 Qiang Zhang
Qiang Zhang Yusi Liu1†
Yusi Liu1† Xuan Liu
Xuan Liu
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Genet. , 29 January 2024
Sec. Genetics of Common and Rare Diseases
Volume 15 - 2024 | https://doi.org/10.3389/fgene.2024.1344682
Hypotonia, Ataxia, Developmental Delay, and Tooth Enamel Defect Syndrome (HADDTS) is an exceptionally rare disorder resulting from a heterozygous variant in the C-terminal binding protein 1 (CTBP1) gene. To date, a mere two variants (14 patients) have been documented on a global scale. The aim of this study was to identify a causative CTBP1 variant in a Chinese patient, and to determine the potential pathogenicity of the identified variant. Here, Whole-exome sequencing (WES) was conducted on the proband to pinpoint the candidate variant. Following this, Sanger sequencing was employed to validate the identified candidate variant and examine its co-segregation within the available family members. Employing both in silico prediction and three-dimensional protein modeling, we conducted an analysis to assess the potential functional implications of the variant on the encoded protein. Our investigation led to the identification of a novel heterozygous variant in the CTBP1 gene, namely, c.371 C>T (p.Ser124Phe), in a Chinese patient. This case represents the first confirmed instance of such a variant in a Chinese patient. When comparing the patient’s clinical symptoms with those reported in the literature, notable distinctions were observed between her primary symptoms and those associated with HADDTS. She showed other signs such as microcephaly, coarse facial features, single transverse palmar crease, visible beard, myopia, coarse toenail and skeletal anomalies. This study enriching the spectrum of genetic variants observed in different ethnic populations and expanding the phenotypic profile associated with this gene. These findings are expected to contribute to the enhancement of future variant-based screening and genetic diagnosis, while also providing further insights into the pathogenic mechanisms underlying CTBP1-related conditions.
C-terminal binding protein 1 (CTBP1), located on chromosome 4p16, functions as a transcriptional co-repressor (Chen, 2021). Additionally, CTBP1 serves as a crucial scaffolding protein that recruits chromatin-modifying enzymes to specific genomic regions, thereby coordinating gene expression programs (Sekiya et al., 2021). CTBP1 plays a regulatory role in gene expression during various processes, including development, apoptosis, and oncogenesis (He et al., 2021). Initially, CTBP1 was discovered as a protein capable of binding to the C-terminus of the human adenovirus E1A protein (Hildebrand and Soriano, 2002; Senyuk et al., 2005). Studies in mice have demonstrated that CTBP1 has specific transcriptional functions during fetal development (Stankiewicz et al., 2014; Blevins et al., 2017). Furthermore, Hu et al. (2019) uncovered the protective effect of CTBP1 on hippocampal and cortical neurons, safeguarding them against degeneration. Notably, high CTBP1 expression has been shown to attenuate apoptosis and enhance neuronal activity in these specific neuronal populations (Stankiewicz et al., 2014). In 2016, it was definitively established that variant in the CTBP1 gene, located at chr4:1,205,233–1,242,918 GRCh37/hg19, cause Hypotonia, Ataxia, Developmental Delay, and Dental Enamel Defect Syndrome (HADDTS, MIM:617915) (Beck et al., 2016). In this study, we present a unique phenotype resulting from a novel de novo variant in the CTBP1 gene, which represents the first confirmed case in a Chinese patient.
The patient underwent a whole-exome sequencing examination, wherein a genomic DNA sample was collected and utilized to construct sequence libraries. The construction of libraries was carried out using the Agilent Sure Select Human Whole Exome V2 Kit (Agilent Technologies, Santa Clara, CA). Subsequently, sequencing of the prepared libraries was performed using the HiSeq2500 System (Illumina, San Diego, CA). The reads obtained from the Burrows-Wheeler Aligner (BWA) software package (v. 0.7.15) were aligned to the human reference genome (GRCh37/hg19). Variant calling and annotation utilized the Genome Analysis Toolkit (GATK), and further variant annotation and prioritization were conducted using TGex software (LifeMap Sciences, Inc. v5.7).
Sanger sequencing was conducted to validate the variant in the proband and her family members. The designed primers, 5′-CTGCCACGTGAACCTAAAGT-3′ and 5′-GTGTGTTATCTGCATGTGCC-3′, obtained from Oligo7, were utilized specifically for c.371 C>T (p.Ser124Phe).
