 Yujing Gong1†
Yujing Gong1† Weijian Zhu2†
Weijian Zhu2† Mianmian Zhu1†
Mianmian Zhu1† Dan Chen1,3
Dan Chen1,3 Sunke Wu1,4
Sunke Wu1,4 Sisi Hu1
Sisi Hu1 Yi Luo1,4
Yi Luo1,4 Yiyi Jiang5
Yiyi Jiang5 Ting Zhu1
Ting Zhu1 Dan Wang1*
Dan Wang1*- 1Department of Pediatrics, The First Affiliated Hospital of Wenzhou Medical University, Wenzhou, Zhejiang, China
- 2Central Laboratory, The First Affiliated Hospital of Wenzhou Medical University, Wenzhou, Zhejiang, China
- 3Department of Pediatrics, Yongjia People’s Hospital, Wenzhou, Zhejiang, China
- 4Department of Pediatrics, Taizhou Woman and Children’s Hospital, Taizhou, Zhejiang, China
- 5Department of Radiography, The First Affiliated Hospital of Wenzhou Medical University, Wenzhou, Zhejiang, China
Background: Stickler syndrome type I (STL1) is an autosomal dominant disorder characterized by ocular, auditory, orofacial, and skeletal anomalies. The main causes of STL1 are variants in the COL2A1 gene, which encodes a type II collagen precursor protein. The specific focus of this study was on a newborn from China diagnosed with STL1, with the aim of providing novel insights into the effects of a newly identified intronic variant in the COL2A1 gene on pre-mRNA splicing.
Methods: Trio whole exome sequencing was used to identify the causative variant in the family. The identified variant was validated using Sanger sequencing. Bioinformatics programs were used to predict the pathogenicity of the candidate variant. Additionally, an in vitro minigene assay was used to investigate the effects of the identified variant on RNA splicing.
Results: The proband with STL1 had a novel heterozygous splicing variant in the intron nine acceptor donor site of COL2A1 (c.655-2A>G). This splice junction variant resulted in aberrant COL2A1 mRNA splicing, leading to the skipping of exon 10 and the production of a shorter protein that may lack the last 18 native amino acids.
Conclusion: The c.655-2A>G variant in the COL2A1 gene leads to STL1 through abnormal splicing. By expanding the spectrum of variants in the COL2A1 gene, this finding improves the clinical understanding of STL1 and provides guidance for early diagnosis and disease counseling.
1 Introduction
Stickler syndrome is a collagen connective tissue disease characterized by ocular, auditory, orofacial, and skeletal anomalies, with dominant inheritance being common and recessive inheritance being rare (Snead and Yates, 1999). The incidence rate of Stickler syndrome in neonates is approximately 1 in 7,500–9,000 (Hoornaert et al., 2010). Based on locus heterogeneity, autosomal dominant hereditary Stickler syndrome is divided into three types: I, II, and III. The most prevalent form, Stickler syndrome type I (STL1, MIM #108300), is predominantly caused by defects in the COL2A1 gene (Faber et al., 2000). STL2 (MIM #604841) is associated with defects in COL11A1 and patients have exhibited the “beaded” type 2 vitreous phenotype (Poulson et al., 2004), (Wilkin et al., 1999). Type III, also known as non-ocular Stickler syndrome or otospondylomegaepiphyseal dysplasia, is caused by pathogenic variants in COL11A2 (Annunen et al., 1999). The symptoms and signs of patients diagnosed with STL exhibit wide variations. STL1 may present with a wide range of findings, including cleft palate, small jaw, hearing loss, or early-onset osteoarthritis (Stickler et al., 2001). STL1 is most commonly associated with ocular complications, but inner ear, skeletal/joint, and craniofacial structures are often involved. The clinical characteristics of STL1 include typical vitreous (membranous) lesions accompanied by retinal detachment; facial abnormalities, such as cleft palate, glossophthalmos, and retropalatal deformity; relatively mild corresponding osteophytes; and early-onset arthropathy. The phenotype of STL1 constantly evolves throughout life, making diagnosis challenging, particularly in sporadic cases. Therefore, it is crucial to diagnose this disease by combining phenotypic and genetic analyses (Boothe et al., 2020).
The COL2A1 gene maps on to 12q13.11-q13.2, contains 54 exons, and encodes the type II collagen precursor protein (Nishimura et al., 2005). Alterations in the amino acid arrangement of type II collagen due to COL2A1 variants can affect the structural stability and function of the protein helix, leading to type II collagenopathies (MIM #120140). (Annunen et al., 1999; Wilkin et al., 1999; Yamamoto et al., 2020). Type II collagenopathies can be broadly classified into five categories: lethal diseases, types mainly affecting spinal deformity, types involving long bones and joints, types primarily associated with ocular manifestations, and unusual types. Although STL1 belongs to the type mainly involving ocular manifestations, it is also characterized by skeletal dysplasia (Zhang et al., 2020). The most frequent manifestation of this disease during the neonatal period is a cleft palate (Hanspal et al., 2012). STL1 is the mildest type of collagen II disease (Barat-Houari et al., 2016; Zhang et al., 2020).
In this study, we present the case of a Chinese infant whose clinical manifestations aligned with those of patients with Stickler syndrome. Through whole exome sequencing, a rare and previously undescribed intron variant of COL2A1 (c.655-2A>G) was identified, broadening. The variant spectrum of COL2A1. Furthermore, we performed in silico analysis to investigate the possible molecular pathogenesis of Stickler syndrome in this patient.
2 Materials and methods
2.1 Participants
The proband and parents were enrolled at the First Affiliated Hospital of Wenzhou Medical University, where the proband was hospitalized because of a palatal deformity discovered during a post-birth physical examination. Relevant clinical records, including physical, routine blood, ultrasound, and oral computed tomography (CT) examination findings, were collected. Written consent was obtained from the parents of the newborn proband before commencing the study, which was approved by the Ethics Committee of the First Affiliated Hospital of Wenzhou Medical University.
