
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Genet., 15 February 2024
Sec. Genetics of Common and Rare Diseases
Volume 15 - 2024 | https://doi.org/10.3389/fgene.2024.1355962
This article is part of the Research TopicInborn errors of Carbohydrate MetabolismView all 15 articles
 Bianca Panis1,2,3†E. Naomi Vos1,2,3,4,5†Ivo Barić6
Bianca Panis1,2,3†E. Naomi Vos1,2,3,4,5†Ivo Barić6 Annet M. Bosch2,3,7Martijn C. G. J. Brouwers2,8
Annet M. Bosch2,3,7Martijn C. G. J. Brouwers2,8 Alberto Burlina2,9David Cassiman10
Alberto Burlina2,9David Cassiman10 David J. Coman11
David J. Coman11 María L. Couce2,12
María L. Couce2,12 Anibh M. Das2,13Didem Demirbas14Aurélie Empain2,15Matthias Gautschi16
Anibh M. Das2,13Didem Demirbas14Aurélie Empain2,15Matthias Gautschi16 Olga Grafakou2,17
Olga Grafakou2,17 Stephanie Grunewald18Sandra D. K. Kingma2,19
Stephanie Grunewald18Sandra D. K. Kingma2,19 Ina Knerr20Elisa Leão-Teles2,21Dorothea Möslinger2,22
Ina Knerr20Elisa Leão-Teles2,21Dorothea Möslinger2,22 Elaine Murphy23
Elaine Murphy23 Katrin Õunap2,24Adriana Pané2,25Sabrina Paci2,26
Katrin Õunap2,24Adriana Pané2,25Sabrina Paci2,26 Rossella Parini2,27Isabel A. Rivera28Sabine Scholl-Bürgi29
Rossella Parini2,27Isabel A. Rivera28Sabine Scholl-Bürgi29 Ida V. D. Schwartz30Triantafyllia Sdogou2,31Loai A. Shakerdi32Anastasia Skouma2,31Karolina M. Stepien33Eileen P. Treacy34Susan Waisbren14
Ida V. D. Schwartz30Triantafyllia Sdogou2,31Loai A. Shakerdi32Anastasia Skouma2,31Karolina M. Stepien33Eileen P. Treacy34Susan Waisbren14 Gerard T. Berry14‡
Gerard T. Berry14‡ M. Estela Rubio-Gozalbo1,2,3,4,5*‡
M. Estela Rubio-Gozalbo1,2,3,4,5*‡Classic galactosemia (CG, OMIM #230400, ORPHA: 79,239) is a hereditary disorder of galactose metabolism that, despite treatment with galactose restriction, affects brain function in 85% of the patients. Problems with cognitive function, neuropsychological/social emotional difficulties, neurological symptoms, and abnormalities in neuroimaging and electrophysiological assessments are frequently reported in this group of patients, with an enormous individual variability. In this review, we describe the role of impaired galactose metabolism on brain dysfunction based on state of the art knowledge. Several proposed disease mechanisms are discussed, as well as the time of damage and potential treatment options. Furthermore, we combine data from longitudinal, cross-sectional and retrospective studies with the observations of specialist teams treating this disease to depict the brain disease course over time. Based on current data and insights, the majority of patients do not exhibit cognitive decline. A subset of patients, often with early onset cerebral and cerebellar volume loss, can nevertheless experience neurological worsening. While a large number of patients with CG suffer from anxiety and depression, the increased complaints about memory loss, anxiety and depression at an older age are likely multifactorial in origin.
Galactose is a natural aldohexose that exists as free galactose and as a component of complex carbohydrates, glycoproteins and glycolipids. Together with glucose, galactose forms lactose, a disaccharide abundantly present in dairy products. Among other functions, it serves as a key source of energy in infants and is important for galactosylation of complex molecules such as galactocerebroside in myelin. On average, 88% of galactose is retained in the liver (Coelho et al., 2015a; Conte et al., 2021).
The Leloir pathway is the main pathway of galactose metabolism and consists of four steps, consecutively mediated by galactose mutarotase (GALM EC 5.1.3.3), galactokinase (GALK1 EC 2.7.1.6), galactose-1-phosphate uridylyltransferase (GALT, EC 2.7.7.2012) and UDP-galactose 4′-epimerase (GALE, EC 5.1.3.7) (Figure 1A). Galactose entering the Leloir pathway either becomes a precursor for glycosylation (as UDP-galactose) or is used in glycolysis and glycogen synthesis pathways (as UDP-glucose) (Conte et al., 2021).
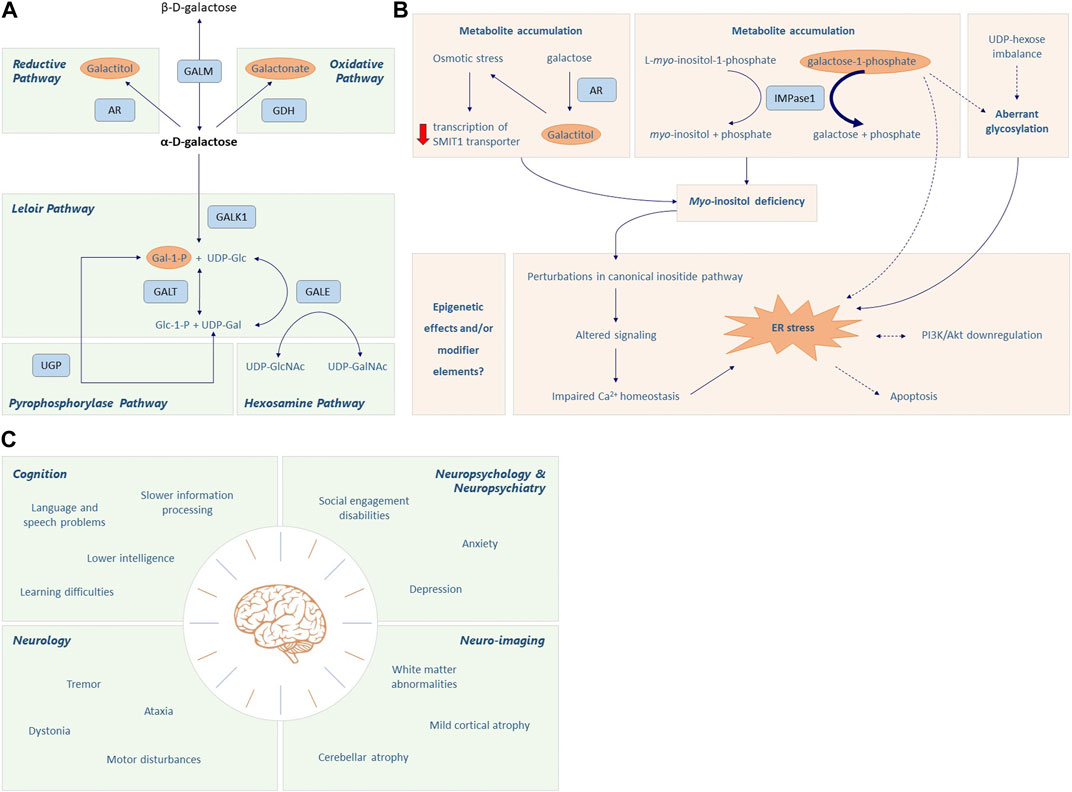
FIGURE 1. Galactose metabolism and CG pathophysiology. (A) The first step of the Leloir pathway involves the conversion of β-D-galactose to its stereoisomer α-D-galactose by galactose mutarotase (GALM). Then, α-D-galactose is phosphorylated to α-D-galactose-1-phosphate (Gal-1-P) by galactokinase (GALK1). Galactose-1-phosphate uridylyltransferase (GALT) catalyzes the 2-step reaction through which Gal-1-P and UDP-glucose are converted to α-D-glucose-1-phosphate and UDP-galactose. Finally, UDP-galactose 4′-epimerase (GALE) mediates the interconversion of UDP-galactose (UDP-gal) and UDP-glucose (UDP-glc). This enzyme is crucial to maintain the steady state UDP-galactose/UDP-glucose ratio in different cells, playing an important role in glycoconjugate formation. Accumulation of α-D-galactose due to GALT deficiency leads to the formation of galactitol and galactonate, mediated by aldose reductase (AR) and galactose dehydrogenase (GALDH), respectively. Additionally, Gal-1-P can be converted into UDP-Gal via the action of UDP-glucose pyrophosphorylase (UGP); however its affinity is much lower when compared to the main substrate, Glc-1-P. Except for GALK1, the enzymes in this pathway can work in both directions, depending on the substrate levels and energy demand of the cell. Please note that there are only two enzymes in humans that are capable of converting Gal-1-P to UDP-galactose, the GALT enzyme and the UGP enzyme. Additionally, while UGP is bidirectional in nature, the reaction usually goes in the direction of Glc-1-P to UDP-Glc because PPi is rapidly hydrolyzed. (B) The accumulation of toxic metabolites, aberrant glycosylation, myo-inositol deficiency, endoplasmic reticulum (ER) stress and oxidative stress, and signaling pathway alterations all seem implicated in the pathophysiological cascade elicited in CG. Increased levels of Gal-1-P can inhibit inositol monophosphatase (IMPase1), which converts L-myo-inositol-1-phosphate to free myo-inositol, thereby limiting the intracellular myo-inositol concentration. Accumulation of galactitol generates osmotic stress which may result in decreased transcription of the myo-inositol cotransporter SMIT1, further aggravating the intracellular myo-inositol deficiency. The myo-inositol deficiency and subsequent alterations in inositide signaling can impair calcium homeostasis and cause ER stress, which is associated with apoptosis and downregulation of PI3K/Akt signaling. Gal-1-P and aberrant glycosylation may also contribute to ER stress. Lastly, the role of epigenetics and modifier genes also needs to be considered in CG pathology. Dotted lines represent associations that are still under debate. (C) Classic Galactosemia (CG) patients can suffer from brain pathology in multiple domains, i.e., cognition, neurology, neuropsychology, neuropsychiatry and neuro-imaging.
