
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Genet. , 26 January 2024
Sec. Livestock Genomics
Volume 15 - 2024 | https://doi.org/10.3389/fgene.2024.1302580
This article is part of the Research Topic Genetics, Genomics and Climatic Adaptation in Tropical Conditions View all 5 articles
 Filippo Cendron1*
Filippo Cendron1* Anel Ledesma-Rodríguez2,3
Anel Ledesma-Rodríguez2,3 Salvatore Mastrangelo4
Salvatore Mastrangelo4 Maria Teresa Sardina4Dervel Felipe Díaz-Herrera2,5Odalys Uffo Reinosa2
Maria Teresa Sardina4Dervel Felipe Díaz-Herrera2,5Odalys Uffo Reinosa2 Martino Cassandro1,6
Martino Cassandro1,6 Mauro Penasa1
Mauro Penasa1Crossbreeding has been employed to address environmental challenges. One successful example is the Siboney de Cuba, developed in response to economic challenges in the 1960s. The aim of this study was to perform the first genomic characterization of the Siboney de Cuba breed, a successful hybrid breed resulting from the crossbreeding of Cuban Zebu and Holstein, using SNP array chip. For this purpose, 48 Siboney de Cuba cattle samples were collected and genotyped with the GGP Bovine 100k BeadChip, resulting in 83,314 SNPs after quality control. The genetic diversity was investigated using observed and expected heterozygosity, inbreeding coefficient, and minor allele frequency. Runs of homozygosity (ROH) analysis provided insights into molecular inbreeding. Additionally, the study investigated copy number variants (CNV), identifying CNV regions and their distribution. The genetic relationship and population structure of Siboney de Cuba were analyzed in comparison with worldwide cattle populations using ADMIXTURE, multidimensional scaling, and phylogenetic analysis. Six ROH islands containing a total of 50 genes were discovered, some of which were uncharacterized loci. Furthermore, 792 CNV with higher occurrence of genetic material loss were observed. The overall genome coverage for CNV regions was 2.16%. The Siboney de Cuba exhibited a good level of genetic variability with high heterozygosity and low inbreeding when compared with other cattle breeds worldwide. Also, the breed shared genetic similarity to hybrids from America and Bos indicus from Africa and highlighted a moderate level of genetic isolation with some overlaps with Bos taurus from America. The breed showed a complex genetic composition, influenced by historical factors. Overall, findings of the present study contribute to the understanding of genomic structure of Siboney de Cuba cattle breed.
The domestication of livestock has significantly impacted the social and economic dynamics of numerous human populations worldwide. Through a combination of genetic drift and natural and artificial selection, several breeds have emerged, showcasing a range of phenotypes such as coat color, body size, behavior, and production characteristics (Diamond, 2002; Gautier et al., 2010). Since the advent of the first Industrial Revolution, the genetic diversity of cattle, in Europe and Asia, has been influenced by selective breeding and adaptation to specific environmental conditions. However, there have been instances where the necessity arose to develop crossbreeds that could encounter highly challenging environmental circumstances (Felius et al., 2014; Pitt et al., 2019).
An example of a hybrid breed that successfully addressed the environmental, economic, and social requirements is the Siboney de Cuba (Figure 1). In the early 1960s, the Cuban cattle population predominantly consisted of Zebu animals (96%), primarily bred for meat production (Ledesma-Rodríguez et al., 2022). However, the economic challenges led to the progressive substitution of the prevailing Zebu population with a breed capable of achieving higher production to meet the increasing demand of milk in Cuba (Acosta et al., 2013).

FIGURE 1. Siboney de Cuba cattle breed.
Through the implementation of artificial insemination, Cuban Zebu was crossed with the most specialized breed for milk production, i.e., Holstein. This resulted in the development of the Siboney de Cuba, which comprises 5/8 Holstein and 3/8 Cuban Zebu (Uffo et al., 2013). The Siboney de Cuba is officially included in the Register of Pure Breeds of Cuba in 1984, with a total of 1,792 males and 68,171 females registered in the herd-book. An official breed standard describes the standard and mandatory identification criteria for the Siboney de Cuba through morphological characteristics to determine whether animals are eligible for registration or not.
The breed demonstrated exceptional adaptability and satisfactory productivity even under extreme climatic conditions (Hernández and Ponce, 2008). The Siboney de Cuba breeding program has been based fundamentally on the improvement of milk production through the identification of the best bulls to be used in the population, without considering other important features such as milk quality, fertility, and longevity.
Several studies have focused on the analysis of productive and reproductive performance of this breed, aiming to identify the best animals for utilization in crossbreeding and genetic improvement programs (Portales et al., 2012; Acosta et al., 2016; García-Díaz et al., 2019). However, to the best of our knowledge, no studies have attempted to assess the genetic structure of the Siboney de Cuba breed using SNP-type molecular markers to provide a comprehensive genetic profile and facilitate direct comparisons with global breeds. The advent of high-throughput SNP genotyping platforms has enabled the implementation of high-density scans using a large number of SNP markers, which can be distributed across the entire genome or concentrated in specific regions in both Bos taurus and Bos indicus. The SNPs serve as valuable tools to investigate genetic diversity, background, and population structure in livestock and thus evaluate breed biodiversity (McKay et al., 2008; Lin et al., 2010).
The aim of the present study is to explore the diversity and genetic background of the Siboney de Cuba breed, providing the first-ever analysis of its population structure through the SNP array chip. Additionally, this research aims to contextualize the Siboney de Cuba within the global cattle heritage.
A total of 48 Siboney de Cuba cattle blood samples (35 females and 13 males, aged between 24 and 30 months) were collected. The DNA was extracted from the blood samples using the phenol–chloroform standard protocol. Animals were chosen on the basis of the viability in the experimental herd of the Centro Nacional de Sanidad Agropecuaria (CENSA, Cuba).
All animals were genotyped using the GGP Bovine 100K BeadChip (B. taurus × B. indicus) containing approximately 100,000 SNPs (Illumina, San Diego, CA, United States). The genomic coordinates for each marker were obtained using the assembled cow genome (ARS-UCD 1.2). SNPs assigned to sexual chromosomes or without position were discarded. PLINK v. 1.9 software (Chang et al., 2015) was used to perform filtering and quality control on the dataset, using the following criteria: i) minor allele frequency (MAF) ≥ 0.05; ii) genotype call rate for an SNP ≥0.95; and iii) individual call rate ≥0.90. After quality check, 48 animals and 83,314 SNPs were retained.
The raw data were merged with the genotypic data of cattle breeds worldwide to investigate the genetic relationship between Siboney de Cuba and other cattle breeds. The genotypes of 212 breeds were retrieved from Mastrangelo et al. (2020) to create a worldwide dataset. All individuals retrieved from Mastrangelo et al. (2020) were genotyped using the Bovine SNP50K BeadChip (v1 and v2). After the merge, the final dataset consisted of 3,331 animals, 16,102 common SNPs (between the GGP Bovine 100K BeadChip and the worldwide dataset), and 213 populations (divided according to the geographical origin).
PLINK v1.9 software (Chang et al., 2015) was used to estimate the observed (HO) and expected heterozygosity (HE), inbreeding coefficient (FHOM), and the MAF to compare the results with those reported in the literature (e.g., Decker et al., 2014; Makina et al., 2016; Mastrangelo et al., 2020).
