Asp. Med. 51, 104–114. doi:10.1016/j.mam.2016.07.001
Ebstein, F., Küry, S., Papendorf, J. J., and Krüger, E. (2021). Neurodevelopmental disorders (NDD) caused by genomic alterations of the ubiquitin-proteasome system (UPS): the possible contribution of immune dysregulation to disease pathogenesis. Front. Mol. Neurosci. 14, 733012. doi:10.3389/fnmol.2021.733012
Ergaz, Z., Weinstein-Fudim, L., and Ornoy, A. (2016). Genetic and non-genetic animal models for autism spectrum disorders (ASD). Reprod. Toxicol. 64, 116–140. doi:10.1016/j.reprotox.2016.04.024
Erlandson, A., Samuelsson, L., Hagberg, B., Kyllerman, M., Vujic, M., and Wahlström, J. (2003). Multiplex ligation-dependent Probe amplification (MLPA) detects large deletions in the MECP2 gene of Swedish Rett syndrome patients. Genet. Test. 7, 329–332. doi:10.1089/109065703322783707
Freitag, C. M. (2007). The genetics of autistic disorders and its clinical relevance: a review of the literature. Mol. Psychiatry 12, 2–22. doi:10.1038/sj.mp.4001896
Fu, J. M., Satterstrom, F. K., Peng, M., Brand, H., Collins, R. L., Dong, S., et al. (2022). Rare coding variation provides insight into the genetic architecture and phenotypic context of autism. Nat. Genet. 54, 1320–1331. doi:10.1038/s41588-022-01104-0
Geetha, B., Sukumar, C., Dhivyadeepa, E., Reddy, J. K., and Balachandar, V. (2019). Autism in India: a case–control study to understand the association between socio-economic and environmental risk factors. Acta Neurol. Belg 119, 393–401. doi:10.1007/s13760-018-01057-4
Glessner, J. T., Wang, K., Cai, G., Korvatska, O., Kim, C. E., Wood, S., et al. (2009). Autism genome-wide copy number variation reveals ubiquitin and neuronal genes. Nature 459, 569–573. doi:10.1038/nature07953
Grabowski, P. A. P., Bello, A. F., Rodrigues, D. L., Forbeci, M. J., Motter, V., and Raskin, S. (2017). Deletion involving the 7q31-32 band at the CADPS2 gene locus in a patient with autism spectrum disorder and recurrent psychotic syndrome triggered by stress. Case Rep. Psychiatry 2017, 4254152–4254154. doi:10.1155/2017/4254152
Graham, J. M., Zadeh, N., Kelley, M., Tan, E. S., Liew, W., Tan, V., et al. (2016). KCNK9 imprinting syndrome—further delineation of a possible treatable disorder. Am. J. Med. Genet. A 170, 2632–2637. doi:10.1002/ajmg.a.37740
Grove, J., Ripke, S., Als, T. D., Mattheisen, M., Walters, R. K., Won, H., et al. (2019). Identification of common genetic risk variants for autism spectrum disorder. Nat. Genet. 51, 431–444. doi:10.1038/s41588-019-0344-8
Guo, D., Yang, X., and Shi, L. (2020). Rho GTPase regulators and effectors in autism spectrum disorders: animal models and insights for therapeutics. Cells 9, 835. doi:10.3390/cells9040835
Guo, H., Peng, Y., Hu, Z., Li, Y., Xun, G., Ou, J., et al. (2017). Genome-wide copy number variation analysis in a Chinese autism spectrum disorder cohort. Sci. Rep. 7, 44155. doi:10.1038/srep44155
Hacohen-Kleiman, G., Sragovich, S., Karmon, G., Gao, A. Y. L., Grigg, I., Pasmanik-Chor, M., et al. (2018). Activity-dependent neuroprotective protein deficiency models synaptic and developmental phenotypes of autism-like syndrome. J. Clin. Investigation 128, 4956–4969. doi:10.1172/JCI98199
Hamdan, F. F., Srour, M., Capo-Chichi, J. M., Daoud, H., Nassif, C., Patry, L., et al. (2014). De novo mutations in moderate or severe intellectual disability. PLoS Genet. 10, e1004772. doi:10.1371/journal.pgen.1004772
