 Lingqi Ma
Lingqi Ma Yuqi Bai3†
Yuqi Bai3†
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Genet., 14 March 2024
Sec. Evolutionary and Genomic Microbiology
Volume 15 - 2024 | https://doi.org/10.3389/fgene.2024.1356956
Prescottella, a distinct genus separate from Rhodococcus, has garnered attention for its adaptability and ecological versatility. In this study, a Gram-stain positive and ovoid-rod shaped the actinobacterium strain R16 was isolated from deep-sea sediment (with a depth of 6,310 m) in the Western Pacific. On the basis of 16S rRNA gene sequence analysis, average nucleotide identity and phylogenomic analysis, strain R16 clearly represents a novel species within the genus Prescottella. Genomic analyses indicate Prescottella sp. R16 contains a circular chromosome of 4,531,251 bp with an average GC content of 68.9%, 4,208 protein-coding genes, 51 tRNA genes, and 12 rRNA operons. Additionally, four CRISPRs and 24 genomic islands are also identified. The presence of rich categories related to catalytic activity, membrane part and metabolic process highlights their involvement in cellular component, biological process, and molecular function. The genome sequence of strain R16 also revealed the presence of 13 putative biosynthetic gene clusters for secondary metabolites, including those for ε-Poly-L-lysine, ectoine, heterobactin, isorenieratene and corynecin, suggesting its potential for antibiotic production and warranting further exploration.
The emergence of Prescottella as a distinct genus, separate from Rhodococcus, has drawn attention due to its adaptability and ecological versatility (Paterson et al., 2019; Kalinowski et al., 2020). Phylogenetically distinct from other rhodococci, strains of Prescottella sp. were reclassified as a new genus through comprehensive taxonomic analysis (Jones et al., 2013; Sangal et al., 2022). Prescottella sp. exhibits extraordinary resilience in extreme environments, thriving in diverse settings such as soil, water, and plant ecosystems. Its remarkable adaptability is further demonstrated by its isolation from varied environmental sources, including animal manure and rock cores (Kalinowski et al., 2020; Ivshina et al., 2022). In biotechnology, Prescottella exhibits significant potential for synthesizing valuable compounds, including biosurfactants and bioflocculants (Cappelletti et al., 2020). It also plays a crucial role in biodegradation (Ivshina et al., 2022) and bioremediation (Kuyukina and Ivshina, 2010). Notably, it excels in degrading pollutants in contaminated ecosystems and transforming pharmaceutical compounds. Its role in biocatalysis, especially in the development of pharmaceutical precursors and new drugs, underscores its importance (Anteneh and Franco, 2019; Busch et al., 2019).
Recent advancements in Whole-genome sequencing (WGS), particularly with third-generation sequencing using PacBio technology, have revolutionized bacterial functional analysis. A complete and highly accurate genome sequence serves as a pivotal foundation for unraveling the genetic features of microorganisms. In this context, high-throughput sequencing technologies, including third-generation sequencing, have played a critical role in obtaining such genome sequences (Didelot et al., 2012; Ma et al., 2016). Particularly, PacBio technology offers an advanced microbial genome assembly method characterized by extended read lengths and reduced GC bias.
In this study, we report the high-quality sequence of a novel strain of Prescottella sp. R16 strain, isolated from marine sediment at a depth of 6,310 m in the Western Pacific. This sequencing was conducted using the Single Molecule Real-Time (SMRT) technique on the PacBio Sequel platform. After assembly, recycling, error correction and annotation, a complete genome map of 4.53 Mb (including 1 plasmid) was obtained. Further bioinformatics analysis revealed its potential in lipid transport, metabolism, and amino acid biosynthesis. The complete genome sequence will provide valuable insight for its application in biotechnological and natural product biosynthesis applications.
The bacterial strain was obtained from deep-sea sediment samples collected from the western Pacific Ocean, located at coordinates N10°54.7′ and E142°19.9′, at a depth of 6,310 m. Isolation was performed by spreading the sediment samples on a specialized isolation medium containing 10 g of glucose, 5 g of peptone, 5 g of yeast extract, 0.2 g of MgSO4·7H2O, 10 g of NaHCO3, 27 g of Na2CO3·10H2O, and 20 g of agar per liter of natural seawater with a pH of 10. Incubation took place at 28°C for a duration of 3 weeks. A single bacterial colony was selected and subsequently streaked onto plates containing marine agar 2216 (MA; Becton Dickinson) to obtain pure cultures. These cultures were cryopreserved at −80°C in a glycerol suspension comprising 15% (v/v) glycerol and 0.5% (w/v) trehalose.