The bioinformatics tools Scale Invariant Feature Transform (SIFT, http://sift.jcvi.org/), Mutation Taster software (https://www.mutationtaster.org/), Provean (http://provean.jcvi.org/seq_submit.php), Combined Annotation Dependent Depletion (CADD, https://cadd.gs.washington.edu/snv) and rare exome variant ensemble learner (REVEL,https://sites.google.com/site/revelgenomics/) were applied to predict the impact of the variant on protein function. The protein 3D structures of CTBP1 were generated by the Swiss-Model server (https://swissmodel.expasy.org/), and the ACMG/AMP variant classification guidelines were employed for variant classification (Li and Wang, 2017).
The Chinese patient arrived at our hospital exhibiting global developmental delay, dwarfism, and distinctive facial features. She was a full-term baby born to non-consanguineous parents, yet she was small for her gestational age (SGA). No signs of neonatal asphyxia were present during delivery, and she achieved perfect Apgar scores of 10/10/10. During the physical examination, we noted her birth data: a height of 46 cm (−2 SD), weight of 2.4 kg (−2 SD), and head circumference of 33 cm (−1.5 SD).
The child displayed unique facial attributes such as microcephaly, a visible beard, synophrys, low-set and protruding ears. She also had other physical characteristics, including a single transverse palmar crease, a short fifth finger, congenital dislocation of the radial head, pectus excavatum, and myopia (Figure 1).
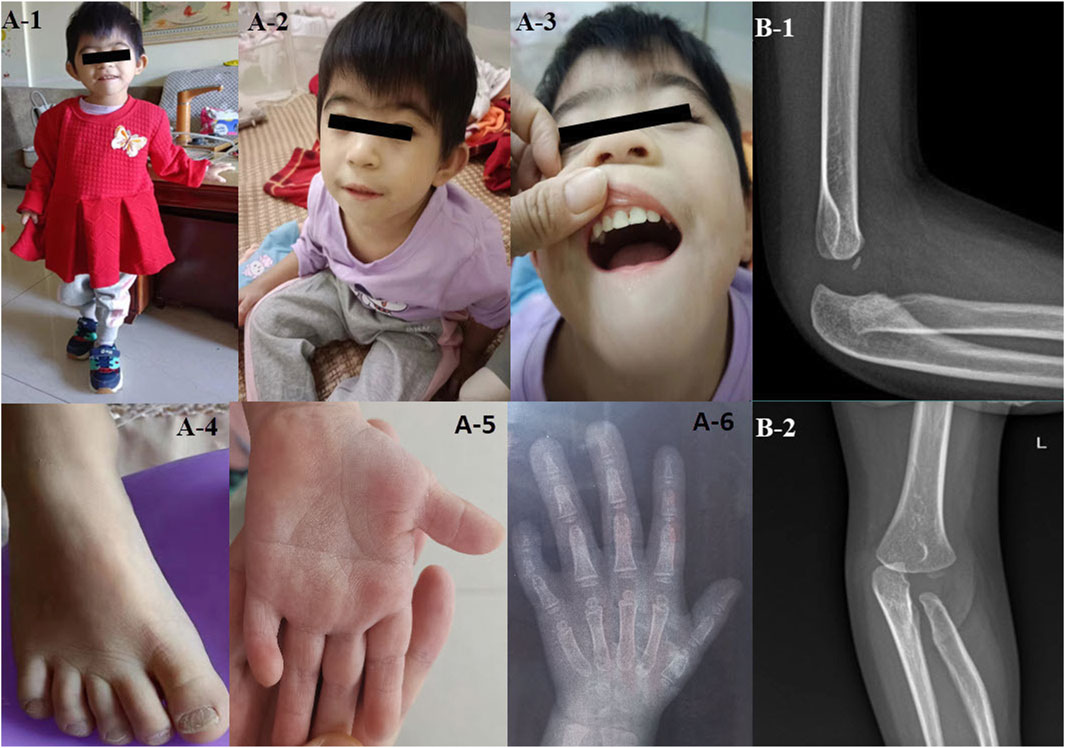
FIGURE 1. The clinical features of the proband with CTBP1 variation (A-1–A-6) illustrate the phenotypic characteristics of the patient: microcephaly, a visible beard, synophrys, low-set ears, protruding ears, coarse toenail, a single transverse palmar crease, and a short fifth finger. (B-1, B-2) depict the congenitally dislocated radial head of the patient.
Assessments using the Gesell Developmental Scale (GDS) (Figure 2C) and the Infant-Junior High School Student’s Social Living Ability Scale (S-M) (with the patient’s score exceeding 8 points) indicated that the child was developmentally delayed and had difficulties adjusting socially. Her intelligence quotient (IQ) score of 49 suggests a moderate level of intellectual disability, as per the Wechsler Preschool and Primary Scale of Intelligence (WPPSI) scale, where moderate intellectual impairment falls within the range of 35–49.