Helsmoortel, C., Vulto-Van Silfhout, A. T., Coe, B. P., Vandeweyer, G., Rooms, L., Van Den Ende, J., et al. (2014). A SWI/SNF-related autism syndrome caused by de novo mutations in ADNP. Nat. Genet. 46, 380–384. doi:10.1038/ng.2899
Hyman, S. L., Levy, S. E., and Myers, S. M.Council on Children with Disabilities, Section On Developmental And Behavioral Pediatrics (2020). Identification, evaluation, and management of children with autism spectrum disorder. Pediatrics 145, e20193447. doi:10.1542/peds.2019-3447
Iossifov, I., O’Roak, B. J., Sanders, S. J., Ronemus, M., Krumm, N., Levy, D., et al. (2014). The contribution of de novo coding mutations to autism spectrum disorder. Nature 515, 216–221. doi:10.1038/nature13908
Irreverre, F., Mudd, S. H., Heizer, W. D., and Laster, L. (1967). Sulfite oxidase deficiency: studies of a patient with mental retardation, dislocated ocular lenses, and abnormal urinary excretion of S-sulfo-l-cysteine, sulfite, and thiosulfate. Biochem. Med. 1, 187–217. doi:10.1016/0006-2944(67)90007-5
Ito, Y., Carss, K. J., Duarte, S. T., Hartley, T., Keren, B., Kurian, M. A., et al. (2018). De novo truncating mutations in WASF1 cause intellectual disability with seizures. Am. J. Hum. Genet. 103, 144–153. doi:10.1016/j.ajhg.2018.06.001
Jiang, Y. H., Sahoo, T., Michaelis, R. C., Bercovich, D., Bressler, J., Kashork, C. D., et al. (2004). A mixed epigenetic/genetic model for oligogenic inheritance of autism with a limited role for UBE3A. Am. J. Med. Genet. A 131, 1–10. doi:10.1002/ajmg.a.30297
Jiang, Y.-H., Yuen, R. K. C., Jin, X., Wang, M., Chen, N., Wu, X., et al. (2013). Detection of clinically relevant genetic variants in autism spectrum disorder by whole-genome sequencing. Am. J. Hum. Genet. 93, 249–263. doi:10.1016/j.ajhg.2013.06.012
Johnson, J. L., Coyne, K. E., Garrett, R. M., Zabot, M. T., Dorche, C., Kisker, C., et al. (2002). Isolated sulfite oxidase deficiency: identification of 12 novel SUOX mutations in 10 patients. Hum. Mutat. 20, 74. doi:10.1002/humu.9038
Kahle, K. T., Rinehart, J., de Los Heros, P., Louvi, A., Meade, P., Vazquez, N., et al. (2005). WNK3 modulates transport of Cl-in and out of cells: implications for control of cell volume and neuronal excitability. Proc. Natl. Acad. Sci. U. S. A. 102, 16783–16788. doi:10.1073/pnas.0508307102
Kalınlı, M., Ekinci, Ö., Güneş, S., and Ekinci, N. (2019). Autistic-like traits in pena–shokeir syndrome. J. Autism Dev. Disord. 49, 1316–1318. doi:10.1007/s10803-018-3824-2
Karczewski, K. J., Francioli, L. C., Tiao, G., Cummings, B. B., Alföldi, J., Wang, Q., et al. (2020). The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 581, 434–443. doi:10.1038/s41586-020-2308-7
Kim, H. G., Rosenfeld, J. A., Scott, D. A., Bénédicte, G., Labonne, J. D., Brown, J., et al. (2019). Disruption of PHF21A causes syndromic intellectual disability with craniofacial anomalies, epilepsy, hypotonia, and neurobehavioral problems including autism. Mol. Autism 10, 35. doi:10.1186/s13229-019-0286-0
Kim, N., Kim, K. H., Lim, W. J., Kim, J., Kim, S. A., and Yoo, H. J. (2021). Whole exome sequencing identifies novel de novo variants interacting with six gene networks in autism spectrum disorder. Genes (Basel) 12, 1–17. doi:10.3390/genes12010001