2.2 Trio whole exome sequencing and sanger sequencing
Genomic DNA from blood samples (1–2 mL) of the proband and their family members was used in this assay. The DNA was first fragmented, and libraries were prepared; subsequently, the DNA in the all exons and the adjacent shear region were captured and enriched using the Roche KAPA HyperExome chip (KAPA Biosystems, Boston, MA, United States of America), and the MGISEQ-2000 sequencing platform (MGI Tech Co., Ltd., Shenzhen, Guangdong, China) was used for variant detection. The quality control index of the sequencing data was as follows: the average sequencing depth of the target region was ≥180X, and the percentage of loci with an average depth >20X in the target region was >95%. Sequenced fragments were mapped to the human reference genome (UCSC hg19) using BWA aligner (Burrows-Wheeler alignment tool, version 0.7.15) to remove duplicates. Base mass value correction for SNV, INDEL, and genotype detection was performed using GATK. ExomeDepth was used to detect copy number variation at the exon level. The main reference databases used in this study included population (ClinVar, ESP6500, and gnomAD) and clinical databases (Online Mendelian Inheritance in Man, Rare Disease Data Center [RDDC], GeneReviews, Orphanet, and Genetic Home Reference). According to the guidelines of the American College of Medical Genetics and Genomics, the pathogenicity of variants is divided into five categories. This is used to determine which category the variant is judged to be in, and further reference is provided for subsequent research. The suspected mutations were subsequently verified using Sanger sequencing (primer sequences: GTCAGAGTTCCTCCACAGCTAG and CCCTCATTTTCTGTTCCGATGC) in the studied family. Sanger sequencing was performed using an ABI 3730xl DNA Analyzer (Applied Biosystems, Carlsbad, CA, United States of America).
2.3 In silico assay
The RDDC database (https://rddc.tsinghua-gd.org/) and varSEAK (https://varseak.bio/) were used to analyze the possible effects of the mutation. SpliceAI Lookup (https://spliceailookup.broadinstitute.org/#) was used for the mutation evaluation. The three-dimensional (3D) structure of the protein template after introducing mutations was predicted using PyMOL. The template protein structure (XP_016874318.1) was obtained from the National Center for Biotechnology Information protein database (https://www.ncbi.nlm.nih.gov/). The prediction method followed the standard procedures.
2.4 Minigene constructs and mutagenesis
An in vitro minigene assay was performed to examine the target gene regions covering COL2A1 exons 10–11, intron 9, and intron 10, which were amplified from the gDNA of the proband. The wild-type COL2A1 gene fragment was obtained by nested polymerase chain reaction (PCR) using genomic DNA as the template and COL2A1-7948-F/COL2A1-9565-R and COL2A1-8186-F/COL2A1-996-R (Table 1) as primers. Three pairs of primers (pcDNA3.1-COL2A1-KpnI-F/COL2A1-MUT-R, COL2A1-MUT-F/pcDNA3.1-COL2A1-EcoRI-R, and pcDNA3.1-COL2A1-KpnI-F/pcDNA3.1-COL2A1-EcoRI-R) were developed to amplify the heterozygous c.655-2A>G mutation site from the product of nested PCR via seamless cloning (Vazyme Biotech Co., Ltd., Nanjing, China). Subsequently, the amplified DNA products were recombined and cloned into two digestion sites (HIKpnI/EcoRI) of the pcDNA3.1 vector (Hitrobio Biotechnology Co., Ltd., Beijing, China). In addition, the recombinant plasmids pcDNA3.1-COL2A1-t (wild-type) and pcDNA3.1-COL2A1-mut (c.655-2A>G) were validated using Sanger sequencing.
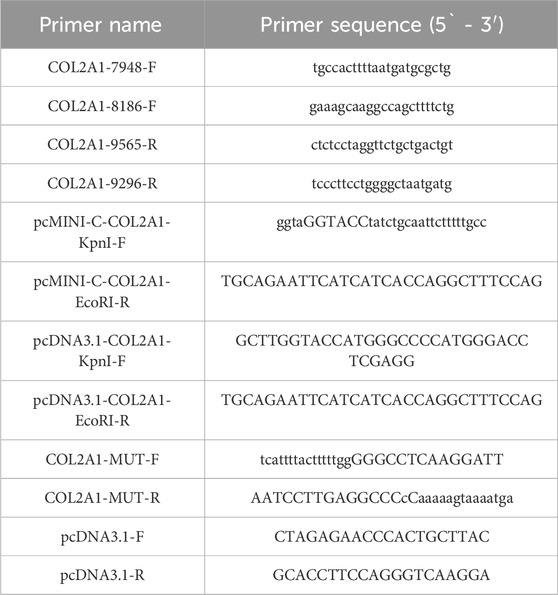
Table 1. Primer sequences used to analyze the variant of the COL2A1 gene and vector pcDNA3.1.
To confirm the conclusions drawn in this study, the assay was repeated using the same experimental procedure with another pcMINI-N vector containing the universal Exon A-intron A-MCS sequence. The cells were transfected, and the ExonA-Exon10-Exon11 splicing pattern was observed to determine whether it was abnormal, as previously described.
2.5 Cell culture and transfection
Human embryonic kidney (HEK293T) and HeLa cell lines (Cell Resource Center of the Chinese Academy of Medical Science, Beijing, China) were maintained at 37°C in a humidified environment using 5% CO2 in Dulbecco’s modified Eagle medium supplemented with 10% fetal bovine serum and 1% penicillin-streptomycin. COL2A1-wt and variant cDNA were inserted into the pcDNA3.1 vector according to the manufacturer’s instructions, and HEK293T and HeLa cells were transfected using Lipo3000 transfection reagent (GlpBio Technology Inc., Montclair, CA, United States of America). The transfected cells were incubated for 48 h prior to RNA extraction.
2.6 RNA extraction, polymerase chain reaction, and sequencing
Total RNA from HEK293T and HeLa cells transfected with small gene plasmids was extracted using the GenElute Mammalian Total RNA Kit (Sigma-Aldrich) according to the manufacturer’s recommendations. DNA was degraded on the columns using DNA enzyme I (Qiagen, Valencia, CA, United States of America). The extracted total RNA was reversely transcribed to cDNA, and a primer was used for the retropolymerase chain reaction of pcDNA3.1-F/pcDNA3.1-R (Table 1). Next, the cDNA products were detected using 1% agarose gel electrophoresis and further validated via Sanger sequencing. The lengths of the wild-type cDNA products were 396 bp in the pcDNA3.1-COL2A1 vector and 537 bp in the pcMINI-N-COL2A1 vector.