In Classic Galactosemia (CG), severe deficiency of GALT (<1% residual activity) fuels several alternative galactose disposal routes. Firstly, aldose reductase (AR, EC 1.1.1.21) converts α-D-galactose into galactitol in a NADPH-dependent reaction. Secondly, galactose is oxidized to galactonate by galactose dehydrogenase (GALDH, EC 1.1.1.48), producing NADH. Galactonate is excreted from the body or converted to D-xylulose 5-phosphate to enter the pentose phosphate pathway (Coelho et al., 2015a; Conte et al., 2021). Lastly, although a poor substrate (low affinity), Gal-1-P can be converted to UDP-galactose by UDP-glucose pyrophosphorylase 2 (UGP2, EC 2.7.7.9) (Coelho et al., 2015a).
The first description of an infant with galactosemia dates from 1908 (von Reuss, 1908). In the following years, more patients were described with hypergalactosemia and neonatal illness, including hepatocellular damage, renal tubular disease, Escherichia coli sepsis, encephalopathy and cataract (Göppert, 1917; Mason and Turner, 1935). A well-recognized phenomenon is brain edema, also called ‘pseudotumor cerebri’, leading to increased intracranial pressure and bulging of the fontanel (Wells et al., 1965; Quan-Ma et al., 1966; Huttenlocher et al., 1970; Belman et al., 1986; Berry et al., 2001). A galactose-restricted diet resolves the acute neonatal symptoms but is insufficient to prevent long-term complications, which have the same prevalency in patients with and without neonatal illness.
Brain impairments occur in 85% of CG patients despite diet (Rubio-Gozalbo et al., 2019) (Figure 1C; Supplementary Table S1). Cognitive problems frequently experienced are global developmental delay and language delay (Komrower and Lee, 1970; Fishler et al., 1980; Waisbren et al., 1983; Waggoner et al., 1990; Schweitzer et al., 1993; Kaufman et al., 1995a; Hansen et al., 1996; Robertson et al., 2000; Antshel et al., 2004; Bosch et al., 2004; Potter et al., 2008; Hughes et al., 2009; Hoffmann et al., 2011; Potter, 2011; Timmers et al., 2011; Timmers et al., 2012; Waisbren et al., 2012; Potter et al., 2013; Demirbas et al., 2019; Kuiper et al., 2019; Welsink-Karssies et al., 2020a; Welsink-Karssies et al., 2020b; Hermans et al., 2023), with a below average mean total intelligence quotient (IQ) of 87 (Coss et al., 2013; Welling et al., 2017a). The language and speech impairments cannot solely be explained by lower cognitive abilities (Waisbren et al., 1983; Waggoner et al., 1990; Schweitzer et al., 1993; Kaufman et al., 1995a; Robertson et al., 2000; Antshel et al., 2004; Potter et al., 2008; Hughes et al., 2009; Potter, 2011; Timmers et al., 2011; Timmers et al., 2012; Waisbren et al., 2012; Potter et al., 2013; Kuiper et al., 2019). Expressive language is mainly affected, with receptive language and comprehension being relatively preserved (Potter et al., 2008; Timmers et al., 2011). Among the speech disorders are verbal dyspraxia (23.5%) and dysarthria (19.9%) (Nelson et al., 1991; Potter et al., 2008; Potter, 2011; Kuiper et al., 2019). Patients require more time to prepare and finish the utterances and make more errors (Timmers et al., 2012), and also recruit additional and more extensive brain regions than control participants (Timmers et al., 2015a).
Approximately half of the CG patients suffer from neurological complications, the most prevalent being tremor (31.0%), which may affect daily life in some cases (Rubio-Gozalbo et al., 2019). Other complications include general motor abnormalities, ataxia, dystonia and epilepsy (Jan and Wilson, 1973; Lo et al., 1984; Bohles et al., 1986; Friedman et al., 1989; Waggoner et al., 1990; Koch et al., 1992; Schweitzer et al., 1993; Robertson et al., 2000; Arn, 2003; Antshel et al., 2004; Martins et al., 2004; Ridel et al., 2005; Potter et al., 2008; Hughes et al., 2009; Shah and Kuchhai, 2009; Waisbren et al., 2012; Rubio-Agusti et al., 2013; Demirbas et al., 2019; Kuiper et al., 2019; Rubio-Gozalbo et al., 2019; Welling et al., 2019; Özgün et al., 2019; Welsink-Karssies et al., 2020a; MacWilliams et al., 2021). Epilepsy is not frequently reported (Friedman et al., 1989; Aydin-Ozemir et al., 2014) and may be the result of brain damage occurring in the neonatal period, or the consequence of unrelated genetic predisposition. Psychiatric and behavioral problems such as depression and anxiety disorder are reported in 44.4% of the patients (Rubio-Gozalbo et al., 2019). Most have a shy and reserved personality (Antshel et al., 2004) and achieve fewer social developmental milestones when compared to healthy controls, which is postulated to be intrinsic to the disease rather than a result of the burden of a chronic disease or lifelong dietary restrictions (Bosch et al., 2009; Gubbels et al., 2011).
Numerous central nervous system (CNS) grey and white matter abnormalities have been reported in CG (Crome, 1962; Haberland et al., 1971; Lo et al., 1984; Choulot et al., 1991; Koch et al., 1992; Nelson et al., 1992; Kaufman et al., 1995b; Hughes et al., 2009; Timmers et al., 2015b; Timmers et al., 2016; Özgün et al., 2019; Ahtam et al., 2020; Welsink-Karssies et al., 2020a; Welsink-Karssies et al., 2020c). Magnetic resonance imaging (MRI) in a cohort of 67 patients showed cerebral and cerebellar atrophy in 22 and 8 patients, respectively, as well as white matter abnormalities in 11 patients (Nelson et al., 1992). In a study that assessed the integrity of myelinated networks, abnormal somatosensory evoked potentials were present in 17 (28%) of 60 CG patients who had electrophysiological testing of the median nerve, and in 26 (77%) of 34 CG patients who had the posterior tibial nerve tested (Kaufman et al., 1995b). Neurite orientation dispersion and density imaging (NODDI) revealed a lower neurite density index (NDI) in bilateral anterior areas and increased orientation dispersion index (ODI) mainly in the left hemisphere (Timmers et al., 2015b). More recent studies showed lower white matter volume and impaired microstructure in the whole brain, especially in the corticospinal tract (Welsink-Karssies et al., 2020c), as well as the left cerebellum, bilateral putamen and left superior temporal sulcus (Ahtam et al., 2020). Additional disturbances in grey matter density have also been described (Nelson et al., 1992; Dubroff et al., 2008; Timmers et al., 2016; Ahtam et al., 2020).
Several studies have attempted to correlate the grey and white matter abnormalities with clinical outcome. The severity of symptoms at the age of diagnosis was associated with abnormal somatosensory evoked potentials (Kaufman et al., 1995b). Additionally, neurocognitive outcome was linked to patients’ resting-state brain connectivity patterns (van Erven et al., 2017), and grey matter density disturbances associated with later initiation of dietary intervention (Timmers et al., 2016). Furthermore, patients with a tremor and/or dystonia had smaller white matter volume, more impaired white matter microstructure and less myelin compared to patients without movement disorders. Patients with IQ < 85 had grey and white matter abnormalities, as well as lower cerebral and cerebellar volume (Welsink-Karssies et al., 2020c). Lastly, language difficulties were correlated with abnormal diffusivity values of the bilateral dorsal and ventral language networks (Ahtam et al., 2020).
Although the aforementioned abnormalities could help explain the neurocognitive profile, the possibility of a coexistent disorder should always be considered, especially in case of unexpected symptoms (Papachristoforou et al., 2014; Neville et al., 2016; Boca and Whone, 2017; Rossi-Espagnet et al., 2021).
Toxic metabolites, aberrant glycosylation, myo-inositol deficiency, endoplasmic reticulum (ER) stress and oxidative stress, signaling pathway alterations, and structural impairment of GALT, all seem implicated in the pathophysiological cascade elicited in CG (Haskovic et al., 2020) (Figure 1B). Additionally, the role of epigenetics and modifier genes needs to be considered. Different mechanisms could be acting synergistically, depending on the tissue type and developmental stage.
Despite diet, the levels of galactose metabolites are persistently increased due to endogenous production of galactose, which is mainly derived from lysosomal hydrolysis of glycolipids, glycoproteins and proteoglycans (Berry et al., 1995a; Berry et al., 1997). The rate of galactose production is higher in infants and children and decreases until adulthood (Berry et al., 2004; Schadewaldt et al., 2004).
Gal-1-P is deemed one of the key pathogenic agents of CG (Gitzelmann, 1995; Leslie, 2003; Lai et al., 2009). Toxicity has been ascribed to the inhibition of enzymes like UGP, phosphoglucomutase, glycogen phosphorylase and inositol monophosphatase, but convincing evidence is still lacking (Gitzelmann, 1995; Lai et al., 2009). Notably, GALK1 deficiency, which causes accumulation of the galactose metabolites except Gal-1-P, does not give rise to the brain and ovarian complications seen in CG (Tang et al., 2010).