Genomic DNA extraction was carried out using a bacterial genomic DNA extraction kit (TaKaRa, Dalian, China). The near-complete 16S rRNA gene of strain R16 was amplified from the genomic DNA and sequenced using universal bacterial primers 27F (5′-AGAGTTTGATCCTGGCTCAG-3′) and 1492R (5′-GGTTACCTTGTTACGACTT-3′). The sequence was compiled using Contig Express software and compared with 16S rRNA gene sequences of valid species from GenBank via the BLAST program and the EzTaxon-e server (Yoon et al., 2017).
Cell morphology of strain R16 was observed with light microscopy after incubation on MA medium at 25°C for 3 days. Standard Gram staining was performed as described by Cerny (1978). Growth was tested at different temperatures (10, 15, 20, 25, 30, 35, 40°C and 45°C) and pH (4.0–12.0, at intervals of 1.0 pH unit) on MA liquid medium. NaCl tolerance (0%–8%, at intervals of 1%, w/v) was also tested using salt-free MA liquid medium as the basal medium. The growth of the strains was observed at 8 h intervals, in extreme conditions, the observation time should be extended to 14 days. Catalase activity was detected by the production of bubbles after the addition of a drop of 3% (v/v) H2O2. Oxidase activity was determined by the oxidation of tetramethyl-p-phenylenediamine. H2S production test and hydrolysis of cellulose and starch were performed as described by Li et al. (2007). Additional physiological and biochemical characteristics of strain R16 were carried out using API ZYM (bioMérieux) and API 20NE (bioMérieux) kits according to the manufacturer’s instructions.
The complete genome of the bacterial strain R16 was sequenced utilizing the PacBio Sequel system (Pacific Biosciences, Menlo Park, CA, USA). Genome assembly was performed following a hierarchical genome-assembly process (HGAP) as described by Chin et al. (2013) using HGAP4 (v. 6.0) software. Open-reading frame (ORF) prediction was accomplished using Prodigal (v. 2.6.3) software with default parameters, and ORFs spanning sequencing gap regions were excluded. The bacterial proteome was annotated using BLAST (v. 2.6.0) software by alignment with the Clusters of Orthologous Groups (COGs) database. Metabolic pathways within the bacterium were reconstructed using the online tool KEGG Mapper (Kanehisa and Sato, 2020). Identification of ribosomal RNA (rRNA) genes and transfer RNA (tRNA) genes was performed using RNAmmer (v. 1.2) and tRNAScanSE (v. 2.0.9) (Lagesen et al., 2007), respectively. Clustered regularly interspaced short palindromic repeats (CRISPR) elements were identified using CRISPRCasFinder (v. 4.3.2) with default parameters (Couvin et al., 2018). Genomic islands were detected using the web tool IslandViewer (v. 4.0) with the independent methods Islander and IslandPath-DIMOB with default parameters (Bertelli et al., 2017). The presence of secondary metabolic gene clusters was assessed using the antiSMASH 5.0 platform with default parameters (Blin et al., 2019). The pan-genome analysis was performed on the IPGA platform v1.09 (https://nmdc.cn/ipga/ (Liu et al., 2022). The parameters of the analysis process are all default values. CheckM (v.1.0.18) was used to assess the completeness and contamination of the reference genomes that used in this paper based on marker gene sets of 111 essential single-copy genes. Average nuclear identities (ANI) and 16S rRNA similarity were evaluated between ten reference genomes utilizing pyani (https://github.com/widdowquinn/pyani) and Blastn with the initial setting.
Utilizing whole-genome sequences represents a promising approach for elucidating the phylogenetic relationships among microorganisms. When it comes to strain identification, the gold standard in evolutionary phylogeny analysis relies on the examination of core genomes, which is more robust than relying on a single gene marker or concatenated sequences of a limited number of genes. With this rationale in mind, we conducted a phylogenomic analysis based on the core genes found in the entirety of the genome sequences of ten typical strains, all of which exhibited genome completeness exceeding 95%. These genome sequences were retrieved from the National Center for Biotechnology Information (NCBI) database.