Runs of homozygosity (ROH) analysis was performed to obtain an estimate of molecular inbreeding using PLINK v1.9 (Chang et al., 2015). To define ROH, the following parameters were fixed: i) the minimum length was set to 1 Mb (–homozig-kb); ii) two missing SNPs and up to one possible heterozygous genotype were allowed in the ROH (–homozyg-window-missing 50 and –homozyg-window-het 1); iii) the minimum number of SNPs that constituted the ROH was set to 50 (–homozyg-snp 50); iv) the minimum SNP density per ROH was set to one SNP every 100 kb (–homozyg-density 100); and v) the maximum gap between consecutive homozygous SNPs was 1,000 kb (–homozyg-gap 1,000). The inbreeding coefficient based on ROH (FROH) for each animal, the mean number of ROH per individual, and the mean length of ROH per individual were estimated. The total percentage of SNPs clustering inside ROH was determined by counting the number of times that each target appeared in ROH and dividing this by the total number of animals (n = 48). To identify regions of high homozygosity, called ROH islands, the top 0.999% of SNPs in the locus homozygosity range was selected. Subsequently, the annotation of gene mapping within ROH islands was also conducted using the list of the cow autosome ARS-UDC 1.2 B. taurus from the UCSC Genome Browser database (https://genome.ucsc.edu/).
Identification of copy number variants (CNV) was performed using PennCNV software (Wang et al., 2007), exploiting the Log R ratio (LRR) and B allele frequency (BAF) (Kumar et al., 2021) obtained from the intensity files of genotyping. The CNV calling at the individual level was carried out through the default parameters of the hidden Markov model: standard deviation of LRR <0.30, BAF drift <0.01, waviness factor value between −0.05 and 0.05, and minimum of 3 SNPs to define CNV. The distribution of CNV per individual spanned from 0 to >100. To avoid the detection of false-positive and/or -negative CNV and outliers, two different “.hmm” (agre.hmm and hh550. hmm) files were used to run PennCNV, as described in Gorla et al. (2017). Indeed, the “.hmm” file may substantially affect the false-positive and false-negative rates. To obtain valid CNV, the common calls from all the hidden Markov models were considered (Gorla et al., 2017). This solved the critical choice about the correct .hmm file to use in order to mapping CNV to control false-positive and -negative calls.
The R package HandyCNV (Zhou et al., 2021) was used on PennCNV output files to summarize CNV and define CNV regions (CNVR). The following package commands were imputed to the analysis: i) cnv_clean () function to convert CNV results into a standard format and make basic summary [the CNV larger than 5 Mb were discarded following Pinto et al. (2011)]; ii) cnv_summarise_plot () to create the CNV distribution, frequency, and length group plot; and iii) call_cnvr () to define CNVR and their frequency (merging CNV that overlapped by at least 1 bp) (Zhou et al., 2022). In the CNVR map and definition, “gain” CNVR indicates the regions that contain more than two copies of CNV, “loss” indicates the regions that contain deleted CNV, and “mixed” indicates the regions that contain at least one duplicated and one deleted CNV. Consensus CNVR were generated with the call_cnvr () command by combining the identified CNVR and the overlapping regions in the final CNVR distribution map (Zhou et al., 2022).
The population structure was investigated by applying the model-based clustering algorithm run in ADMIXTURE for K = 2–50 (Alexander and Lange, 2011). The cross-validation procedure was used to estimate the most likely number of populations. The K-value that minimizes the cross-validation prediction error was then assumed as the most likely. The BITE R package was used to graphically represent the results through the membercoef. circos function and estimate the most likely number of clusters following the cross-validation procedure (Milanesi et al., 2017). Pair-wise genetic relationships within and between the breeds were estimated using a matrix of genome-wide identity-by-state genetic distances in PLINK v1.9 (Chang et al., 2015) and plotted using a multidimensional scaling (MDS) plot that represented the components C1 and C2. Phylogenetic relationships between the breeds were analyzed by determining the Reynolds genetic distances using the R package ape (Paradis and Schliep, 2019). Neighbor networks were constructed from the estimated genetic distances using FigTree (Huson and Bryant, 2006). Graphical representation was created using R software (R Core Team, 2020).
Genetic diversity indices were estimated using different approaches in order to identify the levels of variability of the Siboney de Cuba breed. Descriptive statistics are presented in Table 1. Expected and observed heterozygosity showed high levels of diversity (0.404 ± 0.103 and 0.409 ± 0.404, respectively), and FHOM and FROH highlighted low levels of inbreeding in the population (−0.013 ± 0.033 and 0.004 ± 0.002, respectively).

TABLE 1. Mean and standard deviation of genetic diversity indices estimated in the Siboney de Cuba cattle breed.
A total of 1,202 ROH were identified, ranging from 3 to 48 per individual. The average number of ROH per animal was 25.04, with an average length of 4.4 Mb. The top 0.999% SNPs of percentile distribution were selected and used to define genomic ROH islands. In detail, adjacent SNPs over the threshold were merged into the genomic regions corresponding to ROH islands in the Siboney de Cuba (Figure 2). The Manhattan plot showed few peaks above the minimum value, considering each significant marker with a percentage of occurrence >18%.
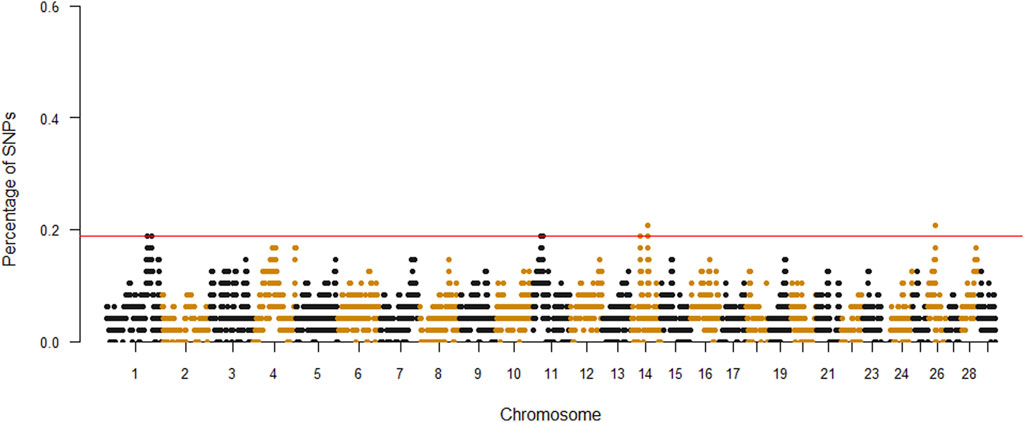
FIGURE 2. Manhattan plot of each single-nucleotide polymorphism (SNP) significance in runs of homozygosity. The red line indicates the top 0.999% of SNPs.
Table 2 provides a complete description of the identified ROH islands. The chromosome (Chr) position, start and end, and number of SNPs of the islands with the annotated genes were reported. A total of six islands were detected: one in B. taurus Chr 1, two in Chr 11 and Chr 14, and one in Chr 26. The islands ranged from 0.03 Mb (Chr 1) to 1.8 Mb (Chr 26). A total of 50 genes were identified, of which 20 were uncharacterized loci (LOC). The region on Chr 26 includes 55 SNPs, while 3 molecular markers are present in Chr 1.