Krumm, N., Turner, T. N., Baker, C., Vives, L., Mohajeri, K., Witherspoon, K., et al. (2015). Excess of rare, inherited truncating mutations in autism. Nat. Genet. 47, 582–588. doi:10.1038/ng.3303
Kuo, P. H., Chuang, L. C., Su, M. H., Chen, C. H., Chen, C. H., Wu, J. Y., et al. (2015). Genome-wide association study for autism spectrum disorder in taiwanese han population. PLoS One 10, e0138695. doi:10.1371/journal.pone.0138695
Latypova, X., Vincent, M., Mollé, A., Adebambo, O. A., Fourgeux, C., Khan, T. N., et al. (2021). Haploinsufficiency of the Sin3/HDAC corepressor complex member SIN3B causes a syndromic intellectual disability/autism spectrum disorder. Am. J. Hum. Genet. 108, 929–941. doi:10.1016/j.ajhg.2021.03.017
Levy, S., Mandell, D., and Schultz, R. (2009). Autism. Lancet 374, 1627–1638. doi:10.1016/S0140-6736(09)61376-3
Lionel, A. C., Tammimies, K., Vaags, A. K., Rosenfeld, J. A., Ahn, J. W., Merico, D., et al. (2014). Disruption of the ASTN2/TRIM32 locus at 9q33.1 is a risk factor in males for autism spectrum disorders, ADHD and other neurodevelopmental phenotypes. Hum. Mol. Genet. 23, 2752–2768. doi:10.1093/hmg/ddt669
Lizio, M., Abugessaisa, I., Noguchi, S., Kondo, A., Hasegawa, A., Hon, C. C., et al. (2019). Update of the FANTOM web resource: expansion to provide additional transcriptome atlases. Nucleic Acids Res. 47, D752–D758. doi:10.1093/nar/gky1099
Lo-Castro, A., and Curatolo, P. (2014). Epilepsy associated with autism and attention deficit hyperactivity disorder: is there a genetic link? Brain Dev. 36, 185–193. doi:10.1016/j.braindev.2013.04.013
Loomes, R., Hull, L., and Mandy, W. P. L. (2017). What is the male-to-female ratio in autism spectrum disorder? A systematic review and meta-analysis. J. Am. Acad. Child. Adolesc. Psychiatry 56, 466–474. doi:10.1016/j.jaac.2017.03.013
López-Doménech, G., Serrat, R., Mirra, S., D’Aniello, S., Somorjai, I., Abad, A., et al. (2012). The Eutherian Armcx genes regulate mitochondrial trafficking in neurons and interact with Miro and Trak2. Nat. Commun. 3, 814. doi:10.1038/ncomms1829
Luo, Y., Eran, A., Palmer, N., Avillach, P., Levy-Moonshine, A., Szolovits, P., et al. (2020). A multidimensional precision medicine approach identifies an autism subtype characterized by dyslipidemia. Nat. Med. 26, 1375–1379. doi:10.1038/s41591-020-1007-0
Ma, D., Salyakina, D., Jaworski, J. M., Konidari, I., Whitehead, P. L., Andersen, A. N., et al. (2009). A genome-wide association study of autism reveals a common novel risk locus at 5p14.1. Ann. Hum. Genet. 73, 263–273. doi:10.1111/j.1469-1809.2009.00523.x
Manoli, I., Golas, G., Westbroek, W., Vilboux, T., Markello, T. C., Introne, W., et al. (2010). Chediak-Higashi syndrome with early developmental delay resulting from paternal heterodisomy of chromosome. Am. J. Med. Genet. A 152, 1474–1483. doi:10.1002/ajmg.a.33389
Marini, A., Ozbič, M., Magni, R., and Valeri, G. (2020). Toward a definition of the linguistic profile of children with autism spectrum disorder. Front. Psychol. 11, 808. doi:10.3389/fpsyg.2020.00808
Marshall, C. R., Noor, A., Vincent, J. B., Lionel, A. C., Feuk, L., Skaug, J., et al. (2008). Structural variation of chromosomes in autism spectrum disorder. J. Hum. Genet. 82, 477–488. doi:10.1016/j.ajhg.2007.12.009
Maydan, G., Noyman, I., Har-Zahav, A., Neriah, Z. B., Pasmanik-Chor, M., Yeheskel, A., et al. (2011). Multiple congenital anomalies-hypotonia-seizures syndrome is caused by a mutation in PIGN. J. Med. Genet. 48, 383–389. doi:10.1136/jmg.2010.087114