3 Results
3.1 Clinical data
The male proband, born to a non-consanguineous 43-year-old mother and a 56-year-old father, was the second child in the family. The mother’s pregnancy was complicated because of connective tissue disease and gestational diabetes mellitus. However, there was no reported family history of genetic diseases. Pregnancy care revealed no exposure to teratogenic or infectious agents. He was delivered at the 38th week of gestation via spontaneous vaginal delivery, weighing 2,800 g (10th–25th percentile) and measuring 50 cm in length (50th–75th percentile). His Apgar scores were 9 and 10 at 1 and 5 min, respectively. Physical examination at birth revealed a small jaw, back tongue root, cleft palate, and intact hard palatal mucosa. Shortly after birth, the proband was hospitalized because of dyspnea and feeding difficulties. CT findings and 3D reconstructions revealed mandibular retraction, glossoptosis, airway stenosis, and incomplete cleft palate (Figures 1A–D). Additionally, abdominal ultrasonography revealed right renal pelvic separation, and an echocardiogram indicated a patent foramen ovale. Additionally, the result of universal newborn hearing screening was positive for hearing loss. Blood biochemical test results for total protein, albumin, alanine aminotransferase, leukocytes, hemoglobin, red blood cells, and platelet counts were all within normal limits. His cleft palate was surgically repaired 8 days after birth. Up to the age of 18 months, he exhibited normal motor and language development without experiencing symptoms of retinal detachment.
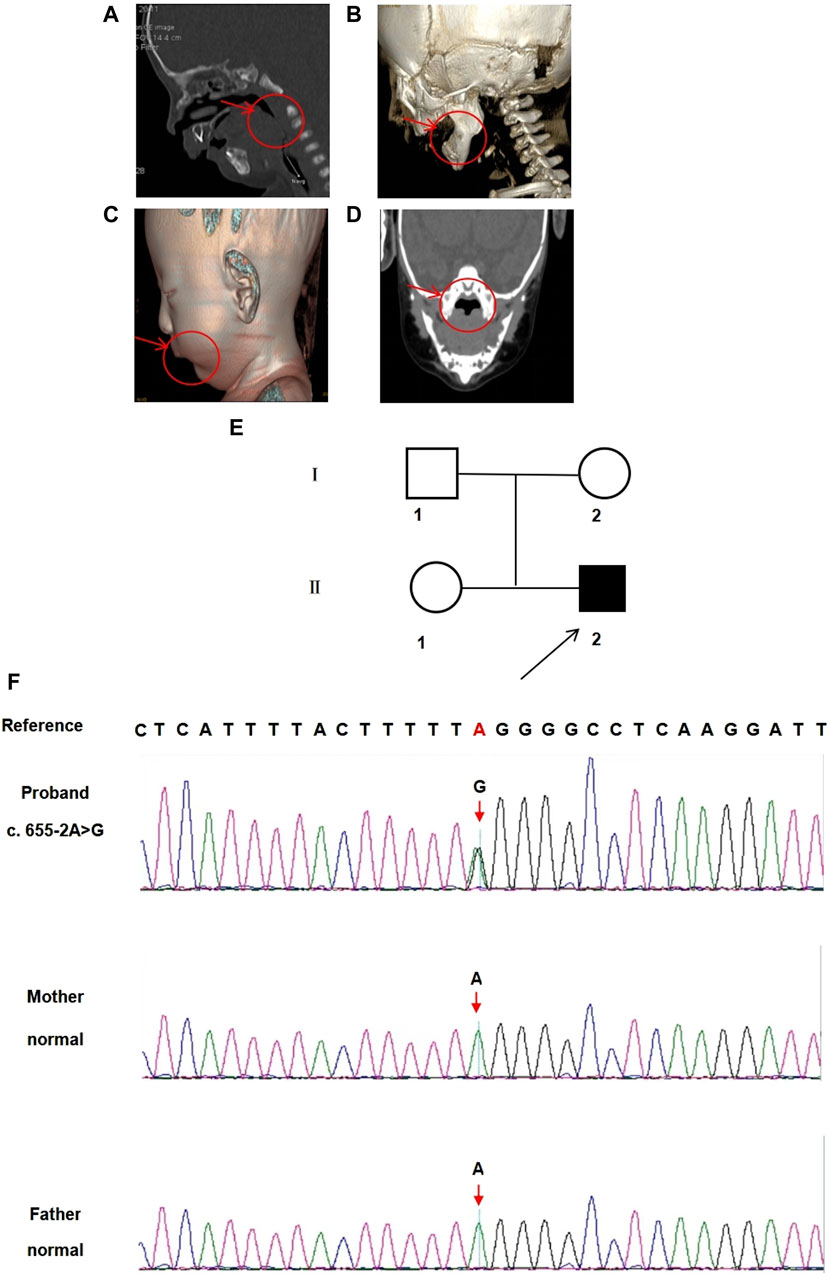
Figure 1. CT images and 3D reconstructions were conducted for this infant. (A) The arrow indicates back tongue root in the CT sagittal axial view. (B) The arrow indicates small jaw in the 3D reconstructions sagittal axial view. (C) The arrow indicates small jaw in the 3D reconstructed sagittal view. (D) The arrow indicates a cleft palate in the CT coronal view. (E) Pedigree of the family. □ represents the normal male; ○ represents the normal female; ■ represents the affected male. Arrow indicates the proband. (F) The sequencing result of the family. Sanger sequencing showing COL2A1 (C).655-2A>G variant. The arrow symbol indicates the mutation site.
3.2 Variant detection
A novel heterozygous point variant in the intron 9 (c.655-2A>G, NM_001844.4) splice donor site of COL2A1 (NC_000012.12) was identified in the proband (II-2, Figure 1E) using trio exome sequencing and confirmed by PCR-based Sanger sequencing (Figure 1F). DNA analyses of the parents (I-1 and I-2) and sister (II-1) were normal, indicating that the variant was de novo in the proband.