Galactitol excretion in urine can be elevated up to 300 times in patients on diet (Krabbi et al., 2011). Studies have reported galactitol elevations in the brains of neonatal and pediatric CG patients (Quan-Ma et al., 1966; Berry et al., 2001; Otaduy et al., 2006; Rossi-Espagnet et al., 2021). Galactitol is poorly diffusible and highly osmotic, and can lead to cell swelling and brain edema. In vivo elevation of brain galactitol was associated with diffuse white matter abnormalities in a newborn with CG and encephalopathy (Berry et al., 2001). Furthermore, increased T2 signal in white matter and areas of restricted diffusion involving the cortex and deep grey matter nuclei, consistent with cytotoxic edema, was observed in 3 patients during neonatal illness and confirmed galactitol accumulation (Rossi-Espagnet et al., 2021).
Little attention has been paid to the possible role of galactonate in CG pathophysiology. Although the metabolite is excreted in urine or used in the pentose phosphate pathway, its toxicity cannot be ruled out completely and requires further study (Berry et al., 1998).
Aberrant glycosylation has been hypothesized to be a major mechanism of disease (Maratha et al., 2017). UDP-hexoses serve as key sugar donors for glycosylation, and deficiency of UDP-galactose and disturbance of the UDP-glucose/UDP-galactose ratio have been described in CG (Charlwood et al., 1998; Lai et al., 2003; Coss et al., 2014; Maratha et al., 2017). Furthermore, Gal-1-P may compete as substrate for other nucleotide sugar reactions.
Systemic glycan assembly defects have been documented in neonatal illness which largely resolve with galactose restriction (Charlwood et al., 1998; Quintana et al., 2009). However, there is evidence of continuing glycan processing abnormalities (Coss et al., 2012; Coss et al., 2014; Maratha et al., 2017). Of interest, in a CG sibling study, marked differences in outcomes of the second born siblings were noted, with early onset cerebellar and cerebral atrophy in 2 sibling pairs (Hughes et al., 2009), and significant differences in N-glycosylation in later life (Coman et al., 2010).
While differences in glycosylation can be identified in CG individuals at older age, with differing tolerances to moderate galactose intake liberalization (Coss et al., 2012; Knerr et al., 2015), the significance of these findings is unknown. Polymorphic glycan modifier genes (MGAT3, FUT8 and ALG9) can influence glycan chain bisecting and fucosylation, and subsequent cell signaling and adhesion (Wahl et al., 2018).
Myelin may be especially vulnerable to disturbed glycosylation, as it is rich in galactocerebrosides (Barnes-Vélez et al., 2023). Low levels in autopsy brain tissue of an untreated patient raised the question of aberrant glycosylation of galactocerebrosides (Haberland et al., 1971; Koch et al., 1992; Lebea and Pretorius, 2005). Glycosylation also plays an important role in the neuromuscular junction (NMJ) (Dani and Broadie, 2012), and GALT was identified as a potent regulator of NMJ structure in Drosophila melanogaster (Jumbo-Lucioni et al., 2014).
Myo-inositol serves a dual role in human physiology. It is a precursor of membrane phospholipids that are important for calcium- and protein kinase C signaling, and serves as a buffer of osmotic balance (Berry et al., 1995b; Berry, 2011). Brain content of myo-inositol peaks prenatally and continues to decline until a postnatal baseline is reached, which is maintained up to a second decline at middle age (Kreis et al., 2002; Buccafusca et al., 2008). Reduction in intracellular myo-inositol has been associated with impaired integrated stress response signaling and ER stress (Wells and Remy, 1965; Slepak et al., 2007; Hagen-Lillevik et al., 2022). The first reports of myo-inositol deficiency in the brain of CG children date back to 1965 (Wells et al., 1965) and 1966 (Quan-Ma et al., 1966). High levels of Gal-1-P may sequester myo-inositol as inositol monophosphate by inhibition of inositol monophosphatase (Slepak et al., 2007). In addition, galactitol accumulation may lead to poor myo-inositol transport into the cell, further decreasing myo-inositol availability (Berry, 2011).
ER stress (Slepak et al., 2007; De-Souza et al., 2014) and oxidative stress (Slepak et al., 2007; Jumbo-Lucioni et al., 2012; Tang et al., 2014) are two other pathological mechanisms. In fibroblasts derived from CG patients (Slepak et al., 2007) and GalT gene-trapped mice (Balakrishnan et al., 2016; Balakrishnan et al., 2017), evidence was found for activation of the unfolded protein response and ER stress. Interestingly, salubrinal (an eIF2α phosphatase inhibitor) administration in these mice reversed the downregulation of PI3K/Akt signaling pathway and significantly slowed down the loss of Purkinje cells in the cerebellum (Balakrishnan et al., 2017). Additionally, administration of purple sweet potato color (PSPC) and myo-inositol, two compounds hypothesized to rescue aberrant signaling pathways in CG partly due to their antioxidant properties, ameliorated dysregulation of cellular pathways in this model (Hagen-Lillevik et al., 2022).
The fundamental biochemical cause of the disease is a severe decrease in enzymatic activity. Some of the pathogenic variants result in a less stable protein that is unable to reach a correct folding, and so has an increased propensity to aggregation and proteolysis (McCorvie et al., 2013; Coelho et al., 2014).
The role of epigenetics and modifier genes needs to be studied more extensively. Genetic modifiers are genetic variants that can influence the phenotypic outcome of a disease-causing variant in another gene, and have repeatedly been postulated to explain the phenotypic variability seen in CG (see also subsection aberrant glycosylation). It is well recognized that genetic modifiers can affect glycosylation pathways in Congenital Disorders of Glycosylation, rendering what was considered to be single gene abnormalities as ‘multifactorial’ (Quelhas et al., 2023). Several genetic modifiers have already been discovered for other rare Mendelian disorders (Rahit and Tarailo-Graovac, 2020).
Intra-uterine toxicity of galactose metabolites has been postulated an important pathogenic factor (Holton, 1995; Segal, 1995). Gal-1-P was elevated in the liver of galactosemic fetuses at 20 weeks gestation, as well as in the cord blood of galactosemic infants born to mothers who abstained from galactose consumption during pregnancy (Gitzelmann, 1995; Holton, 1995).
GALT activity measured in several animal models throughout development (Shin-Buehring et al., 1977; Rogers et al., 1989a; Rogers et al., 1989b; Rogers et al., 1992; Daude et al., 1996) was particularly higher in the early postnatal period relative to adulthood, which has been attributed to the high galactose ingestion and physiological needs during the suckling period (Rogers et al., 1989a). GALT mRNA and protein are already weakly expressed during late embryonic and postnatal development of the brain and peripheral nerve of the rat, with a peak of expression concomitant with myelogenesis (Daude et al., 1996). GALT activity in the late prenatal stage in various organs of a sheep model (Coelho et al., 2017) showed that galactosemia acute target organs–liver, small intestine and kidney–had the highest late prenatal activity, whereas the chronic target organs–brain and ovary–did not exhibit a noticeable pre- or postnatal different activity, in line with the notion that some organs/cells have a greater susceptibility to impaired galactose metabolism.
Supporting an early life injury, disruptions of fiber tracts and brain nuclei formed during embryogenesis and early fetal brain development were reported in ten adult patients (Ahtam et al., 2020). Moreover, a recent study that used retinal neuro-axonal imaging as a surrogate of brain pathology to assess neuronal integrity and monitor neurodegenerative disease progression pointed towards early brain damage (Lotz-Havla et al., 2023).
Some movement disorders, e.g., tremor, are more frequently seen at an older age (Kuiper et al., 2019). It is not clear whether disease-related mechanisms continue to damage structures (striatum/cerebellum) or whether this is the result of a prenatal/perinatal hit with a dying back phenomenon. Vasogenic edema might play a role in the delayed myelination later in life (Rossi-Espagnet et al., 2021). However, disturbances in myelination are also found in children without neonatal illness, suggesting the implication of other disease mechanisms.
Dietary galactose restriction is currently the cornerstone for treatment but does not prevent long-term complications. In 2016, the members of the Galactosemia Network (GalNet) developed an evidence-based and internationally applicable guideline for diagnosis, treatment and follow-up of CG patients (Welling et al., 2017b). The guideline recommends a galactose-restricted diet that eliminates sources of galactose from dairy products but permits galactose from non-milk sources. The natural history study showed that patients with a liberalized diet did not have a worse outcome neurologically (Rubio-Gozalbo et al., 2019). Moderate liberalization of galactose intake improved IgG glycosylation in a small number of patients (Coss et al., 2014; Knerr et al., 2015). The guideline also offers guidance for testing various neurocognitive and psychosocial domains to facilitate tailored interventions as part of the treatment plan.
In search of new therapeutic approaches, extensive research has been performed to limit accumulation of toxic metabolites or increase levels of deficient metabolites (Simard-Duquesne et al., 1985; Ng et al., 1989; Mizisin and Powell, 1993; Tang et al., 2012; Ji et al., 2017; Hu et al., 2019; Mackinnon et al., 2021). GALK1 inhibitors were shown to prevent accumulation of Gal-1-P in cellular models, but remain to be studied in vivo (Tang et al., 2012; Hu et al., 2019; Mackinnon et al., 2021). Uridine supplementation to increase levels of UDP-Glc and UDP-Gal was not able to rescue the biochemical and clinical phenotype (Ng et al., 1989). Safety and effectiveness of the AR inhibitor AT007 is currently being investigated (NCT04902781; NCT05418829).