Meshalkina, D. A., Kizlyk, M. N., Kysil, E. V., Collier, A. D., Echevarria, D. J., Abreu, M. S., et al. (2018). Zebrafish models of autism spectrum disorder. Exp. Neurol. 299, 207–216. doi:10.1016/j.expneurol.2017.02.004
Milunsky, A., Ito, M., Maher, T. A., Flynn, M., and Milunsky, J. M. (2009). Prenatal molecular diagnosis of tuberous sclerosis complex. Am. J. Obstet. Gynecol. 200, 321.e1–e6. doi:10.1016/j.ajog.2008.11.004
Moey, C., Hinze, S. J., Brueton, L., Morton, J., McMullan, D. J., Kamien, B., et al. (2016). Xp11.2 microduplications including IQSEC2, TSPYL2 and KDM5C genes in patients with neurodevelopmental disorders. Eur. J. Hum. Genet. 24, 373–380. doi:10.1038/ejhg.2015.123
Morrow, E. M., Yoo, S.-Y., Flavell, S. W., Kim, T.-K., Lin, Y., Hill, R. S., et al. (2008). Identifying autism loci and genes by tracing recent shared ancestry. Science 321, 218–223. doi:10.1126/science.1157657
Nguyen, M., Roth, A., Kyzar, E. J., Poudel, M. K., Wong, K., Stewart, A. M., et al. (2014). Decoding the contribution of dopaminergic genes and pathways to autism spectrum disorder (ASD). Neurochem. Int. 66, 15–26. doi:10.1016/j.neuint.2014.01.002
Okamoto, N., Hatsukawa, Y., Shimojima, K., and Yamamoto, T. (2011). Submicroscopic deletion in 7q31 encompassing CADPS2 and TSPAN12 in a child with autism spectrum disorder and PHPV. Am. J. Med. Genet. A 155, 1568–1573. doi:10.1002/ajmg.a.34028
O’Roak, B. J., Vives, L., Girirajan, S., Karakoc, E., Krumm, N., Coe, B. P., et al. (2012). Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature 485, 246–250. doi:10.1038/nature10989
Ozonoff, S., Young, G. S., Carter, A., Messinger, D., Yirmiya, N., Zwaigenbaum, L., et al. (2011). Recurrence risk for autism spectrum disorders: a baby siblings research consortium study. Pediatrics 128, e488–e495. doi:10.1542/peds.2010-2825
Pangalos, M. N., Neefs, J. M., Somers, M., Verhasselt, P., Bekkers, M., Van Der Helm, L., et al. (1999). Isolation and expression of novel human glutamate carboxypeptidases with N-acetylated alpha-linked acidic dipeptidase and dipeptidyl peptidase IV activity. J. Biol. Chem. 274, 8470–8483. doi:10.1074/jbc.274.13.8470
Pensado-López, A., Veiga-Rúa, S., Carracedo, Á., Allegue, C., and Sánchez, L. (2020). Experimental models to study autism spectrum disorders: hipscs, rodents and zebrafish. Genes (Basel) 11, 1376. doi:10.3390/genes11111376
Pinto, D., Pagnamenta, A. T., Klie, L., Anney, R., Merico, D., Regan, R., et al. (2010). Functional impact of global rare copy number variation in autism spectrum disorders. Nature 466, 368–372. doi:10.1038/nature09146
Purcell, S., Neale, B., Todd-brown, K., Thomas, L., Ferreira, M. A. R., Bender, D., et al. (2007). PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 81, 559–575. doi:10.1086/519795
Qiao, Y., Liu, X., Harvard, C., Hildebrand, M. J., Rajcan-Separovic, E., Holden, J. J. A., et al. (2008). Autism-associated familial microdeletion of Xp11.22. Clin. Genet. 74, 134–144. doi:10.1111/j.1399-0004.2008.01028.x
Rajab, A., Schuelke, M., Gill, E., Zwirner, A., Seifert, F., Gonzalez, S. M., et al. (2015). Recessive DEAF1 mutation associates with autism, intellectual disability, basal ganglia dysfunction and epilepsy. J. Med. Genet. 52, 607–611. doi:10.1136/jmedgenet-2015-103083