3.3 In silico assay
In the gnomAD database, the COL2A1 variant (c.655-2A>G) is not included and has not been reported previously, indicating that this is a novel variant (Table 2). Functional prediction of the COL2A1 variant was performed using the RDDC platform. The analysis indicated that this variant potentially had pathogenic effects, as suggested by the three different predicted patterns of RNA splicing (Figures 2A–C). For c.655-2A>G, the splice AI algorithm returned a high value (Δ Score = 1.00). This value, which was above the high precision threshold (Δ Score ≥0.8), was used to detect higher sensitivity of splice change variants (Table 3). These results were validated using varSEAK (Figure 2D). Mutations in the 3′acceptor splice site could lead to exon skipping (Table 4). Our analyses suggested that the detected COL2A1 variant (c.655-2A>G) is a novel variant that affects mRNA splicing.
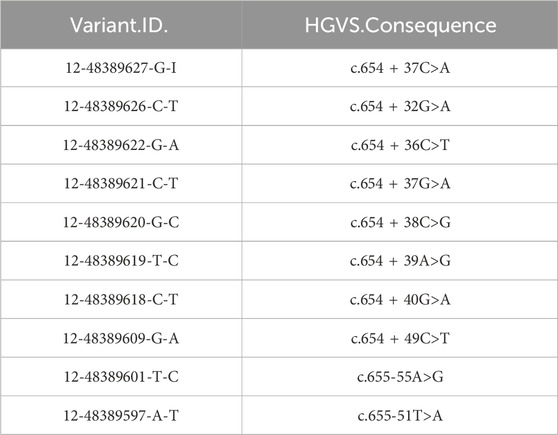
Table 2. Intronic variations of COL2A1 previously reported in gnomAD database.
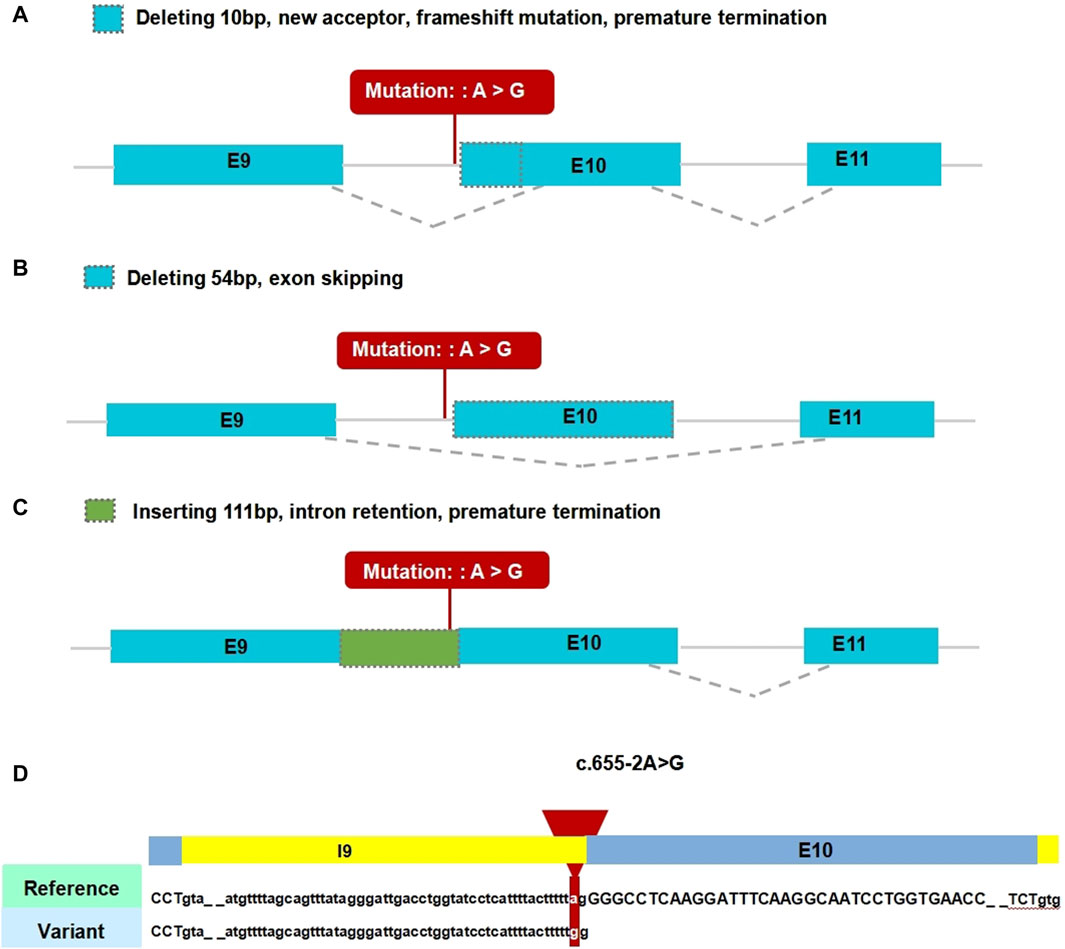
Figure 2. Results of in silico assay. A-C. Three patterns of RNA splicing prediction. (A) Pattern one: Deleting 10 bp, new acceptor, frameshift mutation, premature termination. (B) Pattern two: Deleting 54 bp, exon skipping. (C) Inserting 111 bp, intron retention, premature termination. The minigene results showed that our variant of interest was consistent with pattern two. (D) Result of the prediction in varSEAK online consists of these in silico assays by Human Splicing Finder (HSF). E9: exon 9; E10: exon 10; E11: exon 11.
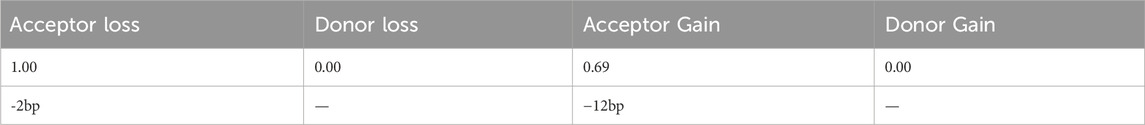
Table 3. Splicing mutation evaluation by SpliceAI.