The extraction of core genes was carried out using the state-of-the-art Bacterial Core Gene (UBCG) pipeline (Na et al., 2018). Subsequently, these genes were concatenated, and a maximum-likelihood tree was constructed employing the Genetic Testing Registry (GTR) model, facilitated by the RAxML (v. 7.0.4) tool (Stamatakis, 2014). The selection of 92 core genes was informed by a comprehensive dataset comprising 1,429 complete genome sequences spanning 28 phyla, ensuring the inclusion of genes that are either widely distributed across genomes or exhibit high conservation as single-copy genes.
Colonies of strain R16 grown on MA medium were observed to be circular, opaque, smooth and slightly convex, with light pink colouration after incubation for 3 days at 25°C. The cells of the organism were found to be Gram-stain positive, ovoid-rod shaped, occurring singly or in pairs. Growth of strain R16 occurred at 20°C–35°C, pH 5-9 and in 0%–4% (w/v) NaCl, with optimal growth occurring at 25°C, pH 7 and in 1% NaCl. Oxidase-negative and catalase-positive. Negative for H2S production and hydrolysis of cellulose and starch. In the API ZYM strip, positive for the activity of alkaline phosphatase, lipase (C14), cystine arylamidase, trypsin, α-chymotrypsin, α-glucosidase, N-acetyl-β-glucosaminidase, β-glucuronidase, β-glucosidase, α-galactosidase, β-galactosidase, α-mannosidase and β-fucosidase; negative for the activity of esterase (C4), leucine arylamidase, acid phosphatase, naphthol-AS-BI-phosphohydrolase, esterase lipase (C8) and valine arylamidase. In the API 20NE strip, positive for reduction of nitrate to nitrite, urease activity and hydrolysis of p-nitrophenyl-β-D-galactopyranoside and esculin; negative for denitrification, indole production, glucose fermentation, arginine dihydrolase activity, hydrolysis of aesculin and gelatin, and assimilation of adipic acid, D-glucose, D-mannose, D-maltose, L-arabinose, N-acetylglucosamine, D-mannitol, potassium gluconate, capric acid, malic acid, trisodium citrate and phenylacetic acid.
The taxonomic placement of Prescottella sp. has long posed a challenge for taxonomists, with considerable controversy surrounding its classification within the genus Rhodococcus. To elucidate its phylogenomic relationships, we employed strain of the Aldersonia genus (specifically, Aldersonia kunmingensis DSM 45001) as an outgroup for comparative analysis. Additionally, we included five strains from the Prescottella genus (Prescottella equi DSSKP-R-001, Prescottella agglutinans CFH S0262, Prescottella defluvii Ca11, Prescottella subtropica C9-28, and Prescottella sp. R16) along with four strains from the Rhodococcus genus (Rhodococcus wratislaviensis WS3308, Rhodococcus pseudokoreensis R79, Rhodococcus triatomae DSM 44893, and Rhodococcus rhodochrous EP4) in the construction of a phylogenomic tree based on their genome sequences (Figure 1).

FIGURE 1. Maximum likelihood tree of 92 concatenated markers and genome similarity heatmap show the affiliations of the Prescottella sp. R16(bold font) and eight other strains of the genera Prescottella and Rhodococcus. The phylogenetic tree was rooted by Aldersonia were used as outgroups. The genome sequence of each strain is available from the NCBI database, and the GenBank accession number is shown in parentheses. Bar, 0.1 substitution per nucleotide position.
The phylogenomic analysis revealed a distinct topological arrangement within the constructed tree (Figure 1), featuring two primary population clusters alongside the outgroup. Notably, Prescottella sp. R16 and four other Prescottella strains formed a novel evolutionary branch, exhibiting the shortest evolutionary distance in comparison to Prescottella subtropica C9-28. Besides, Prescottella sp. R16 shows the highest 16S rRNA similarity value (99.51%) with Prescottella subtropica. However, the ANI clearly differentiated the R16 strain from each other and from their closest relatives, with values ranging from 74.71% to 87.69% for ANI that below the threshold 95%–96% for species delineation (Chun et al., 2018). Consequently, we propose that Prescottella sp. R16 should be classified as a new member within the Prescottella genus, further contributing to the understanding of its taxonomic position.