Roberts, J. L., Hovanes, K., Dasouki, M., Manzardo, A. M., and Butler, M. G. (2014). Chromosomal microarray analysis of consecutive individuals with autism spectrum disorders or learning disability presenting for genetic services. Gene 535, 70–78. doi:10.1016/j.gene.2013.10.020
Rossi, M., El-Khechen, D., Black, M. H., Farwell Hagman, K. D., Tang, S., and Powis, Z. (2017). Outcomes of diagnostic exome sequencing in patients with diagnosed or suspected autism spectrum disorders. Pediatr. Neurol. 70, 34–43. doi:10.1016/j.pediatrneurol.2017.01.033
Rupar, C. A., Gillett, J., Gordon, B. A., Ramsay, D. A., Johnson, J. L., Garrett, R. M., et al. (1996). Isolated sulfite oxidase deficiency. Neuropediatrics 27, 299–304. doi:10.1055/s-2007-973798
Russo, F. B., Brito, A., de Freitas, A. M., Castanha, A., de Freitas, B. C., and Beltrão-Braga, P. C. B. (2019). The use of iPSC technology for modeling Autism Spectrum Disorders. Neurobiol. Dis. 130, 104483. doi:10.1016/j.nbd.2019.104483
Ruzzo, E. K., Pérez-Cano, L., Jung, J. Y., Wang, L. K., Kashef-Haghighi, D., Hartl, C., et al. (2019). Inherited and de novo genetic risk for autism impacts shared networks. Cell 178, 850–866. doi:10.1016/j.cell.2019.07.015
Sadakata, T., Washida, M., Iwayama, Y., Shoji, S., Sato, Y., Ohkura, T., et al. (2007). Autistic-like phenotypes in Cadps2-knockout mice and aberrant CADPS2 splicing in autistic patients. J. Clin. Investigation 117, 931–943. doi:10.1172/JCI29031
Salih, M. A., Tzschach, A., Oystreck, D. T., Hassan, H. H., Aldrees, A., Elmalik, S. A., et al. (2013). A newly recognized autosomal recessive syndrome affecting neurologic function and vision. Am. J. Med. Genet. A 161, 1207–1213. doi:10.1002/ajmg.a.35850
Salpietro, V., Dixon, C. L., Guo, H., Bello, O. D., Vandrovcova, J., Efthymiou, S., et al. (2019). AMPA receptor GluA2 subunit defects are a cause of neurodevelopmental disorders. Nat. Commun. 10, 3094. doi:10.1038/s41467-019-10910-w
Samia, P., Kanana, M., King, J., Donald, K. A., Newton, C. R., and Denckla, C. (2020). Childhood autism spectrum disorder: insights from a tertiary hospital cohort in Kenya. Afr. J. Health Sci. 33, 12–21.
Sanders, S. J., Ercan-sencicek, A. G., Hus, V., Luo, R., Murtha, M. T., Moreno-de-luca, D., et al. (2011). Multiple recurrent de novo CNVs, including duplications of the 7q11.23Williams syndrome region, are strongly associated with autism. Neuron 70, 863–885. doi:10.1016/j.neuron.2011.05.002
Sanders, S. J., Murtha, M. T., Gupta, A. R., Murdoch, J. D., Raubeson, M. J., Willsey, A. J., et al. (2012). De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature 485, 237–241. doi:10.1038/nature10945
Satterstrom, F. K., Kosmicki, J. A., Wang, J., Breen, M. S., De Rubeis, S., An, J. Y., et al. (2020). Large-scale exome sequencing study implicates both developmental and functional changes in the neurobiology of autism. Cell 180, 568–584.e23. doi:10.1016/j.cell.2019.12.036
Schaaf, C. P., Sabo, A., Sakai, Y., Crosby, J., Muzny, D., Hawes, A., et al. (2011). Oligogenic heterozygosity in individuals with high-functioning autism spectrum disorders. Hum. Mol. Genet. 20, 3366–3375. doi:10.1093/hmg/ddr243