Furthermore, to improve GALT activity, chaperone therapy and nucleic acid therapy have been studied. Supplementation of the amino acid arginine as a chaperone had a mutation-specific effect with rescue of human GALT in an E. coli model (Coelho et al., 2015b), but failed to exhibit positive effects in four c.563A>G; p.Gln188Arg homozygous patients (Haskovic et al., 2018). Whether other pathogenic variants are amenable has not been studied. hGALT mRNA therapy and GALT gene therapy restored GALT activity in cellular and animal models of CG (Balakrishnan et al., 2020; Brophy et al., 2022; Daenzer et al., 2022; Delnoy et al., 2022), but many unknowns remain to be answered before these therapies can be applied. Other treatment options could be compounds that target the integrated stress response such as PSPC and myo-inositol (Balakrishnan et al., 2016), which improved brain tissue structures in GalT gene-trapped mice (Hagen-Lillevik et al., 2022). An advantage is their favorable safety profile in humans, which could hasten their application in clinical practice.
In CG, brain function is affected in 85% of the patients, with significant individual variability in severity and symptoms (Rubio-Gozalbo et al., 2019). Since Komrower (Komrower and Lee, 1970) in 1970 reported on physical and mental development in the first cohort of 60 dietary treated CG patients, numerous studies have been performed to shed more light on brain effects in CG (summarized in Supplementary Table S1). While these have expanded our knowledge on the neurocognitive, neuropsychological, neuropsychiatric, social emotional and neurological difficulties associated with the disease, as well as the abnormalities seen in neuroimaging and electrophysiological assessments, information about the disease course in adulthood is still scarce. The majority of studies are cross-sectional, or retrospective using cross-sectional data from different age groups. The few longitudinal studies that have been performed are usually based on small sample sizes, with relatively young patients and limited follow-up time. Although the results from these studies should be treated cautiously, they provide us with the first valuable insights into CG brain function over time.
Nelson et al. (Nelson et al., 1992) performed MRI imaging in 63 CG patients (1 month–42 years of age). Of the 24 patients who underwent follow-up MRI after 1–4 years, abnormal peripheral white matter and ventricle enlargement remained unchanged. One patient showed progression of cerebellar atrophy. Schadewaldt et al. (Schadewaldt et al., 2010) reported TIQ, PIQ and VIQ scores in 23 patients, with the first tests performed at a mean age of 11±5 years and the second tests at a mean age of 26±5 years. The mean TIQ and PIQ did not change significantly over time, whereas the mean VIQ score showed a variable but significant decline at follow-up. However, no consistent changes were found, as a number of participants showed significant increases and other patients decreases of these scores with age. In line with these results, neurocognitive function did not deteriorate in a cohort of 35 patients aged 1 week - 16 years, but the follow-up time was only 2–5 years (Manis et al., 1997). A recent study in a robust dataset of CG patients (mean age of 18 years) concluded that speech/voice/language, cognitive, motor, and psychosocial outcomes are not progressive in most patients, but also here the time between testing was limited (Smith et al., 2023). A pilot study with 10 adult patients (mean age 33 years) and a mean time interval of 3 years and 9 months reported cognitive stability (Hermans et al., 2024).
In addition to the longitudinal studies, Lotz-Havla et al. (Lotz-Havla et al., 2023) studied retinal neuroaxonal function as marker for neurodegeneration in 11 CG patients and 60 controls, and did not find evidence for retinal neuroaxonal degeneration. Moreover, specialist teams within the GalNet that treat CG patients and take part in this review observe improvements in language performance (scores on verbal tests), and absence of cognitive decline. Patients, nevertheless, do complain about motor and social function, and at older ages complaints about tremor, memory issues, anxiety and depression are reported more often.
Although there might be progression concerning signs of early aging, memory issues and depression, “growing into deficits” can play a role. Adult life is often more stressful than childhood, so the features of anxiety and depression (financial worries, loneliness) may be more pronounced with time. There is also a subset of patients that experience neurological worsening, which are very often patients who already had significant neurological issues in childhood. Patients with early onset cerebral or cerebellar atrophy can also show progressive natural senescence effects. The pathophysiological mechanisms therefore seem multifactorial, with individual susceptibility as one of the most important determinants. Studies with sensitive tests for the different affected domains and follow-up of decades in larger cohorts need to be performed to adequately delineate the disease course through adulthood.
In this review, we describe the role of impaired galactose metabolism on brain dysfunction. Our conclusion is that, based on the current data and insights, the majority of patients do not exhibit cognitive decline. A subset of patients experiences neurological worsening, often those patients with early onset cerebral and cerebellar volume loss. At older ages complaints about memory issues, anxiety and depression are seen more often, but are likely multifactorial in origin.
BP: Writing–original draft, Writing–review and editing. EV: Writing–original draft, Writing–review and editing. IB: Writing–review and editing. ABo: Writing–review and editing. MB: Writing–review and editing. ABu: Writing–review and editing. DCa: Writing–review and editing. DCo: Writing–review and editing. MC: Writing–review and editing. AD: Writing–review and editing. DD: Writing–review and editing. AE: Writing–review and editing. MG: Writing–review and editing. OG: Writing–review and editing. SG: Writing–review and editing. SK: Writing–review and editing. IK: Writing–review and editing. EL-T: Writing–review and editing. DM: Writing–review and editing. EM: Writing–review and editing. KÕ: Writing–review and editing. AP: Writing–review and editing. SP: Writing–review and editing. RP: Writing–review and editing. IR: Writing–review and editing. SS-B: Writing–review and editing. IS: Writing–review and editing. TS: Writing–review and editing. LS: Writing–review and editing. AS: Writing–review and editing. KS: Writing–review and editing. ET: Writing–review and editing. SW: Writing–review and editing. GB: Conceptualization, Writing–original draft, Writing–review and editing. MR-G: Conceptualization, Writing–original draft, Writing–review and editing.
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2024.1355962/full#supplementary-material
AR, aldose reductase; CG, classic galactosemia; CNS, central nervous system; ER, endoplasmic reticulum; Gal-1-P, galactose-1-phospate; GALDH, galactose dehydrogenase; GALE, UDP-galactose 4’-epimerase; GALK1, galactokinase; GalNet, Galactosemia Network; GALT, galactose-1-phosphate uridylyltransferase; GALM, galactose mutarotase; IQ, intelligence quotient; MRI: magnetic resonance imaging; NDI, neurite density index; NMJ: neuromuscular junction; NODDI, neurite orientation dispersion and density imaging; ODI, orientation dispersion index; PIQ, performance IQ; PSPC, purple sweet potato color; TIQ, total IQ; UDP, Uridine diphosphate; UGP2, UDP-glucose pyrophosphorylase 2: VIQ, verbal component IQ.
Ahtam, B., Waisbren, S. E., Anastasoaie, V., Berry, G. T., Brown, M., Petrides, S., et al. (2020). Identification of neuronal structures and pathways corresponding to clinical functioning in galactosemia. J. Inherit. Metab. Dis. 43 (6), 1205–1218. doi:10.1002/jimd.12279
Antshel, K. M., Epstein, I. O., and Waisbren, S. E. (2004). Cognitive strengths and weaknesses in children and adolescents homozygous for the galactosemia Q188R mutation: a descriptive study. Neuropsychology 18 (4), 658–664. doi:10.1037/0894-4105.18.4.658
Arn, P. H. (2003). Galactosemia. Curr. Treat. Options Neurol. 5 (4), 343–345. doi:10.1007/s11940-003-0040-x
Aydin-Ozemir, Z., Tekturk, P., Uyguner, Z. O., and Baykan, B. (2014). Galactosemia and phantom absence seizures. J. Pediatr. Neurosci. 9 (3), 253–256. doi:10.4103/1817-1745.147581
Balakrishnan, B., An, D., Nguyen, V., DeAntonis, C., Martini, P. G. V., and Lai, K. (2020). Novel mRNA-based therapy reduces toxic galactose metabolites and overcomes galactose sensitivity in a mouse model of classic galactosemia. Mol. Ther. 28 (1), 304–312. doi:10.1016/j.ymthe.2019.09.018
Balakrishnan, B., Chen, W., Tang, M., Huang, X., Cakici, D. D., Siddiqi, A., et al. (2016). Galactose-1 phosphate uridylyltransferase (GalT) gene: a novel positive regulator of the PI3K/Akt signaling pathway in mouse fibroblasts. Biochem. Biophys. Res. Commun. 470 (1), 205–212. doi:10.1016/j.bbrc.2016.01.036