Table 4. Assessing splicing impact with varSEAK.
3.4 Minigene assay
In vitro transcriptional assays were performed to analyze the effect of intron donor locus variation on COL2A1 mRNA splicing. The pcDNA3.1-COL2A1 small gene was 665 bp long and covered DNA regions, including exon 9 (45 bp), intron 9 (111 bp), exon 10 (54 bp), intron 10 (401 bp), and exon 11 (54 bp, Figures 3A, B). A full-length reverse transcription PCR product of 396 bp, consisting of a partial plasmid sequence of 243 bp and a target gene of 153 bp, was detected in HEK293T and HeLa cells transfected with the pcDNA3.1-COL2A1 minigene (Figure 3C). Sanger sequencing confirmed that the 396 bp band corresponded to normal COL2A1 mRNA, in which exons 9–10–11 were spliced and introns 9–10 were removed between them (Figure 3D). The pcDNA3.1-COL2A1-mut minigene containing the c.655-2A>G variant was transfected into the HeLa and HEK293T cell lines. In contrast, normal COL2A1 mRNA was not detected in the mutant (MUT) swim channel, except for a smaller band (342 bp, Figure 3C). In the MUT lanes, an aberrantly spliced transcript of COL2A1 was observed as a short band of 342 bp, which included only exons nine and 11, with exon 10 being completely skipped. A Sanger DNA chromatogram of the misspelled transcript of COL2A1 is shown in Figure 3D. The results from the assay conducted in the pcMINI-N vector were consistent with previous findings, showing the skipping of exon 10. Translational analysis of the full-length sequence of the misspliced transcript identified by small gene analysis showed that this variant induced complete skipping of exon 10, including 54 bp nucleotides (c.655_708del), resulting in a deletion of 18aa within the shorter protein (p.[Gln221_Pro238del]). These results were consistent with the predictions of in silico assays, and the RNA splicing pattern was consistent with the predictions of splicing pattern II (Figure 3E).
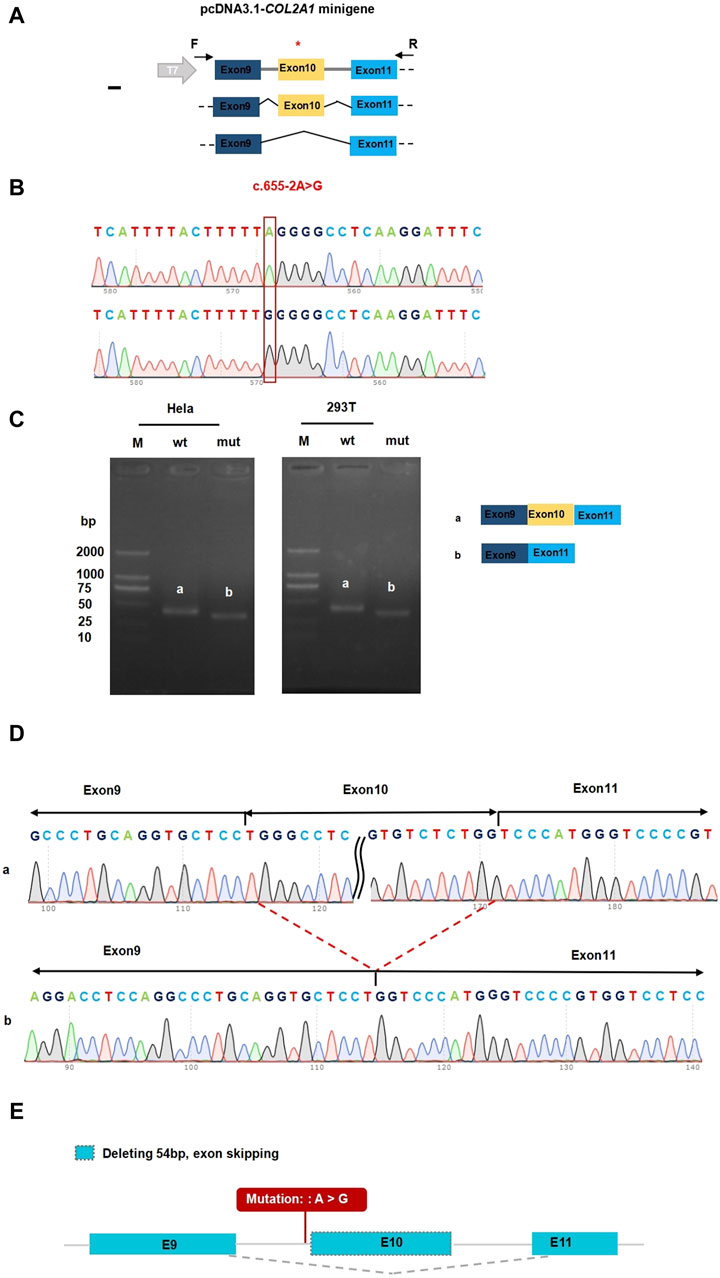
Figure 3. Functional analysis of the COL2A1 variant effect on mRNA splicing. (A) Construction strategy of pcDNA3.1 minigene vector, * representative of the variant site. (B) The sequencing of wildtype COL2A1gene vector and (C).655-2A>G COL2A1 variant gene vector. (C) Gel electrophoresis of reverse transcription polymerase chain reaction products displayed a single band a (estimated 396 bp) from the wild type (wt) and a smaller band b (estimated 342 bp) in the mutant type (mut). (D) Illustration of the sequencing of band a (wild type in HEK293T and Hela cells) and band b (variant c.655-2A>G in HEK293T and Hela cells) products lead to a shorter transcript with deletion of exon 10 including 54 bp. (E) Accurate result of the prediction in silico assay: Deleting 54 bp, exon skipping.
3.5 Three-dimensional structure of the protein
The 3D structure prediction of the variant protein was analyzed using the PyMOL software. Schematic diagrams of the primary and variant models of the COL2A1 protein are shown in Figures 4A–D. Compared with the wild-type sequence, the variant protein had an altered amino acid sequence, resulting in a shorter protein variant with a loss of 18 native amino acids (p.[Gln221_Pro238del]). The loss of 18 native amino acids are predicted to not affect any functional domains (Figure 4E).