The genome characteristics of strain Prescottella sp. R16 and reference strains are presented in Table 1. The complete genome of Prescottella sp. R16 contains a circular chromosome of 4,531,251 bp with an average GC content of 68.9%, which consists of 4,208 protein-coding genes, 51 tRNA genes, and 12 rRNA operons. In addition, a plasmid is included, and a total of 12 protein coding sequences (CDS) are annotated using the SwissProt public database (Supplementary Table S1) and visualized according to their physical locations (Supplementary Figure S1). Four CRISPRs and 24 genomic islands are identified in the genome (Supplementary Tables S2, S3). A circular map of the genome was generated in GCView (Stothard and Wishart, 2005) (Figure 2). Based on the IPGA platform, we analyzed the genomes of Prescottella sp. R16 and reference strains. Pan-genome statistic visualization is showed on the Supplementary Figure S2 and Supplementary Table S4. When more genomes are analyzed, the larger the pan-genome, the fewer the core genes. Among the 26,435 orthologous gene clusters, only 1,450 (5.48%) were core gene clusters (Supplementary Table S5 and Supplementary Figure S3). Considering the genomic contigs will make an impact on the results, so we independent take the most closed Prescottella equi NCTC1621 and Prescottella equi NCTC1621 strains that own less contigs for further compare. Among them, there were 695 unique genes belonged to the strain R16 (Supplementary Figure S4). These results provide evidence at the gene level that strain R16 is highly divergent from other species of the genus Prescottella.

TABLE 1. General features of strain Prescottella sp. R16.
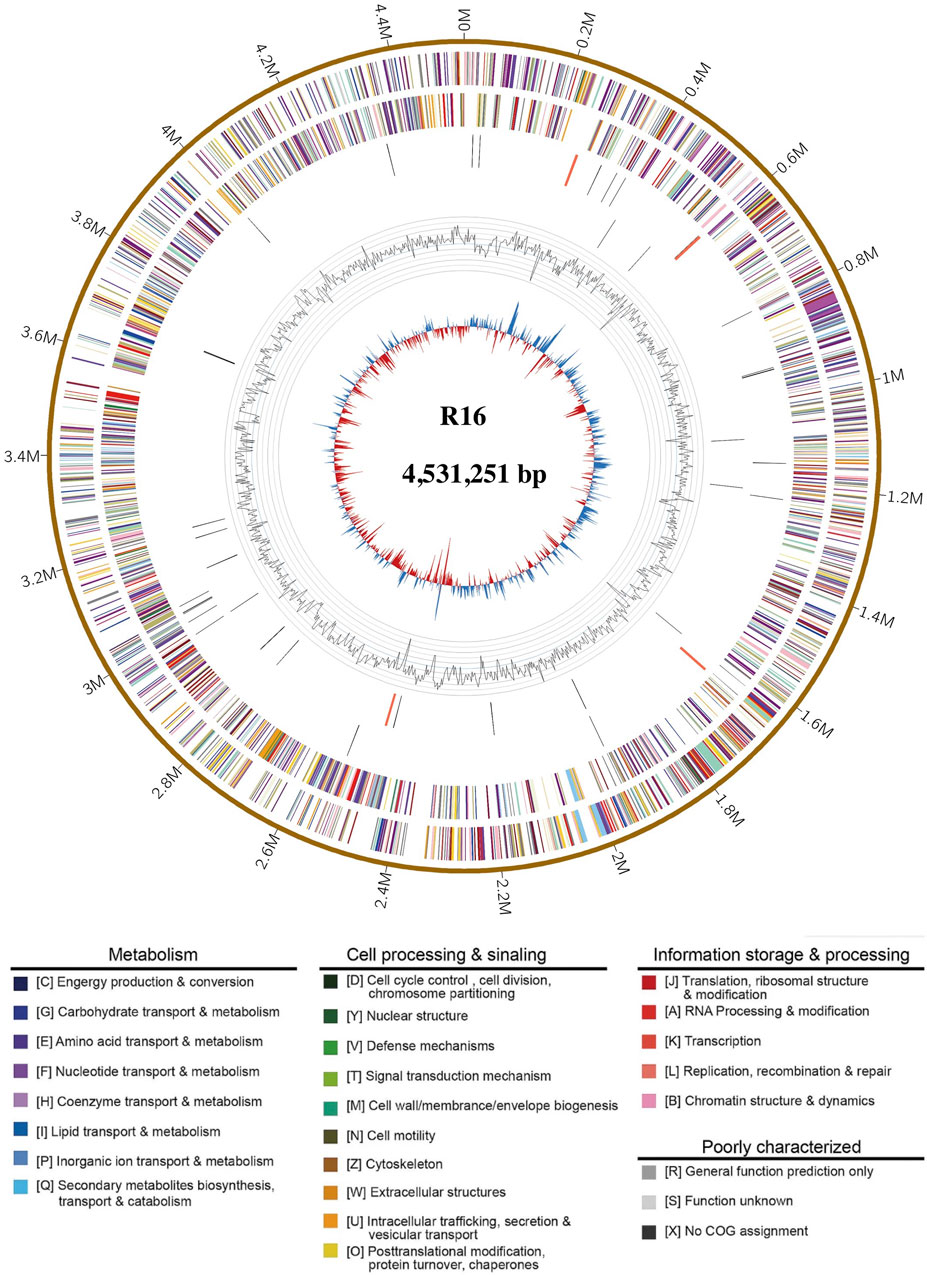
FIGURE 2. Complete genome map of Prescottella sp. R16. Rings from the outside to the center indicate: 1) scale marks for genome size (Mb); 2, 3) protein-coding genes on the forward/reverse strand; 4, 5) tRNA (black) and rRNA (orange) genes on the forward/reverse strand; 6) genomic islands, prediction methods: Integrated (red) and IslandPath-DIMOB (blue); 7) GC content. Protein-coding genes are color coded according to their COG categories (color figure online).
The GO term enrichment analysis of genes provides valuable insights into the potential functions and adaptations of Prescottella sp. R16 (Consortium et al., 2004). The presence of rich categories related to catalytic activity, membrane part, and metabolic process highlights their involvement in cellular component, biological process, and molecular function (Supplementary Figure S5A), respectively. The COG analysis results highlight the functional diversity and adaptation strategies of genes in the Prescottella sp. R16 genome (Koonin, 2002). The