Balakrishnan, B., Nicholas, C., Siddiqi, A., Chen, W., Bales, E., Feng, M., et al. (2017). Reversal of aberrant PI3K/Akt signaling by Salubrinal in a GalT-deficient mouse model. Biochim. Biophys. Acta Mol. Basis Dis. 1863 (12), 3286–3293. doi:10.1016/j.bbadis.2017.08.023
Barnes-Vélez, J. A., Aksoy Yasar, F. B., and Hu, J. (2023). Myelin lipid metabolism and its role in myelination and myelin maintenance. Innov. (Camb). 4 (1), 100360. doi:10.1016/j.xinn.2022.100360
Belman, A. L., Moshe, S. L., and Zimmerman, R. D. (1986). Computed tomographic demonstration of cerebral edema in a child with galactosemia. Pediatrics 78 (4), 606–609. doi:10.1542/peds.78.4.606
Berry, G. T. (2011). Is prenatal myo-inositol deficiency a mechanism of CNS injury in galactosemia? J. Inherit. Metab. Dis. 34 (2), 345–355. doi:10.1007/s10545-010-9260-x
Berry, G. T., Hunter, J. V., Wang, Z., Dreha, S., Mazur, A., Brooks, D. G., et al. (2001). In vivo evidence of brain galactitol accumulation in an infant with galactosemia and encephalopathy. J. Pediatr. 138 (2), 260–262. doi:10.1067/mpd.2001.110423
Berry, G. T., Mallee, J. J., Kwon, H. M., Rim, J. S., Mulla, W. R., Muenke, M., et al. (1995b). The human osmoregulatory Na+/myo-inositol cotransporter gene (SLC5A3): molecular cloning and localization to chromosome 21. Genomics 25 (2), 507–513. doi:10.1016/0888-7543(95)80052-n
Berry, G. T., Moate, P. J., Reynolds, R. A., Yager, C. T., Ning, C., Boston, R. C., et al. (2004). The rate of de novo galactose synthesis in patients with galactose-1-phosphate uridyltransferase deficiency. Mol. Genet. Metab. 81 (1), 22–30. doi:10.1016/j.ymgme.2003.08.026
Berry, G. T., Nissim, I., Gibson, J. B., Mazur, A. T., Lin, Z., Elsas, L. J., et al. (1997). Quantitative assessment of whole body galactose metabolism in galactosemic patients. Eur. J. Pediatr. 156 (Suppl. 1), S43–S49. doi:10.1007/pl00014271
Berry, G. T., Nissim, I., Lin, Z., Mazur, A. T., Gibson, J. B., and Segal, S. (1995a). Endogenous synthesis of galactose in normal men and patients with hereditary galactosaemia. Lancet 346 (8982), 1073–1074. doi:10.1016/s0140-6736(95)91745-4
Berry, G. T., Wehrli, S., Reynolds, R., Palmieri, M., Frangos, M., Williamson, J. R., et al. (1998). Elevation of erythrocyte redox potential linked to galactonate biosynthesis: elimination by Tolrestat. Metabolism 47 (11), 1423–1428. doi:10.1016/s0026-0495(98)90317-1
Boca, M., and Whone, A. (2017). Letter to the editor on "Evidence for dopaminergic denervation in classical galactosemia. Mov. Disord. 32 (12), 1797. doi:10.1002/mds.27187
Bohles, H., Wenzel, D., and Shin, Y. S. (1986). Progressive cerebellar and extrapyramidal motor disturbances in galactosaemic twins. Eur. J. Pediatr. 145 (5), 413–417. doi:10.1007/BF00439251
Bosch, A. M., Grootenhuis, M. A., Bakker, H. D., Heijmans, H. S., Wijburg, F. A., and Last, B. F. (2004). Living with classical galactosemia: health-related quality of life consequences. Pediatrics 113 (5), e423–e428. doi:10.1542/peds.113.5.e423
Bosch, A. M., Maurice-Stam, H., Wijburg, F. A., and Grootenhuis, M. A. (2009). Remarkable differences: the course of life of young adults with galactosaemia and PKU. J. Inherit. Metab. Dis. 32 (6), 706. doi:10.1007/s10545-009-1253-2
Brophy, M. L., Stansfield, J. C., Ahn, Y., Cheng, S. H., Murphy, J. E., and Bell, R. D. (2022). AAV-mediated expression of galactose-1-phosphate uridyltransferase corrects defects of galactose metabolism in classic galactosemia patient fibroblasts. J. Inherit. Metab. Dis. 45 (3), 481–492. doi:10.1002/jimd.12468
Buccafusca, R., Venditti, C. P., Kenyon, L. C., Johanson, R. A., Van Bockstaele, E., Ren, J., et al. (2008). Characterization of the null murine sodium/myo-inositol cotransporter 1 (Smit1 or Slc5a3) phenotype: myo-inositol rescue is independent of expression of its cognate mitochondrial ribosomal protein subunit 6 (Mrps6) gene and of phosphatidylinositol levels in neonatal brain. Mol. Genet. Metab. 95 (1-2), 81–95. doi:10.1016/j.ymgme.2008.05.008
Charlwood, J., Clayton, P., Keir, G., Mian, N., and Winchester, B. (1998). Defective galactosylation of serum transferrin in galactosemia. Glycobiology 8 (4), 351–357. doi:10.1093/glycob/8.4.351
Choulot, J. J., Brivet, M., Virlon, P., Sevely, A., Manelfe, C., Mensire, A., et al. (1991). Severe neurologic course of galactosemia. Default of myelisation caused by deficient synthesis of UDP-galactose? Arch. Fr. Pediatr. 48 (4), 267–269.
Coelho, A. I., Berry, G. T., and Rubio-Gozalbo, M. E. (2015a). Galactose metabolism and health. Curr. Opin. Clin. Nutr. Metab. Care 18 (4), 422–427. doi:10.1097/MCO.0000000000000189
Coelho, A. I., Bierau, J., Lindhout, M., Achten, J., Kramer, B. W., and Rubio-Gozalbo, M. E. (2017). Classic galactosemia: study on the late prenatal development of GALT specific activity in a sheep model. Anat. Rec. Hob. 300 (9), 1570–1575. doi:10.1002/ar.23616
Coelho, A. I., Trabuco, M., Ramos, R., Silva, M. J., Tavares de Almeida, I., Leandro, P., et al. (2014). Functional and structural impact of the most prevalent missense mutations in classic galactosemia. Mol. Genet. Genomic Med. 2 (6), 484–496. doi:10.1002/mgg3.94
Coelho, A. I., Trabuco, M., Silva, M. J., de Almeida, I. T., Leandro, P., Rivera, I., et al. (2015b). Arginine functionally improves clinically relevant human galactose-1-phosphate uridylyltransferase (GALT) variants expressed in a prokaryotic model. JIMD Rep. 23, 1–6. doi:10.1007/8904_2015_420
Coman, D. J., Murray, D. W., Byrne, J. C., Rudd, P. M., Bagaglia, P. M., Doran, P. D., et al. (2010). Galactosemia, a single gene disorder with epigenetic consequences. Pediatr. Res. 67 (3), 286–292. doi:10.1203/PDR.0b013e3181cbd542
Conte, F., van Buuringen, N., Voermans, N. C., and Lefeber, D. J. (2021). Galactose in human metabolism, glycosylation and congenital metabolic diseases: time for a closer look. Biochim. Biophys. Acta Gen. Subj. 1865 (8), 129898. doi:10.1016/j.bbagen.2021.129898
Coss, K. P., Byrne, J. C., Coman, D. J., Adamczyk, B., Abrahams, J. L., Saldova, R., et al. (2012). IgG N-glycans as potential biomarkers for determining galactose tolerance in Classical Galactosaemia. Mol. Genet. Metab. 105 (2), 212–220. doi:10.1016/j.ymgme.2011.10.018
Coss, K. P., Doran, P. P., Owoeye, C., Codd, M. B., Hamid, N., Mayne, P. D., et al. (2013). Classical Galactosaemia in Ireland: incidence, complications and outcomes of treatment. J. Inherit. Metab. Dis. 36 (1), 21–27. doi:10.1007/s10545-012-9507-9
Coss, K. P., Hawkes, C. P., Adamczyk, B., Stockmann, H., Crushell, E., Saldova, R., et al. (2014). N-glycan abnormalities in children with galactosemia. J. Proteome Res. 13 (2), 385–394. doi:10.1021/pr4008305
Crome, L. (1962). A case of galactosaemia with the pathological and neuropathological findings. Arch. Dis. Child. 37 (194), 415–421. doi:10.1136/adc.37.194.415
Daenzer, J. M. I., Rasmussen, S. A., Patel, S., McKenna, J., and Fridovich-Keil, J. L. (2022). Neonatal GALT gene replacement offers metabolic and phenotypic correction through early adulthood in a rat model of classic galactosemia. J. Inherit. Metab. Dis. 45 (2), 203–214. doi:10.1002/jimd.12471
Dani, N., and Broadie, K. (2012). Glycosylated synaptomatrix regulation of trans-synaptic signaling. Dev. Neurobiol. 72 (1), 2–21. doi:10.1002/dneu.20891
Daude, N., Ellie, E., Reichardt, J. K., and Petry, K. G. (1996). In vivo and in vitro expression of rat galactose-1-phosphate uridyltransferase (GALT) in the developing central and peripheral nervous system. Brain Res. Dev. Brain Res. 94 (2), 190–196. doi:10.1016/0165-3806(96)00058-2
Delnoy, B., Haskovic, M., Vanoevelen, J., Steinbusch, L. K. M., Vos, E. N., Knoops, K., et al. (2022). Novel mRNA therapy restores GALT protein and enzyme activity in a zebrafish model of classic galactosemia. J. Inherit. Metab. Dis. 45 (4), 748–758. doi:10.1002/jimd.12512
Demirbas, D., Huang, X., Daesety, V., Feenstra, S., Haskovic, M., Qi, W., et al. (2019). The ability of an LC-MS/MS-based erythrocyte GALT enzyme assay to predict the phenotype in subjects with GALT deficiency. Mol. Genet. Metab. 126 (4), 368–376. doi:10.1016/j.ymgme.2019.01.016
De-Souza, E. A., Pimentel, F. S., Machado, C. M., Martins, L. S., da-Silva, W. S., Montero-Lomeli, M., et al. (2014). The unfolded protein response has a protective role in yeast models of classic galactosemia. Dis. Model Mech. 7 (1), 55–61. doi:10.1242/dmm.012641
Dubroff, J. G., Ficicioglu, C., Segal, S., Wintering, N. A., Alavi, A., and Newberg, A. B. (2008). FDG-PET findings in patients with galactosaemia. J. Inherit. Metab. Dis. 31 (4), 533–539. doi:10.1007/s10545-008-0806-0
Fishler, K., Koch, R., Donnell, G. N., and Wenz, E. (1980). Developmental aspects of galactosemia from infancy to childhood. Clin. Pediatr. (Phila). 19 (1), 38–44. doi:10.1177/000992288001900106
Friedman, J. H., Levy, H. L., and Boustany, R. M. (1989). Late onset of distinct neurologic syndromes in galactosemic siblings. Neurology 39 (5), 741–742. doi:10.1212/wnl.39.5.741
Gitzelmann, R. (1995). Galactose-1-phosphate in the pathophysiology of galactosemia. Eur. J. Pediatr. 154 (7 Suppl. 2), S45–S49. doi:10.1007/BF02143803
Göppert, F. (1917). Galaktosurie nach Milchzuckergabe bei angeborenem, familiaerem chronischem Leberleiden. Klin. Wschr 54, 473–477.