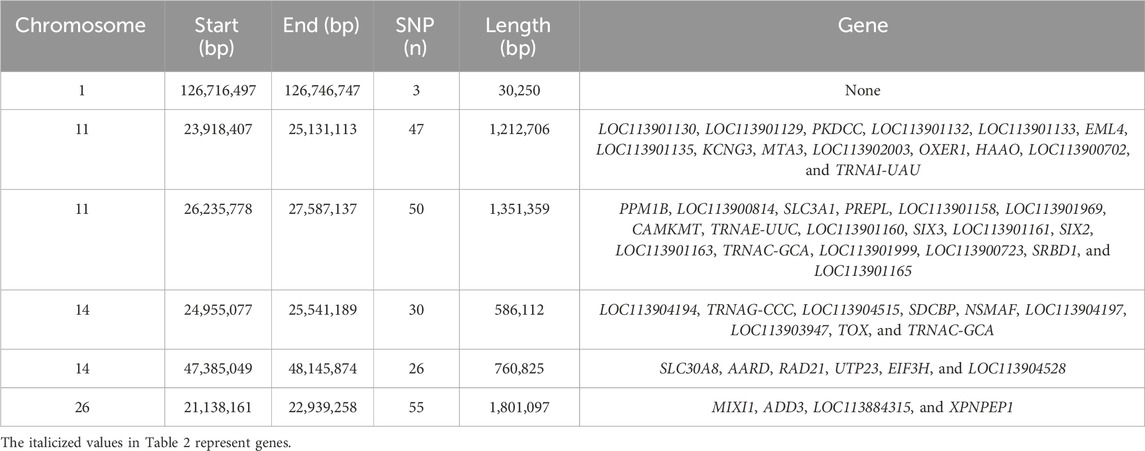
TABLE 2. Runs of homozygosity islands identified in the Siboney de Cuba cattle breed.
A total of 792 CNV were detected in the genome of the Siboney de Cuba (Table 3). The highest number of CNV was associated with the loss of genetic material (type 0 and 1) that covers also the greatest length of the genome. In Table 3, the identified CNVR are also reported, associated with entire length, mean, minimum, and maximum length, and total genome coverage based on the 29 autosomes. A total of 364 CNVR were identified and, as for CNV, the highest number of CNVR was associated with the loss of genomic regions (269), followed by gain (57) and mixed (38). The estimated overall coverage of the genome is 2.16% against the 3.41% found through the CNV. In Figure 3, a summary of CNVR distribution across the 29 chromosomes, divided for each type (loss, gain, and mixed), is reported. A detailed overview of the distribution of CNVR on chromosomes in the studied breed are presented in Supplementary Table S1, where CNVR are divided considering start and end, and the genes inside the regions are annotated.
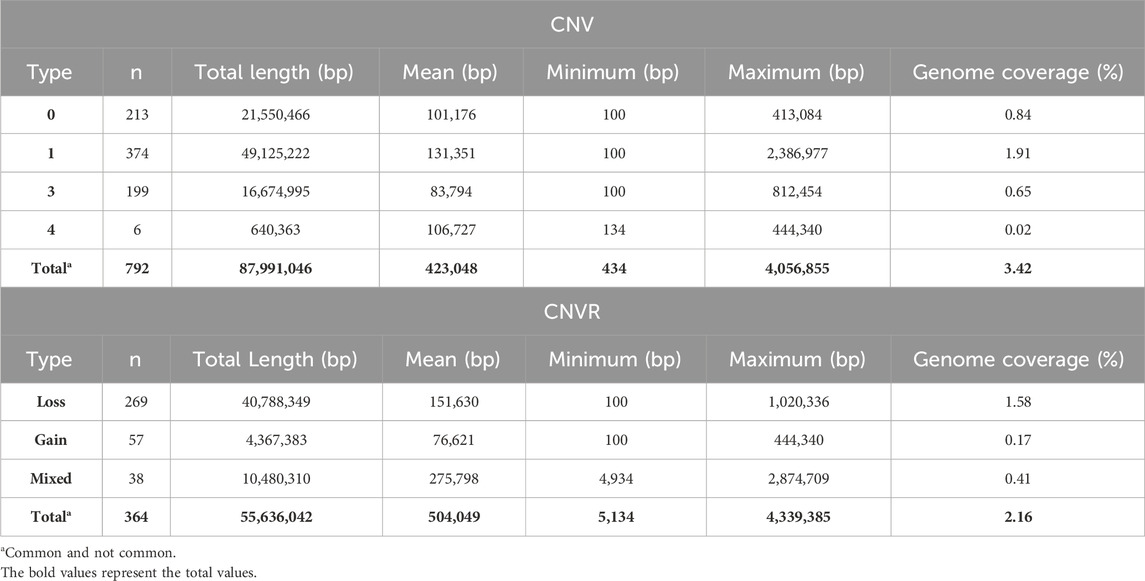
TABLE 3. Descriptive statistics of copy number variants (CNV) and CNV regions in the Siboney de Cuba cattle breed.
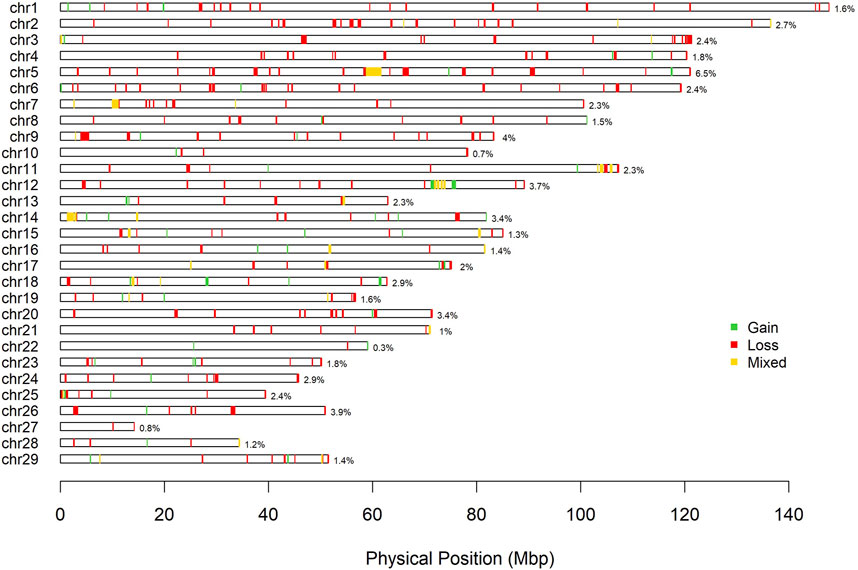
FIGURE 3. Physical distribution of copy number variant regions on chromosomes according to the state (green: gain; red: loss; yellow: mixed).
The admixture analysis, utilizing a range of 2–50 K, provided insights into the population structure in order to identify ancestral components shared among different worldwide bovine populations (Figure 4). To better understand the ancestral component and the genetic relationship, the breeds were clustered considering the geographical distribution. The results at K = 2 highlighted the subdivisions among the domesticated populations by separating B. taurus and B. indicus cattle. The Siboney de Cuba shares the genetic background with B. indicus from Asia, Africa, and America. At K = 12, the genetic background of the Siboney de Cuba shows the marks of B. indicus in general, mixed with B. taurus from Europe, in particular with Holstein. However, at K = 50, the Siboney de Cuba clustered apart from all the other breeds, with all individuals showing a mixed ancestry and genetic composition.