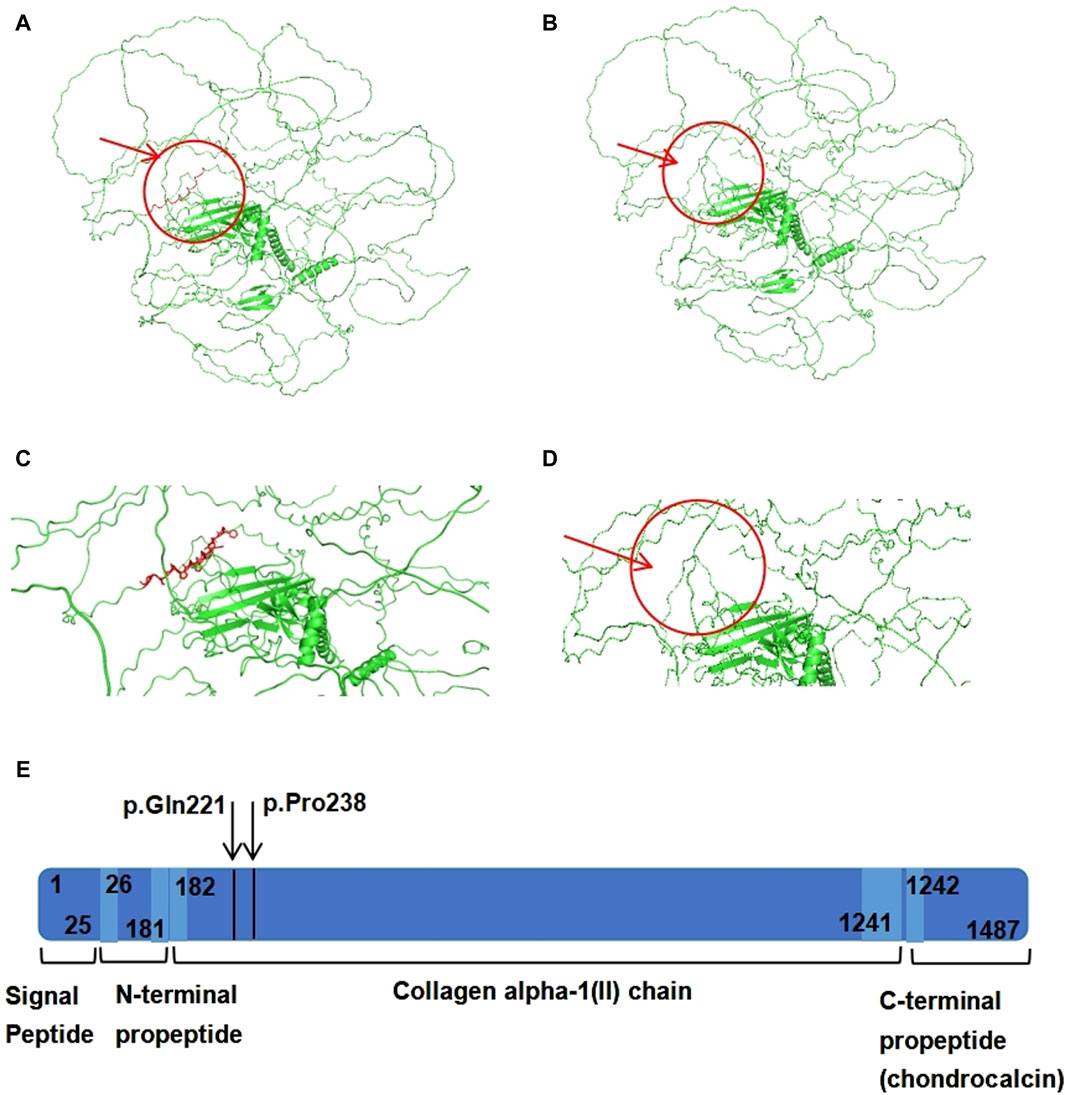
Figure 4. The domain and three-dimensional structures of the wildtype and variant COL2A1 proteins. (A) The 3D structure of wild-type COL2A1 protein. (B) The 3D structure of variant COL2A1 protein. (C) Localized amplification of the 18 amino acids (p.[Gln221_Pro238del]). (D) 3D structure of variant COL2A1 protein. The arrow indicates deletion of 18 amino acids. (E) Schematic representation of COL2A1 protein domains, regions and peptides. The arrow indicates deletion of 18 amino acids.
4 Discussion
In this study, we described the case of a male infant with cleft palate, small lower jaw, glossoptosis, retrognathia, and abnormal hearing. The infant was found to have a novel heterozygous variant, c.655-2A>G in COL2A1, and this had not been previously reported. This splice junction variant led to aberrant COL2A1 mRNA splicing, resulting in the formation of an alternative transcript that produces a shorter COL2A1 protein. The male infant was diagnosed at birth with Pierre Robin sequence (small lower jaw, glossoptosis, cleft palate and airway problems) and did not pass the newborn hearing screening test. Based on the clinical manifestations and imaging results, the proband was diagnosed with STL1.