Gubbels, C. S., Maurice-Stam, H., Berry, G. T., Bosch, A. M., Waisbren, S., Rubio-Gozalbo, M. E., et al. (2011). Psychosocial developmental milestones in men with classic galactosemia. J. Inherit. Metab. Dis. 34 (2), 415–419. doi:10.1007/s10545-011-9290-z
Haberland, C., Perou, M., Brunngraber, E. G., and Hof, H. (1971). The neuropathology of galactosemia. A histopathological and biochemical study. J. Neuropathol. Exp. Neurol. 30 (3), 431–447. doi:10.1097/00005072-197107000-00009
Hagen-Lillevik, S., Johnson, J., Siddiqi, A., Persinger, J., Hale, G., and Lai, K. (2022). Harnessing the power of purple sweet potato color and myo-inositol to treat classic galactosemia. Int. J. Mol. Sci. 23 (15), 8654. doi:10.3390/ijms23158654
Hansen, T. W., Henrichsen, B., Rasmussen, R. K., Carling, A., Andressen, A. B., and Skjeldal, O. (1996). Neuropsychological and linguistic follow-up studies of children with galactosaemia from an unscreened population. Acta Paediatr. 85 (10), 1197–1201. doi:10.1111/j.1651-2227.1996.tb18228.x
Haskovic, M., Coelho, A. I., Bierau, J., Vanoevelen, J. M., Steinbusch, L. K. M., Zimmermann, L. J. I., et al. (2020). Pathophysiology and targets for treatment in hereditary galactosemia: a systematic review of animal and cellular models. J. Inherit. Metab. Dis. 43 (3), 392–408. doi:10.1002/jimd.12202
Haskovic, M., Derks, B., van der Ploeg, L., Trommelen, J., Nyakayiru, J., van Loon, L. J. C., et al. (2018). Arginine does not rescue p.Q188R mutation deleterious effect in classic galactosemia. Orphanet J. Rare Dis. 13 (1), 212. doi:10.1186/s13023-018-0954-8
Hermans, M. E., Geurtsen, G. J., Hollak, C. E. M., and Bosch, A. M. (2024). Neuropsychological stability in classical galactosemia: a pilot study in 10 adult patients. JIMD Rep. doi:10.1002/jmd2.12410
Hermans, M. E., van Oers, H. A., Geurtsen, G. J., Haverman, L., Hollak, C. E. M., Rubio-Gozalbo, M. E., et al. (2023). The challenges of classical galactosemia: HRQoL in pediatric and adult patients. Orphanet J. Rare Dis. 18 (1), 135. doi:10.1186/s13023-023-02749-8
Hoffmann, B., Wendel, U., and Schweitzer-Krantz, S. (2011). Cross-sectional analysis of speech and cognitive performance in 32 patients with classic galactosemia. J. Inherit. Metab. Dis. 34 (2), 421–427. doi:10.1007/s10545-011-9297-5
Holton, J. B. (1995). Effects of galactosemia in utero. Eur. J. Pediatr. 154 (7 Suppl. 2), S77–S81. doi:10.1007/BF02143809
Hu, X., Zhang, Y. Q., Lee, O. W., Liu, L., Tang, M., Lai, K., et al. (2019). Discovery of novel inhibitors of human galactokinase by virtual screening. J. Comput. Aided Mol. Des. 33 (4), 405–417. doi:10.1007/s10822-019-00190-3
Hughes, J., Ryan, S., Lambert, D., Geoghegan, O., Clark, A., Rogers, Y., et al. (2009). Outcomes of siblings with classical galactosemia. J. Pediatr. 154 (5), 721–726. doi:10.1016/j.jpeds.2008.11.052
Huttenlocher, P. R., Hillman, R. E., and Hsia, Y. E. (1970). Pseudotumor cerebri in galactosemia. J. Pediatr. 76 (6), 902–905. doi:10.1016/s0022-3476(70)80373-0
Jan, J. E., and Wilson, R. A. (1973). Unusual late neurological sequelae in galactosaemia. Dev. Med. Child. Neurol. 15 (1), 72–74. doi:10.1111/j.1469-8749.1973.tb04869.x
Ji, L., Cheng, L., and Yang, Z. (2017). Diosgenin, a novel aldose reductase inhibitor, attenuates the galactosemic cataract in rats. J. Diabetes Res. 2017, 7309816. doi:10.1155/2017/7309816
Jumbo-Lucioni, P., Parkinson, W., and Broadie, K. (2014). Overelaborated synaptic architecture and reduced synaptomatrix glycosylation in a Drosophila classic galactosemia disease model. Dis. Model Mech. 7 (12), 1365–1378. doi:10.1242/dmm.017137
Jumbo-Lucioni, P. P., Garber, K., Kiel, J., Baric, I., Berry, G. T., Bosch, A., et al. (2012). Diversity of approaches to classic galactosemia around the world: a comparison of diagnosis, intervention, and outcomes. J. Inherit. Metab. Dis. 35 (6), 1037–1049. doi:10.1007/s10545-012-9477-y
Kaufman, F. R., Horton, E. J., Gott, P., Wolff, J. A., Nelson, M. D., Azen, C., et al. (1995b). Abnormal somatosensory evoked potentials in patients with classic galactosemia: correlation with neurologic outcome. J. Child. Neurol. 10 (1), 32–36. doi:10.1177/088307389501000109
Kaufman, F. R., McBride-Chang, C., Manis, F. R., Wolff, J. A., and Nelson, M. D. (1995a). Cognitive functioning, neurologic status and brain imaging in classical galactosemia. Eur. J. Pediatr. 154 (7 Suppl. 2), S2–S5. doi:10.1007/BF02143794
Knerr, I., Coss, K. P., Kratzsch, J., Crushell, E., Clark, A., Doran, P., et al. (2015). Effects of temporary low-dose galactose supplements in children aged 5-12 y with classical galactosemia: a pilot study. Pediatr. Res. 78 (3), 272–279. doi:10.1038/pr.2015.107
Koch, T. K., Schmidt, K. A., Wagstaff, J. E., Ng, W. G., and Packman, S. (1992). Neurologic complications in galactosemia. Pediatr. Neurol. 8 (3), 217–220. doi:10.1016/0887-8994(92)90072-7
Komrower, G. M., and Lee, D. H. (1970). Long-term follow-up of galactosaemia. Arch. Dis. Child. 45 (241), 367–373. doi:10.1136/adc.45.241.367
Krabbi, K., Uudelepp, M. L., Joost, K., Zordania, R., and Õunap, K. (2011). Long-term complications in Estonian galactosemia patients with a less strict lactose-free diet and metabolic control. Mol. Genet. Metab. 103 (3), 249–253. doi:10.1016/j.ymgme.2011.03.023
Kreis, R., Hofmann, L., Kuhlmann, B., Boesch, C., Bossi, E., and Hüppi, P. S. (2002). Brain metabolite composition during early human brain development as measured by quantitative in vivo 1H magnetic resonance spectroscopy. Magn. Reson Med. 48 (6), 949–958. doi:10.1002/mrm.10304
Kuiper, A., Grunewald, S., Murphy, E., Coenen, M. A., Eggink, H., Zutt, R., et al. (2019). Movement disorders and nonmotor neuropsychological symptoms in children and adults with classical galactosemia. J. Inherit. Metab. Dis. 42 (3), 451–458. doi:10.1002/jimd.12054
Lai, K., Elsas, L. J., and Wierenga, K. J. (2009). Galactose toxicity in animals. IUBMB Life 61 (11), 1063–1074. doi:10.1002/iub.262
Lai, K., Langley, S. D., Khwaja, F. W., Schmitt, E. W., and Elsas, L. J. (2003). GALT deficiency causes UDP-hexose deficit in human galactosemic cells. Glycobiology 13 (4), 285–294. doi:10.1093/glycob/cwg033
Lebea, P. J., and Pretorius, P. J. (2005). The molecular relationship between deficient UDP-galactose uridyl transferase (GALT) and ceramide galactosyltransferase (CGT) enzyme function: a possible cause for poor long-term prognosis in classic galactosemia. Med. Hypotheses 65 (6), 1051–1057. doi:10.1016/j.mehy.2005.06.025
Leslie, N. D. (2003). Insights into the pathogenesis of galactosemia. Annu. Rev. Nutr. 23, 59–80. doi:10.1146/annurev.nutr.23.011702.073135
Lo, W., Packman, S., Nash, S., Schmidt, K., Ireland, S., Diamond, I., et al. (1984). Curious neurologic sequelae in galactosemia. Pediatrics 73 (3), 309–312. doi:10.1542/peds.73.3.309
Lotz-Havla, A. S., Christmann, T., Parhofer, K. G., Maier, E. M., and Havla, J. (2023). Optical coherence tomography: retinal imaging contributes to the understanding of brain pathology in classical galactosemia. J. Clin. Med. 12 (5), 2030. doi:10.3390/jcm12052030
Mackinnon, S. R., Krojer, T., Foster, W. R., Diaz-Saez, L., Tang, M., Huber, K. V. M., et al. (2021). Fragment screening reveals starting points for rational design of galactokinase 1 inhibitors to treat classic galactosemia. ACS Chem. Biol. 16 (4), 586–595. doi:10.1021/acschembio.0c00498