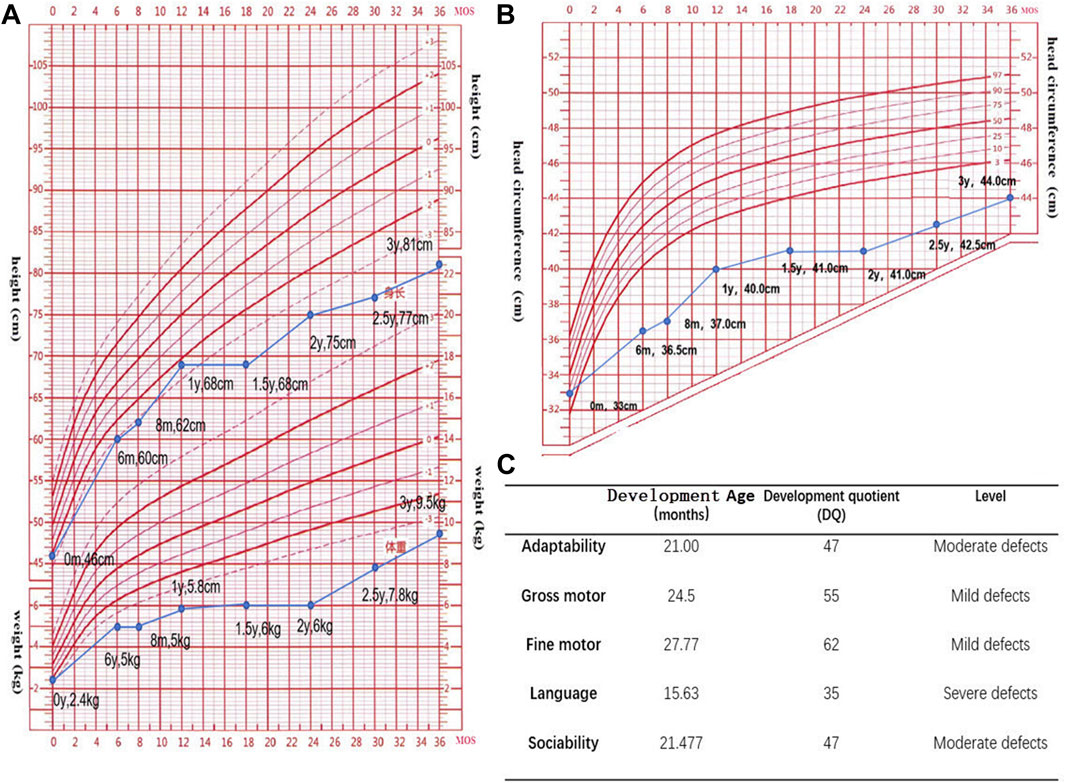
FIGURE 2. (A,B). Standard deviation scores (SDS) for height, weight, and head circumference in the age range of 0–3 years. (The data suggests that the child is experiencing growth and developmental delays, as evidenced by parameters such as height, weight, and head circumference falling below the averages for their age group.) (C). The Gesell Developmental Scale (GDS) has been used to assess neurodevelopment in the patient. The GDS reveals slight shortcomings in motor development, moderate inadequacies in adaptive and social skills, and profound impairments in language development.
Additionally, a cardiac ultrasound revealed mild tricuspid valve regurgitation. However, tests for liver function, kidney function, 25-OH-VD levels, thyroid function, female endocrine function, adrenal gland function, electroencephalogram, blood tandem mass spectrometry, and brain magnetic resonance imaging (MRI) all returned normal results.
We identified a heterozygote variant, c.371 C>T (p.Ser124Phe), in the CTBP1 gene (NM_001328.2, Chr4:1,219,324 in exon 4) in the proband using WES. Sanger sequencing confirmed that the c.371 C>T (p.Ser124Phe) variant was a de novo variant (see Figure 3A). We used five in silico tools to predict the impact of the novel variant (see Figure 3D), which suggested that c.371 C>T (p.Ser124Phe) is a harmful variant. The three-dimensional structure of the variant protein and the pathogenicity of amino acid substitutions and their molecular mechanisms were analyzed using the SWISS-MODEL software and MutPred2 web (Pejaver et al., 2020), respectively (see Figure 3B). It was observed that the variant protein altered ordered interfaces, transmembrane proteins, metal binding, and resulted in the loss of the catalytic site at position N119 and the acquisition of a heterologous site at position P121.