Schneider, A., Puechberty, J., Ng, B. L., Coubes, C., Gatinois, V., Tournaire, M., et al. (2015). Identification of disrupted AUTS2 and EPHA6 genes by array painting in a patient carrying a de novo balanced translocation t(3;7) with intellectual disability and neurodevelopment disorder. Am. J. Med. Genet. A 167, 3031–3037. doi:10.1002/ajmg.a.37350
Scott, E. M., Halees, A., Itan, Y., Spencer, E. G., He, Y., Azab, M. A., et al. (2016). Characterization of Greater Middle Eastern genetic variation for enhanced disease gene discovery. Nat. Genet. 48, 1071–1076. doi:10.1038/ng.3592
Šedivá, M., Laššuthová, P., Zámečník, J., Sedláčková, L., Seeman, P., and Haberlová, J. (2020). Novel variant in the KCNK9 gene in a girl with Birk Barel syndrome. Eur. J. Med. Genet. 63, 103619. doi:10.1016/j.ejmg.2019.01.009
Shen, J., Lincoln, S., and Miller, D. T. (2014). Advances in genetic discovery and implications for counseling of patients and families with autism spectrum disorders. Curr. Genet. Med. Rep. 2, 124–134. doi:10.1007/s40142-014-0047-5
Snijders Blok, L., Hiatt, S. M., Bowling, K. M., Prokop, J. W., Engel, K. L., Cochran, J. N., et al. (2018). De novo mutations in MED13, a component of the Mediator complex, are associated with a novel neurodevelopmental disorder. Hum. Genet. 137, 375–388. doi:10.1007/s00439-018-1887-y
Sofocleous, C., Kolialexi, A., and Mavrou, A. (2009). Molecular diagnosis of Fragile X syndrome. Expert Rev. Mol. Diagn 9, 23–30. doi:10.1586/14737159.9.1.23
Song, S., Tan, B., Dong, X., Yang, Q., Chi, S., Liu, H., et al. (2019). Molecular cloning, characterization and expression analysis of ARMC6, ARMC7, ARMC8 from Pacific white shrimp, Litopenaeus vannamei. Gene 682, 50–66. doi:10.1016/j.gene.2018.10.007
Spataro, N., Trujillo-Quintero, J. P., Manso, C., Gabau, E., Capdevila, N., Martinez-Glez, V., et al. (2023). High performance of a dominant/X-linked gene panel in patients with neurodevelopmental disorders. Genes (Basel) 14, 708. doi:10.3390/GENES14030708
Stefanski, A., Calle-López, Y., Leu, C., Pérez-Palma, E., Pestana-Knight, E., and Lal, D. (2021). Clinical sequencing yield in epilepsy, autism spectrum disorder, and intellectual disability: a systematic review and meta-analysis. Epilepsia 62, 143–151. doi:10.1111/EPI.16755
Stolerman, E. S., Francisco, E., Stallworth, J. L., Jones, J. R., Monaghan, K. G., Keller-Ramey, J., et al. (2019). Genetic variants in the KDM6B gene are associated with neurodevelopmental delays and dysmorphic features. Am. J. Med. Genet. A 179, 1276–1286. doi:10.1002/ajmg.a.61173
Tejada, M. I., Villate, O., Ibarluzea, N., de la Hoz, A. B., Martínez-Bouzas, C., Beristain, E., et al. (2019). Molecular and clinical characterization of a novel nonsense variant in exon 1 of the upf3b gene found in a large Spanish Basque family (Mrx82). Front. Genet. 10, 1074. doi:10.3389/fgene.2019.01074
Tian, M., Chen, J., Li, J., Pan, H., Lei, W., and Shu, X. (2022). Damaging novel mutations in PIGN cause developmental epileptic-dyskinetic encephalopathy: a case report. BMC Pediatr. 22, 1–6. doi:10.1186/s12887-022-03246-w
Toma, C., Torrico, B., Hervás, A., Valdés-Mas, R., Tristán-Noguero, A., Padillo, V., et al. (2014). Exome sequencing in multiplex autism families suggests a major role for heterozygous truncating mutations. Mol. Psychiatry 19, 784–790. doi:10.1038/mp.2013.106
Tran, K. T., Le, V. S., Bui, H. T. P., Do, D. H., Ly, H. T. T., Nguyen, H. T., et al. (2020). Genetic landscape of autism spectrum disorder in Vietnamese children. Sci. Rep. 10, 5034. doi:10.1038/s41598-020-61695-8