enrichment in categories such as general function prediction only, amino acid transport and metabolism, and lipid transport and metabolism underscore the importance of versatility and resource utilization in the survival and success of these bacteria (Supplementary Figure S5B). Further research focused on characterizing specific genes within these categories can provide a deeper understanding of the molecular mechanisms driving their adaptation to different environments and their potential ecological roles.
Biosynthetic clusters encoding potential secondary metabolites were identified using AntiSMASH version 5.0 (Blin et al., 2019). A total of 13 putative biosynthetic gene clusters have been discerned (Supplementary Table S6), each presumed to play a pivotal role in the synthesis of diverse secondary metabolites. Notably, five of these clusters bear a substantial degree of homology with previously documented gene clusters. Specifically, these clusters correspond to ε-Poly-L-lysine (100% homology), ectoine (75% homology), heterobactin B/heterobactin S2 (63% homology), isorenieratene (42% homology), and corynecin III/corynecin I/corynecin II (40% homology). Conversely, the remaining eight putative gene clusters demonstrate relatively lower degrees of similarity, with homologies of 30% or less compared to their closest known counterparts. Notably, three of these gene clusters display no discernible similarity to any previously reported gene clusters, thereby suggesting the possibility that they represent novel genetic pathways implicated in the biosynthesis of secondary metabolites.
Rhodococcus species have garnered significant attention within the field of microbiology, being the subject of extensive research for their utility as biocatalysts in steroid production and their efficacy as bioremediation agents (Van der Geize and Dijkhuizen, 2004). Whole-genome sequencing data has unveiled the substantial capacity of Prescottella for secondary metabolite production. Nonetheless, the isolation of natural products from this genus remains relatively limited. Previous research has elucidated the remarkable chemical diversity inherent in siderophore-related secondary metabolites across the Rhodococcus genus (Bosello et al., 2013). Our recent investigations significantly contribute to the broader comprehension of metabolite diversity within Prescottella, encompassing remarkable compounds such as heterobactin, capreomycin, corynecin, and several others of note (Figures 3A, B). Moreover, our research has unveiled novel gene clusters that offer a promising avenue for the isolation of previously unidentified siderophores, thus advancing our understanding of their variability within this taxonomic group.