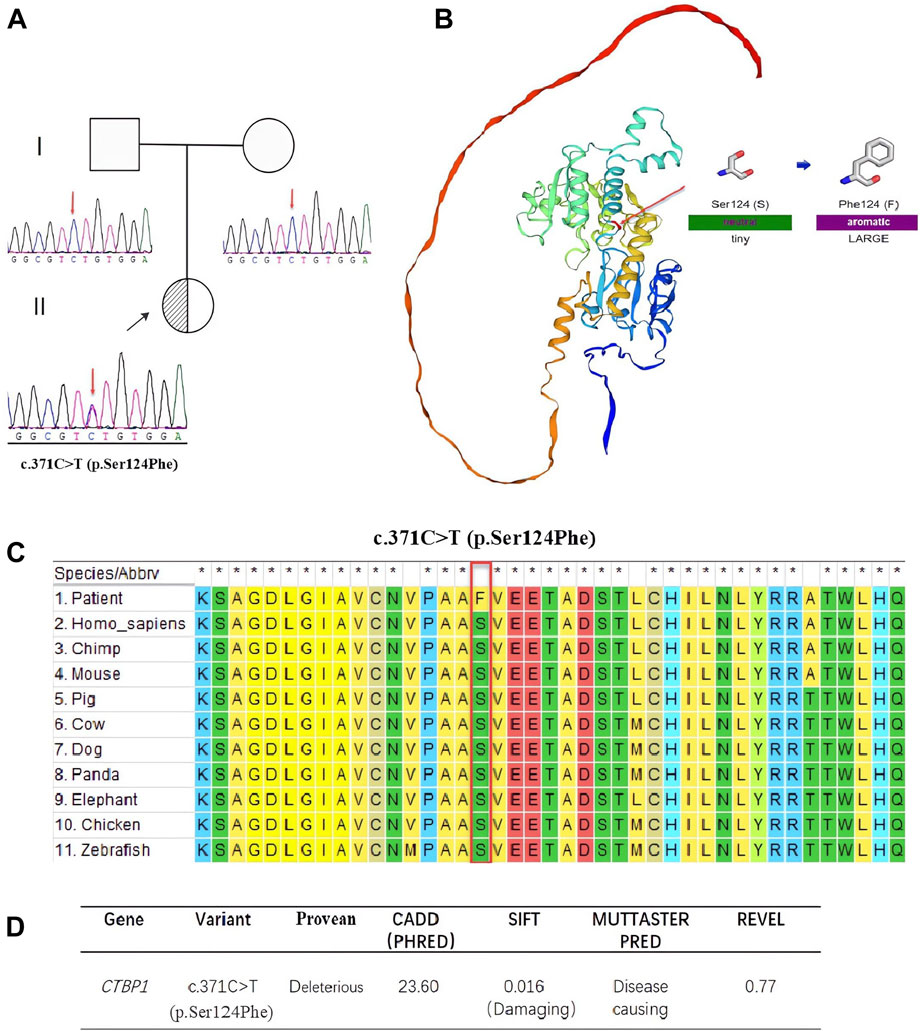
FIGURE 3. (A). Sequencing results of c.371 C>T (p.Ser124Phe) and pedigree chart of the patients’ family. Sanger sequencing DNA chromatograms of CTBP1 indicate that the variant is de novo. The proband is marked with a black arrow. (B). Three-dimensional structures of CTBP1 protein. The residue at sequence position 124 in this protein is serine, which has a neutral side chain. The variant residue is phenylalanine, which has an aromatic side chain. (C). Sequence alignment shows that Ser124Phe is highly conserved across various species. (D). The effects of c.371 C>T (p.Ser124Phe) were predicted using five different software packages.
HADDTS is an extremely rare autosomal dominant disorder caused by a disruptive pathogenic variant of CTBP1. Fourteen cases of pathogenic variants in the CTBP1 gene have been reported globally, in such cases hypotonia, ataxia, developmental delay and dental enamel defects have long been recognized as the core symptoms. Here, we first described a Chinese pedigree with CTBP1 variant, and interestingly the patient developed none of hypotonia, ataxia, and dental enamel defects.