Tsuang, D. W., Millard, S. P., Ely, B., Chi, P., Wang, K., Raskind, W. H., et al. (2010). The effect of algorithms on copy number variant detection. PLoS One 5, e14456. doi:10.1371/journal.pone.0014456
Turner, T. N., Coe, B. P., Dickel, D. E., Hoekzema, K., Nelson, B. J., Zody, M. C., et al. (2017). Genomic patterns of de novo mutation in simplex autism. Cell 171, 710–722. doi:10.1016/j.cell.2017.08.047
Vandeweyer, G., Helsmoortel, C., VanDijck, A., Vulto-Van Silfhout, A. T., Coe, B. P., Bernier, R., et al. (2014). The transcriptional regulator ADNP links the BAF (SWI/SNF) complexes with Autism. Am. J. Med. Genet. C Semin. Med. Genet. 166, 315–326. doi:10.1002/ajmg.c.31413
Vetrini, F., McKee, S., Rosenfeld, J. A., Suri, M., Lewis, A. M., Nugent, K. M., et al. (2019). De novo and inherited TCF20 pathogenic variants are associated with intellectual disability, dysmorphic features, hypotonia, and neurological impairments with similarities to Smith-Magenis syndrome. Genome Med. 11, 12. doi:10.1186/s13073-019-0623-0
Walsh, R., Thomson, K. L., Ware, J. S., Funke, B. H., Woodley, J., McGuire, K. J., et al. (2017). Reassessment of Mendelian gene pathogenicity using 7,855 cardiomyopathy cases and 60,706 reference samples. Genet. Med. 19, 192–203. doi:10.1038/gim.2016.90
Wang, K., Zhang, H., Ma, D., Bucan, M., Glessner, J. T., Abrahams, B. S., et al. (2009). Common genetic variants on 5p14.1 associate with autism spectrum disorders. Nature 459, 528–533. doi:10.1038/nature07999
Wang, Y., Li, M., Luo, Y., Zhao, X., Liao, S., Jiang, L., et al. (2019). Temtamy syndrome caused by a new C12orf57 variant in a Chinese boy, including pedigree analysis and literature review. Exp. Ther. Med. 19, 327–332. doi:10.3892/etm.2019.8183
Weiss, L., Arking, D., Daly, M. J., and Chakravarti, A.The Gene Discovery Project of John Hophins the Autism Consortium (2009). A genome-wide linkage and association scan reveals novel loci for autism. Nature 461, 802–808. doi:10.1038/nature08490
Werling, D. M., and Geschwind, D. H. (2015). Recurrence rates provide evidence for sex-differential, familial genetic liability for autism spectrum disorders in multiplex families and twins. Mol. Autism 6, 27–14. doi:10.1186/s13229-015-0004-5
Xia, L., Ou, J., Li, K., Guo, H., Hu, Z., Bai, T., et al. (2020). Genome-wide association analysis of autism identified multiple loci that have been reported as strong signals for neuropsychiatric disorders. Autism Res. 13, 382–396. doi:10.1002/aur.2229
Yap, C. X., Alvares, G. A., Henders, A. K., Lin, T., Wallace, L., Farrelly, A., et al. (2021). Analysis of common genetic variation and rare CNVs in the Australian Autism Biobank. Mol. Autism 12, 12. doi:10.1186/s13229-020-00407-5
Ye, Y., Cho, M. T., Retterer, K., Alexander, N., Ben-Omran, T., Al-Mureikhi, M., et al. (2015). De novo POGZ mutations are associated with neurodevelopmental disorders and microcephaly. Mol. Case Stud. 1, a000455. doi:10.1101/mcs.a000455
Yoo, H. (2015). Genetics of autism spectrum disorder: current status and possible clinical applications. Exp. Neurobiol. 24, 257–272. doi:10.5607/en.2015.24.4.257
Yousef, A. M., Roshdy, E. H., Abdel Fattah, N. R., Said, R. M., Atia, M. M., Hafez, E. M., et al. (2021). Prevalence and risk factors of autism spectrum disorders in preschool children in Sharkia, Egypt: a community-based study. Middle East Curr. Psychiatry 28, 36–14. doi:10.1186/s43045-021-00114-8