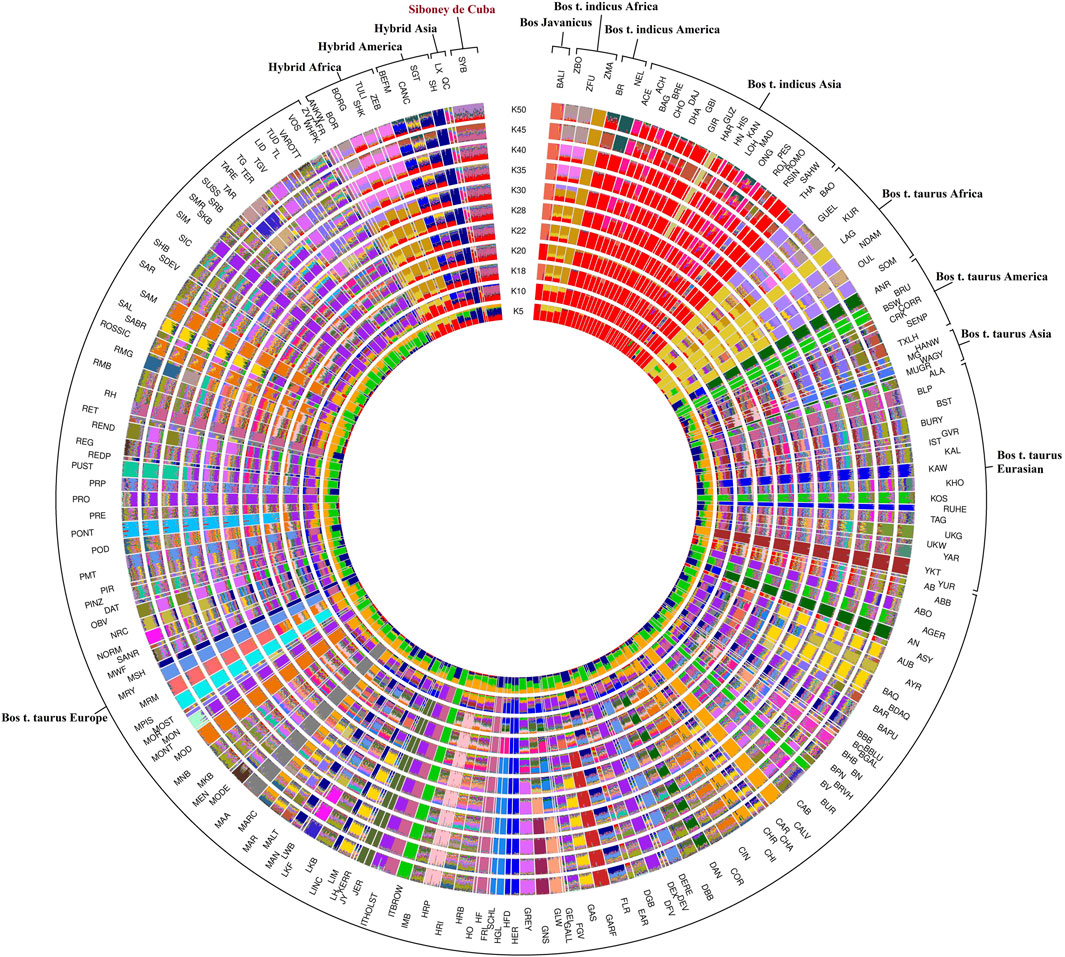
FIGURE 4. Maximum likelihood estimation calculated using the admixture algorithm. The inferred clusters (K) were reported for K = 2–50. For the full definition of breeds’ acronyms, see Supplementary Table S2 (Mastrangelo et al., 2020).
The MDS plot, graphed for components C1 and C2, showed a moderate level of genetic isolation for the Siboney de Cuba cattle breed, which generated a fairly closed cluster. A stronger overlapping with Santa Gertrudis, Beefmaster, and Canchim breeds, belonging to hybrids from America, is clear (Figure 5).
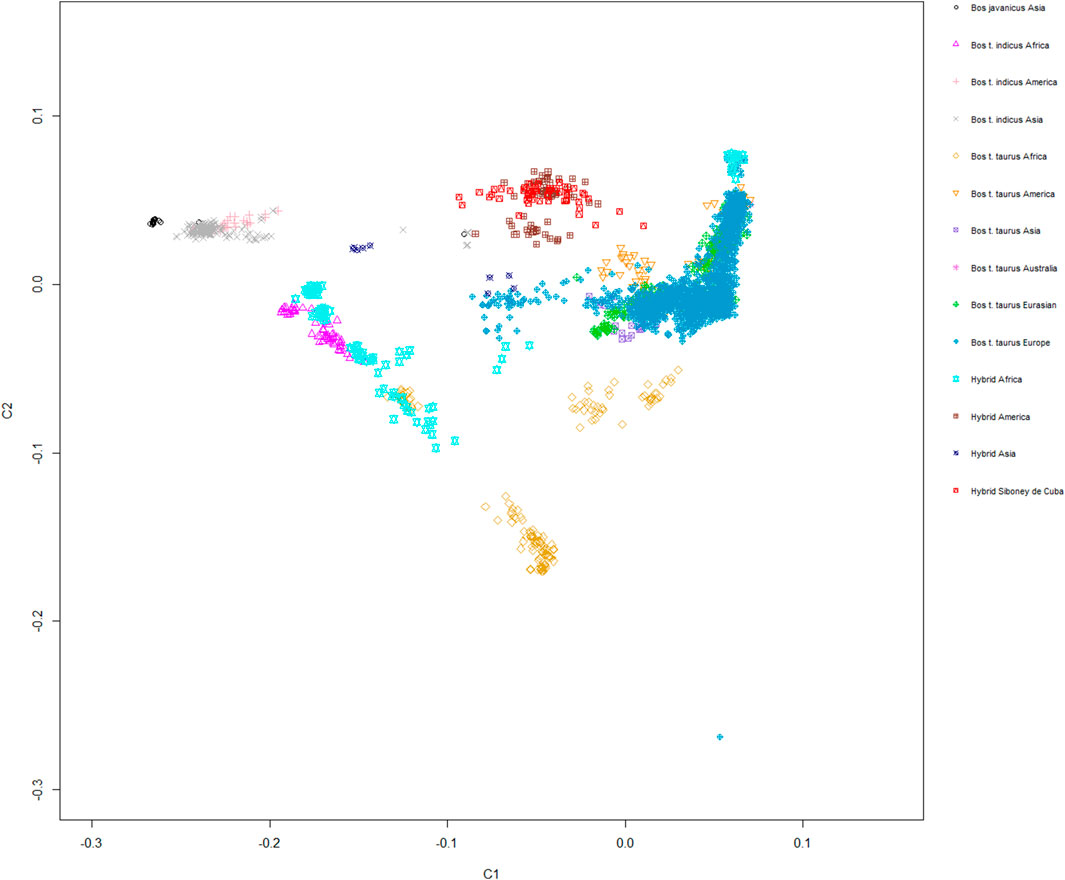
FIGURE 5. Genetic relationship between the Siboney de Cuba and worldwide cattle breeds as inferred by MDS analysis trough C1 and C2 components. Points are colored according to the geographical origin of breeds.
To provide additional insights into the relationships and patterns of divergence, a neighbor-net based on allele sharing distances was constructed (Figure 6). Consistent with the MDS plot, the neighbor-net shows several clusters between the worldwide breeds, with the Siboney de Cuba remaining close to the hybrids from America.