The CTBP1 gene is the only known cause of HADDTS (Bhatia et al., 2020). The gene is widely expressed in brains, livers, lungs, and it may play a key role in controlling the equilibrium between tubular and stacked structures in the Golgi complex, and functioning in brown adipose tissue (BAT) differentiation (Acosta-Baena et al., 2022). The true prevalence of HADDTS is not well-known because only two pathogenic variants (14 patients) in CTBP1 have been found around the world (Jafari Khamirani et al., 2021). A novel variant is described in this study, c.371 C>T (p.Ser124Phe). The variant was de novo and the relatedness was confirmed by paternity testing (PS2). It is a rare Single Nucleotide Polymorphism (SNP), neither found to be present in controls, the 1,000 Genome Project (https://www.internationalgenome.org/) nor the Exome Sequencing Project (https://exome.gs.washington.edu/) (PM2-supporting). Rare SNPs have very broad and deleterious effects on phenotypes when compared with the weak effects of common SNPs (Bomba et al., 2017). Benign missense variant in CTBP1 has a low rate meanwhile missense variants are common mechanisms of disease (Z-score:3.31 > 3.09, PP2), and multiple pathogenicity prediction algorithms support deleterious effects on genes or gene products (Figures 3C, D, PP3). Thus, the novel variant can be classified as likely pathogenic (PS2, PM2-supporting, PP2, PP3) according to ACMG. It is important to note that, despite the ACMG guidelines assessing this variant as potentially pathogenic, further functional studies are necessary to validate its pathogenicity given the significant deviation in phenotype observed in this patient compared to prior reports. In addition, we summarized all the reported mutants that leads to HADDTS (Figure 4). The clinical presentations of HADDTS include intellectual disability, hypotonia, ataxia, developmental delay, failure to thrive, and tooth enamel defects (Vijayalingam et al., 2020). The patient in this study exhibited different phenotype in comparison with the previously reported cases. She had no signs of hypotonia, difficulty in walking or tooth enamel defect. Meanwhile, the child exhibited distinct facial features including microcephaly, a small beard, synophrys, low-set ears, and protruding ears, as well as other physical characteristics such as prominent palms, a short fifth finger, an inability to fully extend the elbow joint, pectus excavatum, and myopia. We compiled the clinical phenotypes, laboratory test results and molecular characteristics of 14 previously reported cases (Table 1). Through extensive literature analysis, we found that the youngest patient among all the enrolled individuals was 3 years old (our study), while the oldest was 26 years old (Table 1-P14). The male-to-female ratio was 2:1. Growth retardation was observed in 93.3% (14/15) of the patients, and developmental regression was present in 40.0% (6/15), with motor and language regression being the most common. Additionally, 84.6% (11/13) had dental defects, 66.7% (10/15) exhibited ataxia, 93.3% (14/15) developed intellectual disability, and 86.7% (13/15) had decreased muscle tone. All patients showed delayed language development (15/15). The genetic variants identified in this study were primarily attributed to de novo variants (14/15), with only one reported case of somatic mosaicism from maternal inheritance (Table 1-P1). Among the 15 patients, 13cases had the same variants (c.991 C > T (p.Arg331Trp) in NM_001012614.1; c.1024 C>T (p.Arg342Trp) in NM_001328.2). These variants were responsible for a high degree of symptom consistency, characterized primarily by developmental delay, intellectual disability, failure to thrive, hypotonia, ataxia, and tooth enamel defects. c.1024 C>T (p.Arg342Trp) is within the C-terminal portion of the PLDLS (Pro Leu-Asp-Leu-Ser) binding cleft, which is the domain through which CTBP1, interacts with chromatin-modifying enzymes and mediates chromatin-dependent gene repression pathways (Quinlan et al., 2006; Beck et al., 2019). The variant discovered in the study, c.371 C>T (p.Ser124Phe), is located in the catalytic domain. Ser124 is a catalytic residue, hence crucial for the protein’s function. The residue at sequence position 124 in this protein is a serine which has a neutral side chain. The variant residue is phenylalanine which has an aromatic side chain (i.e., containing an aromatic ring), that can stack against others. A change from a Ser to a Phe side chain is a very large one, and might well result in a change to the protein’s function. A Ser to Phe residue change is highly unfavoured in terms of conserved amino acid properties (Figure 3C). The newly reported patient is phenotypically different in comparison with the previously reported cases. She had no sign of hypotonia, gait abnormality and dental defect. Initially, we considered the possibility of Cornelia de Lange syndrome of the patient based on her physical features (Sarogni et al., 2020). However, after conducting whole exome sequencing analysis, no genetic variants associated with Cornelia de Lange syndrome, such as NIPBL, SMC1A, SMC3, RAD21 and HDAC8 were found (Helgeson et al., 2018). As a result, this disease was ultimately ruled out. The patient was small for gestational age infant, which suggested the possibility of intrauterine growth retardation. We closely monitored her growth from birth until the age of 3 (Figure 2) and found that the patient’s growth failure remained unimproved throughout. The clinical presentation of the proband in this study was obviously different from the core clinical symptoms (tooth enamel defects, intellectual disability, and dysarthria) of the previously reported. Although CTBP1 variants can lead to mitochondrial complex I and IV activities impairments in skeletal muscle, causing mitochondrial diseases (Sommerville et al., 2017), the patient we described did not exhibit any biochemical changes or muscle abnormalities. The exact pathogenic mechanism of the novel discovered heterozygous variant (c.371 C>T (p.Ser124Phe)) has not been fully elucidated. Further observation is required. It remains to be determined whether this patient will gradually manifest phenotypes of both mitochondrial disease and HADDTS as she ages or if this variant in CTBP1 will result in another disease or subtype alternatively, these symptoms may represent heterogeneous manifestations of HADDTS. Thus, continued monitoring and follow-up is necessary. Further research on patients with pathogenic variants in CTBP1 would greatly contribute to the development of diagnostic and management strategies.