Yu, T. W., Chahrour, M. H., Coulter, M. E., Jiralerspong, S., Okamura-Ikeda, K., Ataman, B., et al. (2013). Using whole-exome sequencing to identify inherited causes of autism. Neuron 77, 259–273. doi:10.1016/j.neuron.2012.11.002
Yuen, R. K. C., Merico, D., Bookman, M., Howe, J. L., Thiruvahindrapuram, B., Patel, R. V., et al. (2017). Whole genome sequencing resource identifies 18 new candidate genes for autism spectrum disorder. Nat. Neurosci. 20, 602–611. doi:10.1038/nn.4524
Yuen, R. K. C. C., Thiruvahindrapuram, B., Merico, D., Walker, S., Tammimies, K., Hoang, N., et al. (2015). Whole-genome sequencing of quartet families with autism spectrum disorder. Nat. Med. 21, 185–191. doi:10.1038/nm.3792
Zahrani, F., Aldahmesh, M. A., Alshammari, M. J., Al-Hazzaa, S. A. F., and Alkuraya, F. S. (2013). Mutations in C12orf57 cause a syndromic form of colobomatous microphthalmia. Am. J. Hum. Genet. 92, 387–391. doi:10.1016/j.ajhg.2013.01.008
Zarrei, M., Burton, C. L., Engchuan, W., Young, E. J., Higginbotham, E. J., MacDonald, J. R., et al. (2019). A large data resource of genomic copy number variation across neurodevelopmental disorders. NPJ Genom Med. 4, 26. doi:10.1038/s41525-019-0098-3
Zhang, W., Hou, T., Niu, C., Song, L., and Zhang, Y. (2015). B3GNT3 expression is a novel marker correlated with pelvic lymph node metastasis and poor clinical outcome in early-stage cervical cancer. PLoS One 10, e0144360. doi:10.1371/journal.pone.0144360
Zheng, F., Kasper, L. H., Bedford, D. C., Lerach, S., Teubner, B. J. W., and Brindle, P. K. (2016). Mutation of the CH1 domain in the histone acetyltransferase CREBBP results in autism-relevant behaviors in mice. PLoS One 11, e0146366. doi:10.1371/journal.pone.0146366
Zhou, X., Feliciano, P., Shu, C., Wang, T., Astrovskaya, I., Hall, J. B., et al. (2022). Integrating de novo and inherited variants in 42,607 autism cases identifies mutations in new moderate-risk genes. Nat. Genet. 54, 1305–1319. doi:10.1038/S41588-022-01148-2
Keywords: autism spectrum disorder (ASD), neurodevelopmental disorders, epilepsy, next-generation sequencing (NGS), copy number variation (CNV), de novo mutation, genetics
Citation: Al-Sarraj Y, Taha RZ, Al-Dous E, Ahram D, Abbasi S, Abuazab E, Shaath H, Habbab W, Errafii K, Bejaoui Y, AlMotawa M, Khattab N, Aqel YA, Shalaby KE, Al-Ansari A, Kambouris M, Abouzohri A, Ghazal I, Tolfat M, Alshaban F, El-Shanti H and Albagha OME (2024) The genetic landscape of autism spectrum disorder in the Middle Eastern population. Front. Genet. 15:1363849. doi: 10.3389/fgene.2024.1363849
Received: 31 December 2023; Accepted: 04 March 2024;
Published: 20 March 2024.
Edited by:
Mehdi Pirooznia, Johnson & Johnson, United StatesReviewed by:
Sheng Wang, University of California, San Francisco, United StatesYonatan Perez, University of California, San Francisco, United States
Boting Ning, Johnson & Johnson, United States
Copyright © 2024 Al-Sarraj, Taha, Al-Dous, Ahram, Abbasi, Abuazab, Shaath, Habbab, Errafii, Bejaoui, AlMotawa, Khattab, Aqel, Shalaby, Al-Ansari, Kambouris, Abouzohri, Ghazal, Tolfat, Alshaban, El-Shanti and Albagha. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yasser Al-Sarraj, eWFsc2FycmFqQHFmLm9yZy5xYQ==; Omar M. E. Albagha, b2FsYmFnaGFAaGJrdS5lZHUucWE=