MacWilliams, J., Patel, S., Carlock, G., Vest, S., Potter, N. L., and Fridovich-Keil, J. L. (2021). Hand fine motor control in classic galactosemia. J. Inherit. Metab. Dis. 44 (4), 871–878. doi:10.1002/jimd.12376
Manis, F. R., Cohn, L. B., McBride-Chang, C., Wolff, J. A., and Kaufman, F. R. (1997). A longitudinal study of cognitive functioning in patients with classical galactosaemia, including a cohort treated with oral uridine. J. Inherit. Metab. Dis. 20 (4), 549–555. doi:10.1023/a:1005357622551
Maratha, A., Colhoun, H. O., Knerr, I., Coss, K. P., Doran, P., and Treacy, E. P. (2017). Classical galactosaemia and cdg, the N-glycosylation interface. A review. JIMD Rep. 34, 33–42. doi:10.1007/8904_2016_5
Martins, E., Teixeira, J., Cardoso, M. L., Lima, M. R., Briones-Godino, P., and Barbot, C. (2004). Galactosemia: the genotype and phenotype of seven patients. Rev. Neurol. 38 (12), 1132–1135. doi:10.33588/rn.3812.2002564
Mason, H. H., and Turner, M. E. (1935). Chronic galactemia, report of case with studies on carbohydrates. Am. J. Dis. Child. 50 (2), 359. doi:10.1001/archpedi.1935.01970080053005
McCorvie, T. J., Gleason, T. J., Fridovich-Keil, J. L., and Timson, D. J. (2013). Misfolding of galactose 1-phosphate uridylyltransferase can result in type I galactosemia. Biochim. Biophys. Acta 1832 (8), 1279–1293. doi:10.1016/j.bbadis.2013.04.004
Mizisin, A. P., and Powell, H. C. (1993). Schwann cell injury is attenuated by aldose reductase inhibition in galactose intoxication. J. Neuropathol. Exp. Neurol. 52 (1), 78–86. doi:10.1097/00005072-199301000-00010
Nelson, C. D., Waggoner, D. D., Donnell, G. N., Tuerck, J. M., and Buist, N. R. (1991). Verbal dyspraxia in treated galactosemia. Pediatrics 88 (2), 346–350. doi:10.1542/peds.88.2.346
Nelson, M. D., Wolff, J. A., Cross, C. A., Donnell, G. N., and Kaufman, F. R. (1992). Galactosemia: evaluation with MR imaging. Radiology 184 (1), 255–261. doi:10.1148/radiology.184.1.1319076
Neville, S., O'Sullivan, S., Sweeney, B., Lynch, B., Hanrahan, D., Knerr, I., et al. (2016). Friedreich ataxia in classical galactosaemia. JIMD Rep. 26, 1–5. doi:10.1007/8904_2015_477
Ng, W. G., Xu, Y. K., Kaufman, F. R., and Donnell, G. N. (1989). Deficit of uridine diphosphate galactose in galactosaemia. J. Inherit. Metab. Dis. 12 (3), 257–266. doi:10.1007/BF01799215
Otaduy, M. C., Leite, C. C., Lacerda, M. T., Costa, M. O., Arita, F., Prado, E., et al. (2006). Proton MR spectroscopy and imaging of a galactosemic patient before and after dietary treatment. AJNR Am. J. Neuroradiol. 27 (1), 204–207.
Özgün, N., Celik, M., Akdeniz, O., Ozbek, M. N., Bulbul, A., and Anlar, B. (2019). Early neurological complications in children with classical galactosemia and p.gln188arg mutation. Int. J. Dev. Neurosci. 78, 92–97. doi:10.1016/j.ijdevneu.2019.07.004
Papachristoforou, R., Petrou, P. P., Sawyer, H., Williams, M., and Drousiotou, A. (2014). A novel large deletion encompassing the whole of the galactose-1-phosphate uridyltransferase (GALT) gene and extending into the adjacent interleukin 11 receptor alpha (IL11RA) gene causes classic galactosemia associated with additional phenotypic abnormalities. JIMD Rep. 12, 91–98. doi:10.1007/8904_2013_249
Potter, N. L. (2011). Voice disorders in children with classic galactosemia. J. Inherit. Metab. Dis. 34 (2), 377–385. doi:10.1007/s10545-010-9213-4
Potter, N. L., Lazarus, J. A., Johnson, J. M., Steiner, R. D., and Shriberg, L. D. (2008). Correlates of language impairment in children with galactosaemia. J. Inherit. Metab. Dis. 31 (4), 524–532. doi:10.1007/s10545-008-0877-y
Potter, N. L., Nievergelt, Y., and Shriberg, L. D. (2013). Motor and speech disorders in classic galactosemia. JIMD Rep. 11, 31–41. doi:10.1007/8904_2013_219
Quan-Ma, R., Wells, H. J., Wells, W. W., Sherman, F. E., and Egan, T. J. (1966). Galactitol in the tissues of a galactosemic child. Am. J. Dis. Child. 112 (5), 477–478. doi:10.1001/archpedi.1966.02090140149018
Quelhas, D., Jaeken, J., and Azevedo, L. (2023). Genetic modifiers in glycosylation pathways: is there a link between PMM2 and PGM1? J. Inherit. Metab. Dis. 46 (1), 1–2. doi:10.1002/jimd.12576
Quintana, E., Navarro-Sastre, A., Hernández-Pérez, J. M., García-Villoria, J., Montero, R., Artuch, R., et al. (2009). Screening for congenital disorders of glycosylation (CDG): transferrin HPLC versus isoelectric focusing (IEF). Clin. Biochem. 42 (4-5), 408–415. doi:10.1016/j.clinbiochem.2008.12.013
Rahit, K., and Tarailo-Graovac, M. (2020). Genetic modifiers and rare mendelian disease. Genes (Basel). 11 (3), 239. doi:10.3390/genes11030239
Ridel, K. R., Leslie, N. D., and Gilbert, D. L. (2005). An updated review of the long-term neurological effects of galactosemia. Pediatr. Neurol. 33 (3), 153–161. doi:10.1016/j.pediatrneurol.2005.02.015
Robertson, A., Singh, R. H., Guerrero, N. V., Hundley, M., and Elsas, L. J. (2000). Outcomes analysis of verbal dyspraxia in classic galactosemia. Genet. Med. 2 (2), 142–148. doi:10.1097/00125817-200003000-00005
Rogers, S., Heidenreich, R., Mallee, J., and Segal, S. (1992). Regional activity of galactose-1-phosphate uridyltransferase in rat brain. Pediatr. Res. 31 (5), 512–515. doi:10.1203/00006450-199205000-00021
Rogers, S. R., Bovee, B. W., Saunders, S. L., and Segal, S. (1989a). Galactose as a regulatory factor of its own metabolism by rat liver. Metabolism 38 (8), 810–815. doi:10.1016/0026-0495(89)90072-3
Rogers, S. R., Bovee, B. W., Saunders, S. L., and Segal, S. (1989b). Activity of hepatic galactose-metabolizing enzymes in the pregnant rat and fetus. Pediatr. Res. 25 (2), 161–166. doi:10.1203/00006450-198902000-00017
Rossi-Espagnet, M. C., Sudhakar, S., Fontana, E., Longo, D., Davison, J., Petengill, A. L., et al. (2021). Neuroradiologic phenotyping of galactosemia: from the neonatal form to the chronic stage. AJNR Am. J. Neuroradiol. 42 (3), 590–596. doi:10.3174/ajnr.A7016
Rubio-Agusti, I., Carecchio, M., Bhatia, K. P., Kojovic, M., Parees, I., Chandrashekar, H. S., et al. (2013). Movement disorders in adult patients with classical galactosemia. Mov. Disord. 28 (6), 804–810. doi:10.1002/mds.25348
Rubio-Gozalbo, M. E., Haskovic, M., Bosch, A. M., Burnyte, B., Coelho, A. I., Cassiman, D., et al. (2019). The natural history of classic galactosemia: lessons from the GalNet registry. Orphanet J. Rare Dis. 14 (1), 86. doi:10.1186/s13023-019-1047-z
Schadewaldt, P., Hoffmann, B., Hammen, H. W., Kamp, G., Schweitzer-Krantz, S., and Wendel, U. (2010). Longitudinal assessment of intellectual achievement in patients with classical galactosemia. Pediatrics 125 (2), e374–e381. doi:10.1542/peds.2008-3325
Schadewaldt, P., Kamalanathan, L., Hammen, H. W., and Wendel, U. (2004). Age dependence of endogenous galactose formation in Q188R homozygous galactosemic patients. Mol. Genet. Metab. 81 (1), 31–44. doi:10.1016/j.ymgme.2003.10.007
Schweitzer, S., Shin, Y., Jakobs, C., and Brodehl, J. (1993). Long-term outcome in 134 patients with galactosaemia. Eur. J. Pediatr. 152 (1), 36–43. doi:10.1007/BF02072514
Segal, S. (1995). In utero galactose intoxication in animals. Eur. J. Pediatr. 154 (7 2), S82–S86. doi:10.1007/BF02143810
Shah, P. A., and Kuchhai, F. A. (2009). Galactosemia with chorea--an unusual presentation. Indian J. Pediatr. 76 (1), 97–98. doi:10.1007/s12098-009-0037-x
Shin-Buehring, Y. S., Beier, T., Tan, A., Osang, M., and Schaub, J. (1977). The activity of galactose-1-phosphate uridyltransferase and galactokinase in human fetal organs. Pediatr. Res. 11 (10 Pt 1), 1045–1051. doi:10.1203/00006450-197710000-00004