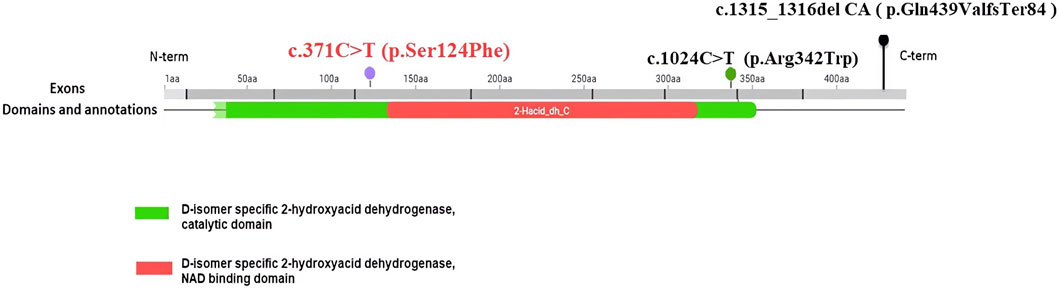
FIGURE 4. The CTBP1 variants that were reported. c.371 C>T (p.Ser124Phe) in exon 4 and D-isomer specific2-hydroxyacid dehydrogenase, catalytic domain.
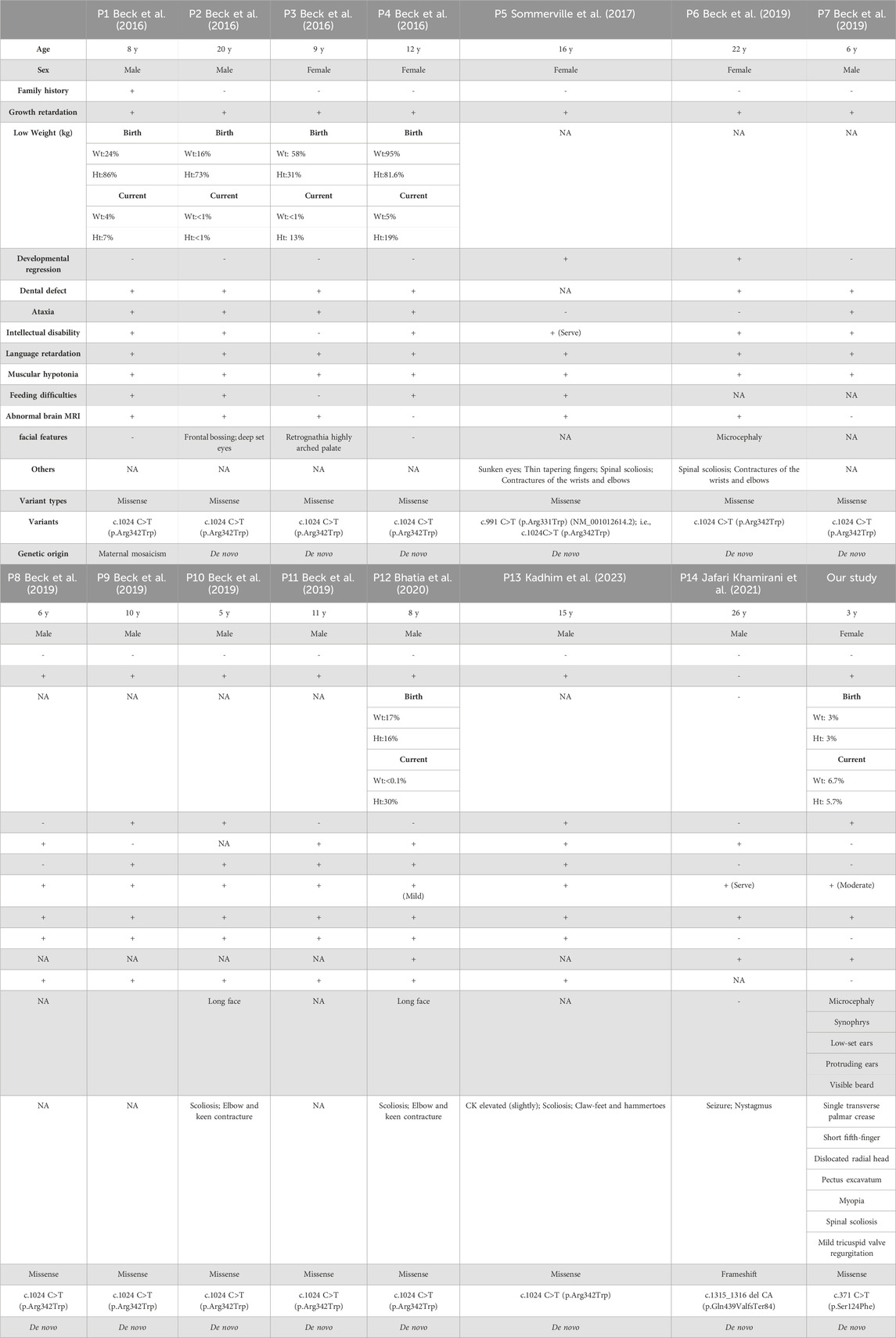
TABLE 1. Summary of clinical and molecular features of all patients with CTBP1 variants.