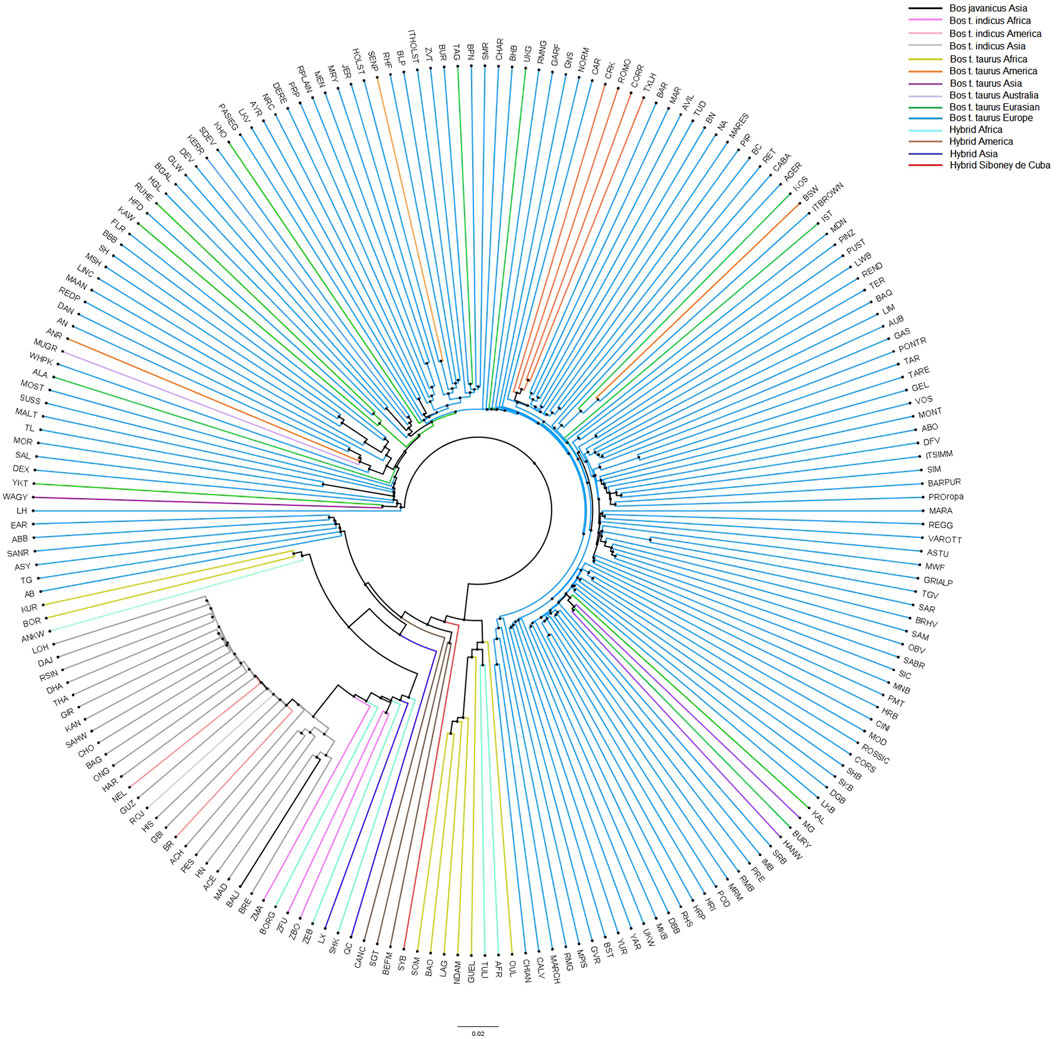
FIGURE 6. Neighbor-joining tree constructed on the allele sharing distances for the Siboney de Cuba and worldwide breeds. Colors represent the breeds and geographical distribution. For the full definition of breeds’ acronyms, see Supplementary Table S2 (Mastrangelo et al., 2020).
The genetic diversity of hybrid populations is an important resource for food security, sustainable rural development, and mitigation of the climate change (Strandén et al., 2019; McIntosh et al., 2023). The ability to create new hybrid breeds that are able to adapt to environmental variations and product demand is important to satisfy human needs. However, hybrid breeds can have different genetic deficits or variations compared to pure or indigenous breeds (McIntosh et al., 2023).
In the present study, a full genetic characterization of the Siboney de Cuba cattle breed is presented. To the best of our knowledge, this is the first study regarding the genomic structure of this breed. In the past, the Siboney de Cuba was genetically characterized by Acosta et al. (2013) through microsatellite molecular markers. The utilization of SNP chips enhances genome characterization compared to microsatellite molecular markers for several reasons. SNP chips offer a higher density of markers across the genome, providing a more comprehensive and detailed view of genetic variations, thus allowing for more precise mapping of genetic traits and a finer resolution in identifying variations associated with specific traits. Furthermore, SNPs are bi-allelic markers, offering a simpler and more straightforward interpretation of genetic data compared to the multi-allelic nature of microsatellite markers, which can be more challenging to analyze accurately (Bovine HapMap Consortium et al., 2009).
The genetic diversity indices (Table 1) underline a good variability associated with the recent history of the Siboney de Cuba (Acosta et al., 2013). However, the estimated HE and HO through SNP showed lower values compared with those reported by Acosta et al. (2013) through microsatellites. The lower values of the indices estimated from SNPs commonly arise because of inherent dissimilarities in marker characteristics. Bi-allelic SNPs generally exhibit reduced allelic diversity than multi-allelic microsatellites, which leads to less heterozygosity. Moreover, factors such as the genomic distribution of SNPs, technical biases, genotyping errors, and variations in sample sizes or studied populations all play a role in the noted discrepancies observed in heterozygosity values (Zimmerman et al., 2020).
The genetic diversity indices presented in the current study are comparable with those reported in previous studies for composite cattle (Marson et al., 2005; Decker et al., 2014) and pure breeds (Engelsma et al., 2012; Senczuk et al., 2020). Makina et al. (2014), in a study on composite breeds (B. taurus x B. indicus), reported lower genetic diversity indices compared to the Siboney de Cuba, with lower HE, which suggests that the Siboney de Cuba has maintained a higher level of genetic diversity, likely because of its more recent history.
Considering that Siboney de Cuba cattle is a hybrid breed with genetic characteristics from both B. taurus and B. indicus, it was anticipated that the average MAF would be higher when including indicine-derived SNPs. In this study, the average MAF (0.317) was higher than the MAF of composite breeds reported by Edea et al. (2015). The utilization of indicine-derived SNPs appeared to reduce the ascertainment bias, as the GGP Bovine BeadChip was intentionally developed to incorporate a greater number of SNPs of indicine origin, as reported in Edea et al. (2020) and Ferraz et al. (2020).
Monitoring and controlling inbreeding is important to limit the potential impact of deleterious alleles, inbreeding depression, and loss of genetic variation. Currently, the method based on ROH is considered one of the most powerful approaches to estimate the genomic inbreeding (Mastrangelo et al., 2017). The genomic inbreeding showed a relevant low homozygosity in accordance with Acosta et al. (2013). The maintenance of low levels of inbreeding is also desirable in composite breeds, as an advantage of crossbreeding is heterosis.
Overall, the results indicate a strong genetic variability for Siboney de Cuba cattle, which corroborates the findings of Uffo et al. (2013) on the same breed through microsatellites. These outcomes are useful when considering the possibility of response of this breed when submitted to genetic selection programs. Therefore, the current study on the genomic characterization of the Siboney de Cuba breed provides feedback about successful efforts to keep the inbreeding level within the population low.
The genomic locations of ROH are important genomic footprints of information on the demographic history of livestock species (Bosse et al., 2012).
ROH islands might be indicative of genomic regions that underwent natural and/or artificial selection. Five out of the six islands allowed us to identify different known genes potentially under selection. Several uncharacterized LOC genes were observed, reflecting the selection action on uncharacterized regulatory regions or simply the fixation of non-coding DNA by genetic drift due to a mild selection (Bosse et al., 2012). In contrast, several coding genes are located inside the ROH islands. Some of these genes are worth mentioning because they show a particular relationship with the productive traits. The PPM1B (protein tyrosine phosphatase, Mg2+/Mn2+-dependent 1B) gene on Chr 11 was negatively associated with muscle atrophy; indeed, PPM1B expression gradually decreases when muscle atrophy increases (Wei and Liang, 2012). On Chr 14, the NSMAF (neutral sphingomyelinase activation-associated factor) gene seems to be involved in cattle growth, development, feed efficiency, and carcass traits in the Red Angus breed (Smith et al., 2022). The TOX (thymocyte selection-associated high mobility group box) gene acts as a transcription factor in the hypothalamus and plays a key role in the development of puberty in Brahman cattle (Fortes et al., 2012). Causal variants of TOX were associated with reproductive traits in Nellore cattle (De Camargo et al., 2015); however, it seems to play a role in the quality of the carcass traits in Korean Hanwoo cattle (Bhuiyan et al., 2018).