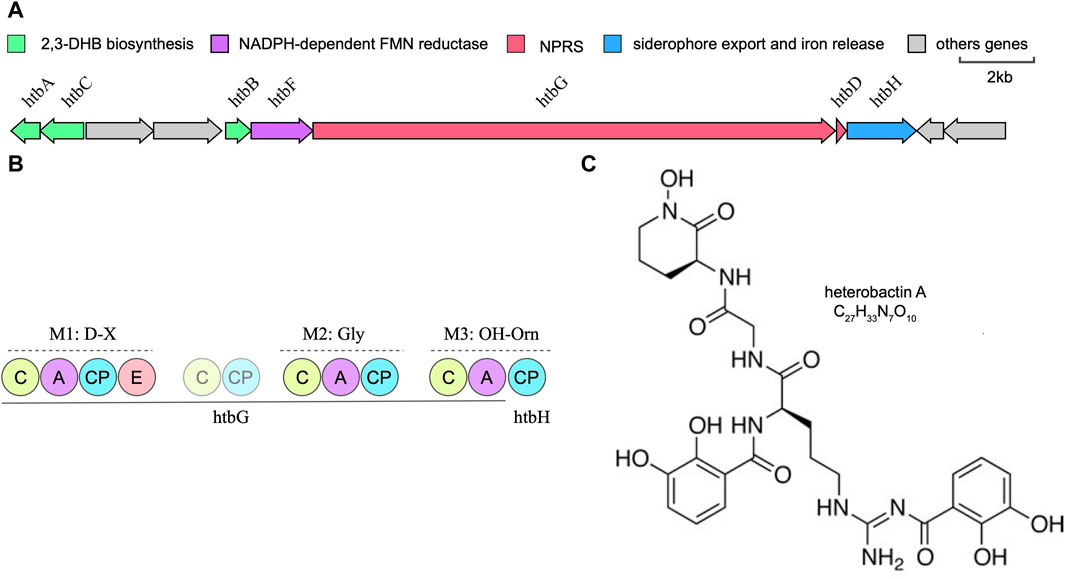
FIGURE 3. Bioinformatic overview of the heterobactin biosynthetic gene cluster. (A) Arrangement of the htb gene cluster in strain Prescottella sp. R16. (B) Domain organization and the adenylation domain substrate predictions of the NRPSs htbG and htbH. The filled circle means domain in a complete module and the transparent circle mean domain in an incomplete module or outside modules. C:Condensation; A: AMP-binding; CP: PP-binding; E:Epomerization. (C) Chemical structure of the heterobatin A.
Heterobactin A is a siderophore composed of the tripeptide sequence (N-OH)-L-Orn-Gly-D-Orn-(delta-N-dihydroyxbenzoate) (Figure 3C). The structure of heterobactin A typically includes functional groups that have a strong affinity for iron, such as hydroxamic acid or catechol moieties. These groups form coordination bonds with iron ions, creating stable complexes. The formation of these complexes enhances the solubility of iron in the extracellular environment and allows bacteria to acquire iron for their metabolic needs (Bosello et al., 2013). Iron is a key nutrient for bacterial growth and survival, and studies of heterobactin A’s structure and its relationship with siderophores provides valuable insights into the mechanisms by which bacteria acquire iron.
Here, we report a complete genome assembly of strain Prescottella sp. R16 from marine sediments at 6,310 m depth in the western Pacific Ocean based on PacBio technology. The genome size is 4.53Mb (including 1 plasmid), and a total of 4208 coding genes are annotated. Further analysis has confirmed the presence of a secondary metabolite biosynthetic gene cluster in the genome sequence, revealing its potential in lipid transport, metabolism, and amino acid biosynthesis. This research is of great significance for expanding microbial genome resources and will provide valuable insights into understanding microbial diversity, biotechnology, and applications in natural product biosynthesis.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
LM: Writing–review and editing, Writing–original draft, Project administration, Formal Analysis, Conceptualization. YB: Data curation, Formal analysis, Visualization, Writing–review and editing. WW: Writing–review and editing, Resources, Methodology, Investigation, Formal Analysis. SP: Writing–review and editing, Software, Methodology, Data curation. GZ: Writing–review and editing, Resources, Project administration, Funding acquisition, Conceptualization.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was funded by the National Natural Science Foundation of China through grant 42206140, the China Postdoctoral Science Foundation through grant 2021M702728, the Cross-Strait Postdoctoral Fellowship Plan through grant 2021B001, and the Outstanding Postdoctoral Scholarship, State Key Laboratory of Marine Environmental Science at Xiamen University.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2024.1356956/full#supplementary-material
Anteneh, Y. S., and Franco, C. M. M. (2019). Whole cell actinobacteria as biocatalysts. Front. Microbiol. 10, 77. doi:10.3389/fmicb.2019.00077
Aus der Beek, T., Weber, F. A., Bergmann, A., Hickmann, S., Ebert, I., Hein, A., et al. (2016). Pharmaceuticals in the environment—global occurrences and perspectives. Environ. Toxicol. Chem. 35 (4), 823–835. doi:10.1002/etc.3339
Bertelli, C., Laird, M. R., Williams, K. P., Lau, B. Y., Hoad, G., Winsor, G. L., et al. (2017). IslandViewer 4: expanded prediction of genomic islands for larger-scale datasets. Nucleic acids Res. 45, W30–W35. W1. doi:10.1093/nar/gkx343