In this study, a novel pathogenic variant was identified in the CTBP1 gene of a Chinese patient. The patient exhibited distinctive facial features, growth delay, delayed speech and language development, intellectual disability, and skeletal anomalies. The variant described in the study has expanded the phenotypic spectrum among patients from diverse ethnic backgrounds. While the mechanisms underlying these differences remain unclear, they contribute to the exploration of the biological mechanisms involved in CTBP1 pathogenesis.
The original contributions presented in the study are publicly available. The data are deposited in the NCBI Sequence Read Archive repository, accession number PRJNA1019228 (https://www.ncbi.nlm.nih.gov/sra/ PRJNA1019228).
The studies involving humans were approved by The study was approved by the Institutional Review Board and Ethics Committee of Guangxi Maternal and Child Health Hospital. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants’ legal guardians/next of kin. Written informed consent was obtained from the individual(s), and minor(s)’ legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
QZ: Funding acquisition, Writing–original draft, Data curation, Investigation, Visualization, Writing–review and editing. YL: Data curation, Writing–review and editing, Resources. XL: Software, Writing–review and editing, Data curation. YZ: Writing–review and editing, Data curation, Validation, Visualization. JZ: Project administration, Supervision, Writing–review and editing, Investigation, Resources.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. The research received financial support from the Guangxi Zhuang Region Health Department (Z20190311, Z-A 20230305, Z20220256.) and Liaoning Province Applied Basic Research Program Project (2023JH2/101300044).
We sincerely thank all the family members for their support during this study.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acosta-Baena, N., Tejada-Moreno, J. A., Arcos-Burgos, M., and Villegas-Lanau, C. A. (2022). CTBP1 and CTBP2 mutations underpinning neurological disorders: a systematic review. Neurogenetics 23, 231–240. doi:10.1007/s10048-022-00700-w
Beck, D. B., Cho, M. T., Millan, F., Yates, C., Hannibal, M., O’Connor, B., et al. (2016). A recurrent de novo CTBP1 mutation is associated with developmental delay, hypotonia, ataxia, and tooth enamel defects. Neurogenetics 17, 173–178. doi:10.1007/s10048-016-0482-4
Beck, D. B., Subramanian, T., Vijayalingam, S., Ezekiel, U. R., Donkervoort, S., Yang, M. L., et al. (2019). A pathogenic CtBP1 missense mutation causes altered cofactor binding and transcriptional activity. Neurogenetics 20, 129–143. doi:10.1007/s10048-019-00578-1
Bhatia, S. K., Arora, V., and Verma, I. C. (2020). A further case of hypotonia, ataxia, developmental delay and tooth enamel defect syndrome due to a recurrent C-terminal binding protein 1 mutation. Clin. Dysmorphol. 29, 148–151. doi:10.1097/MCD.0000000000000321
Blevins, M. A., Huang, M., and Zhao, R. (2017). The role of CtBP1 in oncogenic processes and its potential as a therapeutic target. Mol. Cancer Ther. 16, 981–990. doi:10.1158/1535-7163.MCT-16-0592
Bomba, L., Walter, K., and Soranzo, N. (2017). The impact of rare and low-frequency genetic variants in common disease. Genome Biol. 18, 77. doi:10.1186/s13059-017-1212-4
Chen, Z. (2021). The transrepression and transactivation roles of CtBPs in the pathogenesis of different diseases. J. Mol. Med. 99, 1335–1347. doi:10.1007/s00109-021-02107-w
He, Y., He, Z., Lin, J., Chen, C., Chen, Y., and Liu, S. (2021). CtBP1/2 differentially regulate genomic stability and DNA repair pathway in high-grade serous ovarian cancer cell. Oncogenesis 10, 49. doi:10.1038/s41389-021-00344-9