The RAD21 (RAD21 cohesin complex component) gene was also identified in the Siboney de Cuba and its results were associated with carcass weight and eye muscle area development in cattle. The gene is located on Chr 14 and belongs to the sixth island in ROH analysis. Inside the last region identified, the ADD3 (adducin 3) gene on Chr 26 was identified; it is related to the development and growth traits in cattle (Huang et al., 2019). It is worth noting that the presented analysis also found an interesting gene on Chr 26, XPNPEP1, (X-prolyl aminopeptidase 1) related to milk production traits (Ibeagha-Awemu et al., 2016). In detail, Ibeagha-Awemu et al. (2016) conducted GWAS and found that this gene is related to the presence of tridecylic acid (C13:0) in milk.
Considering the growing body of evidence across diverse mammalian species, which suggests that CNV have the capacity to directly influence phenotypic variation, our objective was to conduct a comprehensive investigation of CNV prevalence in Siboney de Cuba cattle (Keel et al., 2016). This study represents the first CNV scan in the Siboney de Cuba utilizing a medium-density SNP chip, offering valuable insights into structural genomic variations that contribute to the enhancement of the genome Bovine CNV map. A major finding is that the total proportion of the genome spanned by CNV is lower (3.41%) in the Siboney de Cuba compared with other cattle breeds such as Valdostana, Holstein-Friesian, and Xin Jiang Brown (Durán Aguilar et al., 2017; Strillacci et al., 2018; Zhou et al., 2022).
A lower count of duplications (gain state) than deletions (loss state) in the Siboney de Cuba were observed. This finding corroborates previous reports based on SNP analyses (Jiang et al., 2013) and whole-genome sequencing studies (Gao et al., 2017), indicating the presence of substantial genetic variability in the bovine breed under investigation. Some of the candidate genes identified in ROH islands are also found in CNVR, such as PKDCC and EML4 in CNVR 176 and SLC3A1 in CNVR 281 (Supplementary Table S1). This supports the possibility of major selection pressure on these genes; however, their function is associated with several marginal biological processes not related with quantitative/qualitative traits. For instance, the gene PKCDD (protein kinase domain-containing, cytoplasmic) seems to be involved in peptidyl-tyrosine phosphorylation and skeletal system development (Vitorino et al., 2015). EML4 (echinoderm microtubule-associated protein-like 4) is a member of the echinoderm microtubule-associated protein-like family conserved among different species as the mouse, monkey, human, and cattle (Sabir et al., 2017). Finally, the gene SLC3A1 (solute carrier family 3 member 1) provides instructions for producing one part (subunit) of a protein made primarily in the kidneys. During the process of urine formation in the kidneys, this protein complex absorbs particular protein building blocks (amino acids) back into the blood. In particular, the amino acids such as cysteine, ornithine, arginine, and lysine are absorbed back into the blood through the mechanism in which the encoded protein is involved (Calonge et al., 1995).
Population structure analysis allowed us to distinguish the Siboney de Cuba from other cattle populations. The MDS plot (Figure 5) shows a strong overlap with the breeds of the cluster of American hybrids and proximity with B. taurus from America. This result was expected because of the historical and political situation in Cuba (Portes, 2007). The presence of an embargo has probably limited the possibility of developing a genetic reservoir composed by animals from other parts of the world (Carucci, 2000). The genetic situation of the Siboney de Cuba is confirmed by the phylogenetic tree that corroborates the results from the MDS plot. Curiously, the influence of B. taurus from Africa is clear in the phylogenetic tree. However, components C1 and C2 of the MDS plot classify separately the African populations from the Siboney de Cuba. Probably, some genetic traces of B. taurus from Africa may be present in the Siboney de Cuba due to the events during the 17th century (Guerra, 2020).
Admixture analysis corroborates the results from the MDS plot; however, it also shows that B. indicus from America seems to contribute to the genetic background of the breed under investigation as well as the Holstein from Europe. Although the results from admixture analysis confirm the cluster analysis obtained through MDS, the genetic background of the Cuban hybrid breed is confused.
As described by Mastrangelo et al. (2020), some hybrid populations from America showed a close correlation with the European breeds. The presence of European Holstein genetic ancestry components in American Holstein cattle and their integration into the genome of the Siboney de Cuba suggests the potential for migratory flow or commercial exchange between Europe and America. This phenomenon highlights the historical interactions and genetic exchanges that have influenced the genetic composition of Siboney de Cuba cattle; indeed, due to the Cuban embargo restricting the importation of Holstein cattle from America and considering that these cattle comprise genetic elements of European origin, their traces persist within the genome of Siboney de Cuba. It is worth noting that a study of migration flows could account for most of these findings, as reported by Scheu et al. (2015).
Interestingly, as described in Acosta et al. (2013), the Siboney de Cuba has a strong correlation with the other breeds, both local and hybrid, from the Cuban cattle population, highlighting a close system of breeding, confined to the island. Although this aspect was expected, the results showed that the animals belonging to the group of American hybrids strongly overlap with the Siboney de Cuba; however, admixture analysis showed that the genetic background differs between these populations. In this way, the Siboney de Cuba is far from the breeds of the European and Asiatic context, and it seems to have a closer genetic relationship with the breeds from Africa and America.
Most of the endeavors aimed at enhancing the genetic potential of the Siboney de Cuba have traditionally relied on conventional approaches. In the present paper, the genomic characterization of the Siboney de Cuba through SNP molecular markers was performed. Nonetheless, the application of genomic techniques will have a positive impact on the genetic improvement of the breed as it will contribute to reduce the generation interval and inbreeding. Undoubtedly, genomic selection is one of the crucial tools that the Cuban livestock industry needs to fully exploit the desired characteristics of the breed. By harnessing genomic data, it is possible to strategically design breeding programs aimed at selecting the individuals which can contribute to improve the traits of interest for the industry. Moreover, the insights garnered from genomic characterization allow the implementation of measures to monitor and control the inbreeding levels in the population. Therefore, the application of genomic techniques not only contributes to better characterize the Siboney de Cuba breed but also has the potential to effectively contribute to the enhancement of traits of economic relevance in the Cuban livestock industry.
The data presented in the study are deposited in the Research Data Unipd repository, Item ID 1057 (https://researchdata.cab.unipd.it/1057).
The animal study was approved by Centro Nacional de Sanidad Agropecuaria (CENSA, Cuba). The study was conducted in accordance with the local legislation and institutional requirements.
FC: conceptualization, formal analysis, investigation, and writing–original draft. AL-R: methodology and writing–review and editing. SM: conceptualization, data curation, and writing–review and editing. MS: data curation, formal analysis, and writing–review and editing. DD-H: writing–review and editing. OU: project administration and writing–review and editing. MC: funding acquisition, project administration, and writing–review and editing. MP: conceptualization, funding acquisition, project administration, and writing–review and editing.
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. The research was part of the project funded by the Sectoral Program of Animal and Plant Health, which responds to the Science and Innovation Policies of the Ministry of Science, Technology and Environment (CITMA) of Cuba.