Blin, K., Shaw, S., Steinke, K., Villebro, R., Ziemert, N., Lee, S. Y., et al. (2019). antiSMASH 5.0: updates to the secondary metabolite genome mining pipeline. Nucleic acids Res. 47, W81–W87. W1. doi:10.1093/nar/gkz310
Bosello, M., Zeyadi, M., Kraas, F. I., Linne, U., Xie, X., and Marahiel, M. A. (2013). Structural characterization of the heterobactin siderophores from Rhodococcus erythropolis PR4 and elucidation of their biosynthetic machinery. J. Nat. Prod. 76 (12), 2282–2290. doi:10.1021/np4006579
Busch, H., Hagedoorn, P.-L., and Hanefeld, U. (2019). Rhodococcus as a versatile biocatalyst in organic synthesis. Int. J. Mol. Sci. 20 (19), 4787. doi:10.3390/ijms20194787
Cappelletti, M., Presentato, A., Piacenza, E., Firrincieli, A., Turner, R. J., and Zannoni, D. (2020). Biotechnology of Rhodococcus for the production of valuable compounds. Appl. Microbiol. Biotechnol. 104, 8567–8594. doi:10.1007/s00253-020-10861-z
Carrano, C. J., Jordan, M., Drechsel, H., Schmid, D. G., and Winkelmann, G. (2001). Heterobactins: a new class of siderophores from Rhodococcus erythropolis IGTS8 containing both hydroxamate and catecholate donor groups. Biometals 14, 119–125. doi:10.1023/a:1016633529461
Cerny, G. (1978). Studies on the aminopeptidase test for the distinction of gram-negative from gram-positive bacteria. Appl. Microbiol. Biotechnol. 5, 113–122. doi:10.1007/bf00498805
Chin, C. S., Alexander, D. H., Marks, P., Klammer, A. A., Drake, J., Heiner, C., et al. (2013). Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nat. methods 10 (6), 563–569. doi:10.1038/nmeth.2474
Chun, J., Oren, A., Ventosa, A., Christensen, H., Arahal, D. R., da Costa, M. S., et al. (2018). Proposed minimal standards for the use of genome data for the taxonomy of prokaryotes. Int. J. Syst. Evol. Microbiol. 68 (1), 461–466. doi:10.1099/ijsem.0.002516
Consortium, G. O., Clark, J., Ireland, A., Lomax, J., Ashburner, M., Foulger, R., et al. (2004). The Gene Ontology (GO) database and informatics resource. Nucleic acids Res. 32, D258–D261. suppl_1. doi:10.1093/nar/gkh036
Couvin, D., Bernheim, A., Toffano-Nioche, C., Touchon, M., Michalik, J., Néron, B., et al. (2018). CRISPRCasFinder, an update of CRISRFinder, includes a portable version, enhanced performance and integrates search for Cas proteins. Nucleic acids Res. 46, W246–W251. W1. doi:10.1093/nar/gky425
Didelot, X., Bowden, R., Wilson, D. J., Peto, T. E., and Crook, D. W. (2012). Transforming clinical microbiology with bacterial genome sequencing. Nat. Rev. Genet. 13 (9), 601–612. doi:10.1038/nrg3226
Ivshina, I., Bazhutin, G., and Tyumina, E. (2022). Rhodococcus strains as a good biotool for neutralizing pharmaceutical pollutants and obtaining therapeutically valuable products: through the past into the future. Front. Microbiol. 13, 967127. doi:10.3389/fmicb.2022.967127
Jones, A. L., Sutcliffe, I. C., and Goodfellow, M. (2013). Prescottia equi gen. nov., comb. nov.: a new home for an old pathogen. Ant. Van Leeuwenhoek 103, 655–671. doi:10.1007/s10482-012-9850-8
Kalinowski, M., Jarosz, Ł., and Grądzki, Z. (2020). Assessment of antimicrobial susceptibility of virulent strains of Rhodococcus equi isolated from foals and soil of horse breeding farms with and without endemic infections. J. equine veterinary Sci. 91, 103114. doi:10.1016/j.jevs.2020.103114
Kanehisa, M., and Sato, Y. (2020). KEGG Mapper for inferring cellular functions from protein sequences. Protein Sci. 29 (1), 28–35. doi:10.1002/pro.3711
Koonin, E. V. (2002). “The Clusters of Orthologous Groups (COGs) Database: phylogenetic classification of proteins from complete genomes,” in The NCBI handbook.