The COL2A1 gene is located on 12q13.11 and contains 54 exons. The gene’s helix region, encoded by codons 201–1,214, consists of a core repeat of three residues, all beginning with a glycine (Gly-X-Y), where “X” typically denotes proline and “Y” represents hydroxyproline residues (Barat-Houari et al., 2016). The coding sequence of this gene is highly conserved among different species. (Dale and Topczewski, 2011). The COL2A1 gene encodes the α-1 chain of type II procollagen. Type II collagen is a homotrimer of three α-1(II) procollagen chains (Terhal et al., 2012; Nenna et al., 2019). Type II collagen is the primary component of the extracellular matrix in transparent cartilage, intervertebral disc nucleus, inner ear structures, and vitreous body. It acts as an autocrine factor for proliferation and differentiation through various downstream effectors and inhibits chondrocyte apoptosis via the suppression of BMP-SMAD1 pathway activity (Lian et al., 2019), thereby playing a crucial role in cartilage formation and growth, especially in relation to bone development within the cartilage. Various pathogenic or potentially pathogenic mutations in the COL2A1 gene have been reported, including missense and loss-of-function mutations (Hoornaert et al., 2010). Most of these lead to the introduction of a premature stop codon and nonsense-mediated decay, resulting in the production of abnormal type II collagen fibers (Stickler et al., 2001; Hoornaert et al., 2010). This study focused on a novel splice-site receptor variant of COL2A1 in intron 10, emphasizing the pathogenicity of intronic variants. The two nucleotides directly adjacent to the intron–exon junction in the splicing region are highly conserved, with the bases at the 5'(donor site) and 3'(acceptor site) ends of the intron almost always being GT and AG, respectively, according to the GT-AG rule. In the mammalian genome, the probability of a splicing site conforming to the classical GT-AG combination is 98.71% (Mercuri et al., 2005). Consequently, a splice site receptor variant in this conserved region is likely to be pathogenic. Our analysis, using various algorithms and minigene assays, demonstrated that the novel variant disrupted splicing receptor function, causing complete skipping of exon 10 and resulting in the deletion of 18 amino acids within the shorter protein (Figures 4A–D). Therefore, we believe that this novel splice-site variant is pathogenic.
Pathogenic variants in the COL2A1 gene lead to a series of diseases collectively referred to as type II collagenopathies, which are characterized by skeletal dysplasia (Stickler et al., 2001; Barat-Houari et al., 2016). Variations in the COL2A1 gene can lead to abnormal phenotypes in the skeletal, craniofacial, auditory, and visual systems. Based on severity, the clinical phenotypes of COL2A1 gene variations can be classified into lethal, severe, and mild types. Stickler syndrome is classified as mild type (Zhang et al., 2020). STL1, also known as hereditary progressive osteo-ophthalmic disease, is a type II collagenopathy phenotype associated with retinal detachment, facial abnormalities, cleft palate, and mild abnormalities in the development of spinal bone growth centers (Čopíková et al., 2020). The phenotypes of STL1 are diverse. Physical examination of the proband revealed a small jaw, posterior lingual base, and cleft palate. He was hospitalized shortly after birth because of breathing and feeding difficulties. The patient was diagnosed at birth using the Pierre Robin sequence. Similar to the proband, characteristics of midfacial hypoplasia were evident in the infant. Infants with the Pierre Robin sequence experience difficulties in breathing and feeding during early infancy exclusively (Stickler et al., 2001). Variations affecting splicing can lead to nonsense-mediated decay or produce an in-frame protein with a small deletion (Bruni et al., 2021). It is generally believed that the production of an in-frame protein with a small deletion results in severe phenotypes. A recent study showed that variants with deletions of ≤18 amino acids were associated with less severe phenotypes (Bruni et al., 2021). In this case, the detected a splice site variant that results site variant resulted in an internal deletion of 18 amino acids but with a milder phenotype. This suggests that the STL1 phenotype may be related to the size of the in-frame deletion. It has been suggested that midfacial hypoplasia in neonates should be considered during STL1. Based on the clinical presentation of the patient and the results of trio whole exome sequencing and an in vitro minigene assay, we consider the patient to be diagnosed with Stickler syndrome type I.
In conclusion, we identified a de novo heterozygous intronic variant, c.655-2A>G, in COL2A1 and verified its pathogenicity using an in vitro in COL2A1 using whole exome sequencing and an in vitro minigene assay. Abnormalities in intronic splicing sites are essential for predicting splicing variants, and in this case, they lead to aberrant COL2A1 mRNA splicing, resulting in the complete skipping of exon 10 (c.655_708del) and deletion of 18 amino acids (p.[Gln221_Pro238del]). This suggests that the phenotypes of STL1 may be associated with the size of in-frame deletion. Furthermore, our findings expand the spectrum of COL2A1 variants. Therefore, genetic testing should be performed on any individual suspected of having STL1, as it plays a crucial role in guiding patient management (ACMG Board of Directors, 2015).
Data availability statement
The original contributions presented in the study are publicly available. This data can be found here: https://www.ncbi.nlm.nih.gov/clinvar/variation/2627375/?oq=SUB13902533&m=NM_001844.5(COL2A1):c.655-2A%3EG.