The author Anel Ledesma-Rodríguez would like to acknowledge the financial support through the scholarship IILA-M.A.E.C.I./DGCS 2020-2021 to carry out her research and study period at the Department of Agronomy, Food, Natural resources, Animals and Environment (University of Padova, Italy).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2024.1302580/full#supplementary-material
Acosta, A., Sanz-Fernández, A., Ronda-Martínez, R., Osta-Pinzolas, R., Rodellar-Penella, C., Martin-Burriel, I., et al. (2016). Efecto de polimorfismos genéticos en la producción de leche del ganado Siboney de Cuba. Rev. Salud Anim. 3, 142–148.
Acosta, A. C., Uffo, O., Sanz, A., Ronda, R., Osta, R., Rodellar, C., et al. (2013). Genetic diversity and differentiation of five Cuban cattle breeds using 30 microsatellite loci. J. Anim. Breed. Genet. 130, 79–86. doi:10.1111/j.1439-0388.2012.00988.x
Alexander, D. H., and Lange, K. (2011). Enhancements to the ADMIXTURE algorithm for individual ancestry estimation. BMC Bioinform 12, 246. doi:10.1186/1471-2105-12-246
Bhuiyan, M. S., Lim, D., Park, M., Lee, S., Kim, Y., Gondro, C., et al. (2018). Functional partitioning of genomic variance and genome-wide association study for carcass traits in Korean Hanwoo cattle using imputed sequence level SNP data. Front. Genet. 9, 217. doi:10.3389/fgene.2018.00217
Bosse, M., Megens, H. J., Madsen, O., Paudel, Y., Frantz, L. A., Schook, L. B., et al. (2012). Regions of homozygosity in the porcine genome: consequence of demography and the recombination landscape. PLoS Genet. 8, e1003100. doi:10.1371/journal.pgen.1003100
Bovine HapMap Consortium Gibbs, R. A., Taylor, J. F., Van Tassell, C. P., Barendse, W., Eversole, K. A., et al. (2009). Genome-wide survey of SNP variation uncovers the genetic structure of cattle breeds. Science 324 (5926), 528–532. doi:10.1126/science.1167936
Calonge, M. J., Volpini, V., Bisceglia, L., Rousaud, F., de Sanctis, L., Beccia, E., et al. (1995). Genetic heterogeneity in cystinuria: the SLC3A1 gene is linked to type I but not to type III cystinuria. PNAS 92, 9667–9671. doi:10.1073/pnas.92.21.9667
Carucci, A. (2000). Environmental effects of economic sanctions: the Cuban experience. Honors Theses. Available at: https://digitalcommons.colby.edu/honorstheses/49.
Chang, C. C., Chow, C. C., Tellier, L. C., Vattikuti, S., Purcell, S. M., and Lee, J. J. (2015). Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience 4, 7. doi:10.1186/s13742-015-0047-8
De Camargo, G. M., Costa, R. B., de Albuquerque, L. G., Regitano, L. C., Baldi, F., and Tonhati, H. (2015). Polymorphisms in TOX and NCOA2 genes and their associations with reproductive traits in cattle. Reprod. Fertil. 27, 523–528. doi:10.1071/RD13360
Decker, J. E., McKay, S. D., Rolf, M. M., Kim, J., Molina Alcalá, A., Sonstegard, T. S., et al. (2014). Worldwide patterns of ancestry, divergence, and admixture in domesticated cattle. PLoS Genet. 10, e1004254. doi:10.1371/journal.pgen.1004254
Diamond, J. (2002). Evolution, consequences and future of plant and animal domestication. Nature 418, 700–707. doi:10.1038/nature01019
Durán Aguilar, M., Román Ponce, S. I., Ruiz López, F. J., González Padilla, E., Vásquez Peláez, C. G., Bagnato, A., et al. (2017). Genome-wide association study for milk somatic cell score in holstein cattle using copy number variation as markers. J. Anim. Breed. Genet. 134, 49–59. doi:10.1111/jbg.12238
Edea, Z., Bhuiyan, M. S. A., Dessie, T., Rothschild, M. F., Dadi, H., and Kim, K. S. (2015). Genome-wide genetic diversity, population structure and admixture analysis in African and Asian cattle breeds. Animal 9, 218–226. doi:10.1017/S1751731114002560
Edea, Z., Jung, K. S., Shin, S. S., Yoo, S. W., Choi, J. W., and Kim, K. S. (2020). Signatures of positive selection underlying beef production traits in Korean cattle breeds. J. Anim. Sci. Technol. 62 (3), 293. doi:10.5187/jast.2020.62.3.293
Engelsma, K. A., Veerkamp, R. F., Calus, M. P. L., Bijma, P., and Windig, J. J. (2012). Pedigree-and marker-based methods in the estimation of genetic diversity in small groups of Holstein cattle. J. Anim. Breed. Genet. 129, 195–205. doi:10.1111/j.1439-0388.2012.00987.x
Felius, M., Beerling, M. L., Buchanan, D. S., Theunissen, B., Koolmees, P. A., and Lenstra, J. A. (2014). On the history of cattle genetic resources. Diversity 6, 705–750. doi:10.3390/d6040705
Ferraz, J. B. S., Wu, X. L., Li, H., Xu, J., Ferretti, R., Simpson, B., et al. (2020). Development and evaluation of a low-density single-nucleotide polymorphism chip specific to Bos indicus cattle. Anim. Prod. Sci. 60, 1769–1776. doi:10.1071/AN19396
Fortes, M. R. S., Lehnert, S. A., Bolormaa, S., Reich, C., Fordyce, G., Corbet, N. J., et al. (2012). Finding genes for economically important traits: Brahman cattle puberty. Anim. Prod. Sci. 52, 143–150. doi:10.1071/AN11165
François, L., Wijnrocx, K., Colinet, F. G., Gengler, N., Hulsegge, B., Windig, J. J., et al. (2017). Genomics of a revived breed: case study of the Belgian campine cattle. PLoS One 12, e0175916. doi:10.1371/journal.pone.0175916
Gao, Y., Jiang, J., Yang, S., Hou, Y., Liu, G. E., Zhang, S., et al. (2017). CNV discovery for milk composition traits in dairy cattle using whole genome resequencing. BMC Genom 18, 265. doi:10.1186/s12864-017-3636-3
García-Díaz, J. R., Noval-Artiles, E., Quiñones-Ramos, R., Pérez-Bello, A., and Hernández-Barreto, M. (2019). Principales indicadores reproductivos y factores ambientales que afectan a vacas de los genotipos Siboney y Mambí de Cuba. Rev. Argent. Prod. Anim. 2, 34–43.
Gautier, M., Laloë, D., and Moazami-Goudarzi, K. (2010). Insights into the genetic history of French cattle from dense SNP data on worldwide breeds. PLoS One 6, 10. doi:10.1371/journal.pone.0013038
Gorla, E., Cozzi, M. C., Román-Ponce, S. I., Ruiz López, F. J., Vega-Murillo, V. E., Cerolini, S., et al. (2017). Genomic variability in Mexican chicken population using copy number variants. BMC Genet. 18, 61. doi:10.1186/s12863-017-0524-4
Hernández, R., and Ponce, P. (2008). Caracterización de la curva de lactancia y componentes lácteos del genotipo Siboney de Cuba en una granja ganadera de la provincia de la Habana. Rev. Cient. (Maracaibo) 18, 291–295.