Helgeson, M., Keller-Ramey, J., Knight Johnson, A., Lee, J. A., Magner, D. B., Deml, B., et al. (2018). Molecular characterization of HDAC8 deletions in individuals with atypical Cornelia de Lange syndrome. J. Hum. Genet. 63, 349–356. doi:10.1038/s10038-017-0387-6
Hildebrand, J. D., and Soriano, P. (2002). Overlapping and unique roles for C-terminal binding protein 1 (CtBP1) and CtBP2 during mouse development. Mol. Cell. Biol. 22, 5296–5307. doi:10.1128/MCB.22.15.5296-5307.2002
Hu, K., Li, Y., Yu, H., and Hu, Y. (2019). CTBP1 confers protection for hippocampal and cortical neurons in rat models of alzheimer’s disease. Neuroimmunomodulation 26, 139–152. doi:10.1159/000500942
Jafari Khamirani, H., Zoghi, S., Saber Sichani, A., Dianatpour, M., Mohammadi, S., Mohammad Bagher Tabei, S., et al. (2021). Exome sequencing identified a de novo frameshift pathogenic variant of CTBP1 in an extremely rare case of HADDTS. J. Genet. 100, 68. doi:10.1007/s12041-021-01315-0
Kadhim, H., El-Howayek, E., Coppens, S., Duff, J., Topf, A., Kaleeta, J.-P., et al. (2023). A pathogenic CTBP1 variant featuring HADDTS with dystrophic myopathology. Neuromuscul. Disord. 33, 410–416. doi:10.1016/j.nmd.2023.03.007
Pejaver, V., Urresti, J., Lugo-Martinez, J., Pagel, K. A., Lin, G. N., Nam, H.-J., et al. (2020). Inferring the molecular and phenotypic impact of amino acid variants with MutPred2. Nat. Commun. 11, 5918. doi:10.1038/s41467-020-19669-x
Quinlan, K. G. R., Verger, A., Kwok, A., Lee, S. H. Y., Perdomo, J., Nardini, M., et al. (2006). Role of the C-terminal binding protein PXDLS motif binding cleft in protein interactions and transcriptional repression. Mol. Cell Biol. 26, 8202–8213. doi:10.1128/MCB.00445-06
Sarogni, P., Pallotta, M. M., and Musio, A. (2020). Cornelia de Lange syndrome: from molecular diagnosis to therapeutic approach. J. Med. Genet. 57, 289–295. doi:10.1136/jmedgenet-2019-106277
Sekiya, M., Kainoh, K., Sugasawa, T., Yoshino, R., Hirokawa, T., Tokiwa, H., et al. (2021). The transcriptional corepressor CtBP2 serves as a metabolite sensor orchestrating hepatic glucose and lipid homeostasis. Nat. Commun. 12, 6315. doi:10.1038/s41467-021-26638-5
Senyuk, V., Sinha, K. K., and Nucifora, G. (2005). Corepressor CtBP1 interacts with and specifically inhibits CBP activity. Archives Biochem. biophysics 441, 168–173. doi:10.1016/j.abb.2005.06.024
Sommerville, E. W., Alston, C. L., Pyle, A., He, L., Falkous, G., Naismith, K., et al. (2017). De novo CTBP1 variant is associated with decreased mitochondrial respiratory chain activities. Neurol. Genet. 3, e187. doi:10.1212/NXG.0000000000000187
Stankiewicz, T. R., Gray, J. J., Winter, A. N., and Linseman, D. A. (2014). C-terminal binding proteins: central players in development and disease. Biomol. Concepts 5, 489–511. doi:10.1515/bmc-2014-0027
Keywords: C-terminal binding protein 1 (CTBP1), HADDTS, de novo variant, whole-exome sequencing, clinical heterogeneity
Citation: Zhang Q, Liu Y, Liu X, Zhao Y and Zhang J (2024) A novel CTBP1 variant in a Chinese pediatric patient with a phenotype distinct from hypotonia, ataxia, developmental delay, and tooth enamel defect syndrome. Front. Genet. 15:1344682. doi: 10.3389/fgene.2024.1344682
Received: 26 November 2023; Accepted: 10 January 2024;
Published: 29 January 2024.
Edited by:
Ji Hyun Lee, Kyung Hee University, Republic of KoreaReviewed by:
Afagh Alavi, University of Social Welfare and Rehabilitation Sciences, IranCopyright © 2024 Zhang, Liu, Liu, Zhao and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jihong Zhang, MTI3MDMxOTA4NUBxcS5jb20=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.