Simard-Duquesne, N., Greselin, E., Dubuc, J., and Dvornik, D. (1985). The effects of a new aldose reductase inhibitor (tolrestat) in galactosemic and diabetic rats. Metabolism 34 (10), 885–892. doi:10.1016/0026-0495(85)90133-7
Slepak, T. I., Tang, M., Slepak, V. Z., and Lai, K. (2007). Involvement of endoplasmic reticulum stress in a novel Classic Galactosemia model. Mol. Genet. Metab. 92 (1-2), 78–87. doi:10.1016/j.ymgme.2007.06.005
Smith, N. H., Hendrickson, E. T., Garrett, O. S., Chernoff, R. A., Orloff, D. H., Druss, J. J., et al. (2023). Long-term complications in classic galactosemia are not progressive. Mol. Genet. Metab. 140 (3), 107708. doi:10.1016/j.ymgme.2023.107708
Tang, M., Odejinmi, S. I., Vankayalapati, H., Wierenga, K. J., and Lai, K. (2012). Innovative therapy for Classic Galactosemia - tale of two HTS. Mol. Genet. Metab. 105 (1), 44–55. doi:10.1016/j.ymgme.2011.09.028
Tang, M., Siddiqi, A., Witt, B., Yuzyuk, T., Johnson, B., Fraser, N., et al. (2014). Subfertility and growth restriction in a new galactose-1 phosphate uridylyltransferase (GALT) - deficient mouse model. Eur. J. Hum. Genet. 22 (10), 1172–1179. doi:10.1038/ejhg.2014.12
Tang, M., Wierenga, K., Elsas, L. J., and Lai, K. (2010). Molecular and biochemical characterization of human galactokinase and its small molecule inhibitors. Chem. Biol. Interact. 188 (3), 376–385. doi:10.1016/j.cbi.2010.07.025
Timmers, I., Jansma, B. M., and Rubio-Gozalbo, M. E. (2012). From mind to mouth: event related potentials of sentence production in classic galactosemia. PLoS One 7 (12), e52826. doi:10.1371/journal.pone.0052826
Timmers, I., van den Hurk, J., Di Salle, F., Rubio-Gozalbo, M. E., and Jansma, B. M. (2011). Language production and working memory in classic galactosemia from a cognitive neuroscience perspective: future research directions. J. Inherit. Metab. Dis. 34 (2), 367–376. doi:10.1007/s10545-010-9266-4
Timmers, I., van den Hurk, J., Hofman, P. A., Zimmermann, L. J., Uludağ, K., Jansma, B. M., et al. (2015a). Affected functional networks associated with sentence production in classic galactosemia. Brain Res. 1616, 166–176. doi:10.1016/j.brainres.2015.05.007
Timmers, I., van der Korput, L. D., Jansma, B. M., and Rubio-Gozalbo, M. E. (2016). Grey matter density decreases as well as increases in patients with classic galactosemia: a voxel-based morphometry study. Brain Res. 1648, 339–344. doi:10.1016/j.brainres.2016.08.005
Timmers, I., Zhang, H., Bastiani, M., Jansma, B. M., Roebroeck, A., and Rubio-Gozalbo, M. E. (2015b). White matter microstructure pathology in classic galactosemia revealed by neurite orientation dispersion and density imaging. J. Inherit. Metab. Dis. 38 (2), 295–304. doi:10.1007/s10545-014-9780-x
van Erven, B., Jansma, B. M., Rubio-Gozalbo, M. E., and Timmers, I. (2017). Exploration of the brain in rest: resting-state functional MRI abnormalities in patients with classic galactosemia. Sci. Rep. 7 (1), 9095. doi:10.1038/s41598-017-09242-w
Waggoner, D. D., Buist, N. R., and Donnell, G. N. (1990). Long-term prognosis in galactosaemia: results of a survey of 350 cases. J. Inherit. Metab. Dis. 13 (6), 802–818. doi:10.1007/BF01800204
Wahl, A., van den Akker, E., Klaric, L., Štambuk, J., Benedetti, E., Plomp, R., et al. (2018). Genome-wide association study on immunoglobulin G glycosylation patterns. Front. Immunol. 9, 277. doi:10.3389/fimmu.2018.00277
Waisbren, S. E., Norman, T. R., Schnell, R. R., and Levy, H. L. (1983). Speech and language deficits in early-treated children with galactosemia. J. Pediatr. 102 (1), 75–77. doi:10.1016/s0022-3476(83)80292-3
Waisbren, S. E., Potter, N. L., Gordon, C. M., Green, R. C., Greenstein, P., Gubbels, C. S., et al. (2012). The adult galactosemic phenotype. J. Inherit. Metab. Dis. 35 (2), 279–286. doi:10.1007/s10545-011-9372-y
Welling, L., Bernstein, L. E., Berry, G. T., Burlina, A. B., Eyskens, F., Gautschi, M., et al. (2017b). International clinical guideline for the management of classical galactosemia: diagnosis, treatment, and follow-up. J. Inherit. Metab. Dis. 40 (2), 171–176. doi:10.1007/s10545-016-9990-5
Welling, L., Meester-Delver, A., Derks, T. G., Janssen, M. C. H., Hollak, C. E. M., de Vries, M., et al. (2019). The need for additional care in patients with classical galactosaemia. Disabil. Rehabil. 41 (22), 2663–2668. doi:10.1080/09638288.2018.1475514
Welling, L., Waisbren, S. E., Antshel, K. M., Colhoun, H. O., Gautschi, M., Grunewald, S., et al. (2017a). Systematic review and meta-analysis of intelligence quotient in early-treated individuals with classical galactosemia. JIMD Rep. 37, 115–123. doi:10.1007/8904_2017_22
Wells, I. C., and Remy, C. N. (1965). Choline metabolism in normal and choline-deficient rats of different ages. Arch. Biochem. Biophys. 112 (1), 201–206. doi:10.1016/0003-9861(65)90030-5
Wells, W. W., Pittman, T. A., Wells, H. J., and Egan, T. J. (1965). The isolation and identification of galactitol from the brains of galactosemia patients. J. Biol. Chem. 240, 1002–1004. doi:10.1016/s0021-9258(18)97527-7
Welsink-Karssies, M. M., Ferdinandusse, S., Geurtsen, G. J., Hollak, C. E. M., Huidekoper, H. H., Janssen, M. C. H., et al. (2020a). Deep phenotyping classical galactosemia: clinical outcomes and biochemical markers. Brain Commun. 2 (1), fcaa006. doi:10.1093/braincomms/fcaa006
Welsink-Karssies, M. M., Oostrom, K. J., Hermans, M. E., Hollak, C. E. M., Janssen, M. C. H., Langendonk, J. G., et al. (2020b). Classical galactosemia: neuropsychological and psychosocial functioning beyond intellectual abilities. Orphanet J. Rare Dis. 15 (1), 42. doi:10.1186/s13023-019-1277-0
Welsink-Karssies, M. M., Schrantee, A., Caan, M. W. A., Hollak, C. E. M., Janssen, M. C. H., Oussoren, E., et al. (2020c). Gray and white matter are both affected in classical galactosemia: an explorative study on the association between neuroimaging and clinical outcome. Mol. Genet. Metab. 131 (4), 370–379. doi:10.1016/j.ymgme.2020.11.001
Keywords: classic galactosemia, brain, galactose, cognitive problems, neurodevelopment, movement disorders, neuropsychiatry
Citation: Panis B, Vos EN, Barić I, Bosch AM, Brouwers MCGJ, Burlina A, Cassiman D, Coman DJ, Couce ML, Das AM, Demirbas D, Empain A, Gautschi M, Grafakou O, Grunewald S, Kingma SDK, Knerr I, Leão-Teles E, Möslinger D, Murphy E, Õunap K, Pané A, Paci S, Parini R, Rivera IA, Scholl-Bürgi S, Schwartz IVD, Sdogou T, Shakerdi LA, Skouma A, Stepien KM, Treacy EP, Waisbren S, Berry GT and Rubio-Gozalbo ME (2024) Brain function in classic galactosemia, a galactosemia network (GalNet) members review. Front. Genet. 15:1355962. doi: 10.3389/fgene.2024.1355962
Received: 14 December 2023; Accepted: 24 January 2024;
Published: 15 February 2024.
Edited by:
Pranoot Tanpaiboon, Quest Diagnostics, United StatesReviewed by:
Andrea Lynne Gropman, Children’s National Hospital, United StatesCopyright © 2024 Panis, Vos, Barić, Bosch, Brouwers, Burlina, Cassiman, Coman, Couce, Das, Demirbas, Empain, Gautschi, Grafakou, Grunewald, Kingma, Knerr, Leão-Teles, Möslinger, Murphy, Õunap, Pané, Paci, Parini, Rivera, Scholl-Bürgi, Schwartz, Sdogou, Shakerdi, Skouma, Stepien, Treacy, Waisbren, Berry and Rubio-Gozalbo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: M. Estela Rubio-Gozalbo, ZXN0ZWxhLnJ1YmlvQG11bWMubmw=
†These authors have contributed equally to this work and share first authorship
‡These authors share senior authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.