Kuyukina, M. S., and Ivshina, I. B. (2010). Application of Rhodococcus in bioremediation of contaminated environments. Biol. Rhodococcus, 231–262. doi:10.1007/978-3-642-12937-7_9
Lagesen, K., Hallin, P., Rødland, E. A., Stærfeldt, H.-H., Rognes, T., and Ussery, D. W. (2007). RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic acids Res. 35 (9), 3100–3108. doi:10.1093/nar/gkm160
Li, W. J., Xu, L. H., Liu, Z. H., and Jiang, C. L. (2007). Actinomycete systematic——principle, methods and practice. Beijing: Science press.
Liu, D., Zhang, Y., Fan, G., Sun, D., Zhang, X., Yu, Z., et al. (2022). IPGA: a handy integrated prokaryotes genome and pan-genome analysis web service. iMeta 1 (4), e55. doi:10.1002/imt2.55
Ma, T., Zhou, Y., Li, X., Zhu, F., Cheng, Y., Liu, Y., et al. (2016). Genome mining of astaxanthin biosynthetic genes from Sphingomonas sp. ATCC 55669 for heterologous overproduction in Escherichia coli. Biotechnol. J. 11 (2), 228–237. doi:10.1002/biot.201400827
Na, S.-I., Kim, Y. O., Yoon, S.-H., Ha, S.-m., Baek, I., and Chun, J. (2018). UBCG: up-to-date bacterial core gene set and pipeline for phylogenomic tree reconstruction. J. Microbiol. 56, 280–285. doi:10.1007/s12275-018-8014-6
Paterson, M. L., Ranasinghe, D., Blom, J., Dover, L. G., Sutcliffe, I. C., Lopes, B., et al. (2019). Genomic analysis of a novel Rhodococcus (Prescottella) equi isolate from a bovine host. Archives Microbiol. 201, 1317–1321. doi:10.1007/s00203-019-01695-z
Sangal, V., Goodfellow, M., Jones, A. L., and Sutcliffe, I. C. (2022). A stable home for an equine pathogen: valid publication of the binomial Prescottella equi gen. nov., comb. nov., and reclassification of four rhodococcal species into the genus Prescottella. Int. J. Syst. Evol. Microbiol. 72 (9), 005551. doi:10.1099/ijsem.0.005551
Stamatakis, A. (2014). RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30 (9), 1312–1313. doi:10.1093/bioinformatics/btu033
Stothard, P., and Wishart, D. S. (2005). Circular genome visualization and exploration using CGView. Bioinformatics 21 (4), 537–539. doi:10.1093/bioinformatics/bti054
Van der Geize, R., and Dijkhuizen, L. (2004). Harnessing the catabolic diversity of rhodococci for environmental and biotechnological applications. Curr. Opin. Microbiol. 7 (3), 255–261. doi:10.1016/j.mib.2004.04.001
Keywords: Prescottella, whole-genome sequencing, phylogenetic analysis, biotechnological applications, extreme environments
Citation: Ma L, Bai Y, Wang W, Pei S and Zhang G (2024) Complete genome sequence of a novel Prescottella sp. R16 isolate from deep-sea sediments in the western Pacific. Front. Genet. 15:1356956. doi: 10.3389/fgene.2024.1356956
Received: 16 December 2023; Accepted: 22 February 2024;
Published: 14 March 2024.
Edited by:
David W. Ussery, University of Arkansas for Medical Sciences, United StatesReviewed by:
Sankarasubramanian Jagadesan, University of Nebraska Medical Center, United StatesCopyright © 2024 Ma, Bai, Wang, Pei and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gaiyun Zhang, emhneXVuQHRpby5vcmcuY24=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.