Ethics statement
The studies involving humans were approved by the ethics committee of the First Affiliated Hospital of Wenzhou Medical University. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants’ legal guardians/next of kin. Written informed consent was obtained from the individual(s), and minor(s)’ legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
Author contributions
YG: Data curation, Supervision, Writing–original draft. WZ: Methodology, Writing–original draft. MZ: Supervision, Writing–original draft DC: Data curation, Supervision, Writing–review and editing. SW: Supervision, Resources, Writing–review and editing. SH: Conceptualization, Writing–original draft. YL: Data curation, Writing–original draft. YJ: Resources, Writing–original draft. TZ: Resources, Writing–original draft. Resources, Writing–review and editing. DW: Fund acquisition, Resources, Writing–review and editing.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This work was supported by Zhejiang Medical Association (2020ZYC-B23) and National Natural Science Foundation of China (82171701).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
ACMG Board of Directors (2015). Clinical utility of genetic and genomic services: a position statement of the American College of Medical Genetics and Genomics. Genet. Med. 17 (6), 505–507. doi:10.1038/gim.2015.41
Annunen, S., Körkkö, J., Czarny, M., Warman, M. L., Brunner, H. G., Kääriäinen, H., et al. (1999). Splicing mutations of 54-bp exons in the COL11A1 gene cause Marshall syndrome, but other mutations cause overlapping Marshall/Stickler phenotypes. Am. J. Hum. Genet. 65 (4), 974–983. doi:10.1086/302585
Barat-Houari, M., Sarrabay, G., Gatinois, V., Fabre, A., Dumont, B., Genevieve, D., et al. (2016). Mutation update for COL2A1 gene variants associated with type II collagenopathies. Hum. Mutat. 37 (1), 7–15. doi:10.1002/humu.22915
Boothe, M., Morris, R., and Robin, N. (2020). Stickler syndrome: a review of clinical manifestations and the genetics evaluation. J. Pers. Med. 10 (3), 105. doi:10.3390/jpm10030105
Bruni, V., Spoleti, C. B., La Barbera, A., Dattilo, V., Colao, E., Votino, C., et al. (2021). A novel splicing variant of COL2A1 in a fetus with achondrogenesis type II: interpretation of pathogenicity of in-frame deletions. Genes (Basel) 12 (9), 1395. doi:10.3390/genes12091395
Čopíková, J., Paděrová, J., Románková, V., Havlovicová, M., Balaščáková, M., Zelinová, M., et al. (2020). Expanding the phenotype spectrum associated with pathogenic variants in the COL2A1 and COL11A1 genes. Ann. Hum. Genet. 84 (5), 380–392. doi:10.1111/ahg.12386
Dale, R. M., and Topczewski, J. (2011). Identification of an evolutionarily conserved regulatory element of the zebrafish col2a1a gene. Dev. Biol. 357 (2), 518–531. doi:10.1016/j.ydbio.2011.06.020
Faber, J., Winterpacht, A., Zabel, B., Gnoinski, W., Schinzel, A., Steinmann, B., et al. (2000). Clinical variability of Stickler syndrome with a COL2A1 haploinsufficiency mutation: implications for genetic counselling. J. Med. Genet. 37 (4), 318–320. doi:10.1136/jmg.37.4.318
Hanspal, I., McNinch, A., Richards, A., and Snead, M. (2012). Clinical phenotypes associated with type II collagen mutations. J. Paediatr. Child. Health 48 (10), 949–950. doi:10.1111/j.1440-1754.2012.02570.x
Hoornaert, K. P., Vereecke, I., Dewinter, C., Rosenberg, T., Beemer, F. A., Leroy, J. G., et al. (2010). Stickler syndrome caused by COL2A1 mutations: genotype-phenotype correlation in a series of 100 patients. Eur. J. Hum. Genet. 18 (8), 872–880. doi:10.1038/ejhg.2010.23
Lian, C., Wang, X., Qiu, X., Wu, Z., Gao, B., Liu, L., et al. (2019). Collagen type II suppresses articular chondrocyte hypertrophy and osteoarthritis progression by promoting integrin β1-SMAD1 interaction. Bone Res. 7, 8. doi:10.1038/s41413-019-0046-y
Mercuri, E., Lampe, A., Allsop, J., Knight, R., Pane, M., Kinali, M., et al. (2005). Muscle MRI in Ullrich congenital muscular dystrophy and Bethlem myopathy. Neuromuscul. Disord. 15 (4), 303–310. doi:10.1016/j.nmd.2005.01.004
Nenna, R., Turchetti, A., Mastrogiorgio, G., and Midulla, F. (2019). COL2A1 gene mutations: mechanisms of spondyloepiphyseal dysplasia congenita. Appl. Clin. Genet. 12, 235–238. doi:10.2147/tacg.S197205
Nishimura, G., Haga, N., Kitoh, H., Tanaka, Y., Sonoda, T., Kitamura, M., et al. (2005). The phenotypic spectrum of COL2A1 mutations. Hum. Mutat. 26 (1), 36–43. doi:10.1002/humu.20179
Poulson, A. V., Hooymans, J. M., Richards, A. J., Bearcroft, P., Murthy, R., Baguley, D. M., et al. (2004). Clinical features of type 2 Stickler syndrome. J. Med. Genet. 41 (8), e107. doi:10.1136/jmg.2004.018382
Snead, M. P., and Yates, J. R. (1999). Clinical and molecular genetics of stickler syndrome. J. Med. Genet. 36 (5), 353–359. doi:10.1136/jmg.36.5.353
Stickler, G. B., Hughes, W., and Houchin, P. (2001). Clinical features of hereditary progressive arthro-ophthalmopathy (Stickler syndrome): a survey. Genet. Med. 3 (3), 192–196. doi:10.1097/00125817-200105000-00008
Terhal, P. A., van Dommelen, P., Le Merrer, M., Zankl, A., Simon, M. E. H., Smithson, S. F., et al. (2012). Mutation-based growth charts for SEDC and other COL2A1 related dysplasias. Am. J. Med. Genet. C Semin. Med. Genet. 160c (3), 205–216. doi:10.1002/ajmg.c.31332
Wilkin, D. J., Artz, A. S., South, S., Lachman, R. S., Rimoin, D. L., Wilcox, W. R., et al. (1999). Small deletions in the type II collagen triple helix produce kniest dysplasia. Am. J. Med. Genet. 85 (2), 105–112. doi:10.1002/(sici)1096-8628(19990716)85:2<105::aid-ajmg2>3.0.co;2-z
Yamamoto, K., Kubota, T., Takeyari, S., Kitaoka, T., Miyata, K., Nakano, Y., et al. (2020). Parental somatogonadal COL2A1 mosaicism contributes to intrafamilial recurrence in a family with type 2 collagenopathy. Am. J. Med. Genet. A 182 (3), 454–460. doi:10.1002/ajmg.a.61422
Keywords: Stickler syndrome, functional analysis, COL2A1 gene, intron heterozygote variant, type II collagenopathies
Citation: Gong Y, Zhu W, Zhu M, Chen D, Wu S, Hu S, Luo Y, Jiang Y, Zhu T and Wang D (2024) Identification and functional characteristics of a novel splicing heterozygote variant of COL2A1 associated with Stickler syndrome type I. Front. Genet. 15:1308737. doi: 10.3389/fgene.2024.1308737
Received: 07 October 2023; Accepted: 10 June 2024;
Published: 10 July 2024.
Edited by:
Claudia Gonzaga-Jauregui, Universidad Nacional Autónoma de México, MexicoReviewed by:
Anna Sowińska-Seidler, Poznan University of Medical Sciences, PolandLauren Wasson, Harvard Medical School, United States
Copyright © 2024 Gong, Zhu, Zhu, Chen, Wu, Hu, Luo, Jiang, Zhu and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dan Wang, d2Q2MDgwNDRAd211LmVkdS5jbg==
†These authors have contributed equally to this work