Huang, Y. Z., Qian, L. N., Wang, J., Zhang, C. L., Fang, X. T., Lei, C. Z., et al. (2019). Genetic variants in ADD1 gene and their associations with growth traits in cattle. Anim. Biotechnol. 30, 7–12. doi:10.1080/10495398.2017.1398754
Huson, D. H., and Bryant, D. (2006). Application of phylogenetic networks in evolutionary studies. Mol. Biol. Evol. 23, 254–267. doi:10.1093/molbev/msj030
Ibeagha-Awemu, E. M., Peters, S. O., Akwanji, K. A., Imumorin, I. G., and Zhao, X. (2016). High density genome wide genotyping-by-sequencing and association identifies common and low frequency SNPs, and novel candidate genes influencing cow milk traits. Sci. Rep. 6, 31109. doi:10.1038/srep31109
Jiang, L., Jiang, J., Yang, J., Liu, X., Wang, J., Wang, H., et al. (2013). Genome-wide detection of copy number variations using high-density SNP genotyping platforms in Holsteins. BMC Genom 14, 131. doi:10.1186/1471-2164-14-131
Keel, B. N., Lindholm-Perry, A. K., and Snelling, W. M. (2016). Evolutionary and functional features of copy number variation in the cattle genome. Front. Genet. 7, 207. doi:10.3389/fgene.2016.00207
Kumar, H., Panigrahi, M., Saravanan, K. A., Rajawat, D., Parida, S., Bhushan, B., et al. (2021). Genome-wide detection of copy number variations in Tharparkar cattle. Anim. Biotechnol. 34, 448–455. doi:10.1080/10495398.2021.1942027
Ledesma-Rodríguez, A., Cendron, F., Díaz-Herrera, D. F., Fonseca-Rodríguez, Y., Reinosa, O. U., and Penasa, M. (2022). The Siboney de Cuba cattle: a review of studies conducted on the breed. Livest. Res. Rural. Dev. 34, 101.
Lin, B. Z., Sasazaki, S., and Mannen, H. (2010). Genetic diversity and structure in Bos taurus and Bos indicus populations analyzed by SNP markers. Anim. Sci. J. 81, 281–289. doi:10.1111/j.1740-0929.2010.00744.x
Makina, S. O., Muchadeyi, F. C., van Marle-Köster, E., MacNeil, M. D., and Maiwashe, A. (2014). Genetic diversity and population structure among six cattle breeds in South Africa using a whole genome SNP panel. Front. Genet. 5, 333. doi:10.3389/fgene.2014.00333
Makina, S. O., Whitacre, L. K., Decker, J. E., Taylor, J. F., MacNeil, M. D., Scholtz, M. M., et al. (2016). Insight into the genetic composition of South African Sanga cattle using SNP data from cattle breeds worldwide. Genet. Sel. Evol. 48, 88. doi:10.1186/s12711-016-0266-1
Marson, E. P., Ferraz, J. B. S., Meirelles, F. V., Balieiro, J. D. C., Eler, J. P., Figueiredo, L. G. G., et al. (2005). Genetic characterization of European-Zebu composite bovine using RFLP markers. Genet. Mol. Res. 4, 496–505.
Mastrangelo, S., Sardina, M. T., Tolone, M., Di Gerlando, R., Sutera, A. M., Fontanesi, L., et al. (2018). Genome-wide identification of runs of homozygosity islands and associated genes in local dairy cattle breeds. Animal 12, 2480–2488. doi:10.1017/S1751731118000629
Mastrangelo, S., Tolone, M., Ben Jemaa, S., Sottile, G., Di Gerlando, R., Cortés, O., et al. (2020). Refining the genetic structure and relationships of European cattle breeds through meta-analysis of worldwide genomic SNP data, focusing on Italian cattle. Sci. Rep. 10, 14522. doi:10.1038/s41598-020-71375-2
Mastrangelo, S., Tolone, M., Sardina, M. T., Sottile, G., Sutera, A. M., Di Gerlando, R., et al. (2017). Genome-wide scan for runs of homozygosity identifies potential candidate genes associated with local adaptation in Valle del Belice sheep. Genet. Sel. Evol. 49, 84. doi:10.1186/s12711-017-0360-z
McIntosh, M. M., Spiegal, S. A., McIntosh, S. Z., Sanchez, J. C., Estell, R. E., Steele, C. M., et al. (2023). Matching beef cattle breeds to the environment for desired outcomes in a changing climate: a systematic review. J. Arid. Environ. 211, 104905. doi:10.1016/j.jaridenv.2022.104905
McKay, S., Schnabel, R., Murdoch, B., Matukumalli, L., Aerts, J., Coppieters, W., et al. (2008). An assessment of population structure in eight breeds of cattle using a whole genome SNP panel. BMC Genet. 9, 37. doi:10.1186/1471-2156-9-37
Milanesi, M., Capomaccio, S., Vajana, E., Bomba, L., Garcia, J. F., Ajmone-Marsan, P., et al. (2017). BITE: an R package for biodiversity analyses. BioRxiv, 181610. doi:10.1101/181610
Paradis, E., and Schliep, K. (2019). Ape 5.0: an environment for modern phylogenetics and evolutionary analyses in R. Bioinform 35, 526–528. doi:10.1093/bioinformatics/bty633
Pinto, D., Darvishi, K., Shi, X., Rajan, D., Rigler, D., Fitzgerald, T., et al. (2011). Comprehensive assessment of array-based platforms and calling algorithms for detection of copy number variants. Nat. Biotechnol. 29, 512–520. doi:10.1038/nbt.1852
Pitt, D., Sevane, N., Nicolazzi, E. L., MacHugh, D. E., Park, S. D., Colli, L., et al. (2019). Domestication of cattle: two or three events? Evol. Appl. 12, 123–136. doi:10.1111/eva.12674
Portales, A., González-Peña, D., Guerra, D., Évora, J. C., and Acosta, M. (2012). Parámetros genéticos de las características de leche, incorporación y parto en ganado Siboney de Cuba. Cienc. Tecnol. Ganad. 1, 27–33.
Portes, A. (2007). “The Cuban-American Political Machine: reflections on its origins and perpetuation,” in In debating Cuban exceptionalism (New York: Palgrave Macmillan US), 123–137.
Ramljak, J., Bunevski, G., Bytyqi, H., Marković, B., Brka, M., Ivanković, A., et al. (2018). Conservation of a domestic metapopulation structured into related and partly admixed strains. Mol. Ecol. 27, 1633–1650. doi:10.1111/mec.14555
R Core Team (2020). R: a language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing. Available online: https://www.R-project.org/(accessed on July 1, 2023).
Sabir, S. R., Yeoh, S., Jackson, G., and Bayliss, R. (2017). EML4-ALK variants: biological and molecular properties, and the implications for patients. Cancers 9, 118. doi:10.3390/cancers9090118
Scheu, A., Powell, A., Bollongino, R., Vigne, J. D., Tresset, A., Çakırlar, C., et al. (2015). The genetic prehistory of domesticated cattle from their origin to the spread across Europe. BMC Genet. 16, 54. doi:10.1186/s12863-015-0203-2
Senczuk, G., Mastrangelo, S., Ciani, E., Battaglini, L., Cendron, F., Ciampolini, R., et al. (2020). The genetic heritage of Alpine local cattle breeds using genomic SNP data. Genet. Sel. Evol. 52, 40. doi:10.1186/s12711-020-00559-1
Smith, J. L., Wilson, M. L., Nilson, S. M., Rowan, T. N., Schnabel, R. D., Decker, J. E., et al. (2022). Genome-wide association and genotype by environment interactions for growth traits in US Red Angus cattle. BMC Genom 23, 517. doi:10.1186/s12864-022-08667-6
Strandén, I., Kantanen, J., Russo, I. R. M., Orozco-terWengel, P., Bruford, M. W., and Climgen, C. (2019). Genomic selection strategies for breeding adaptation and production in dairy cattle under climate change. Heredity 123, 307–317. doi:10.1038/s41437-019-0207-1
Strillacci, M. G., Gorla, E., Cozzi, M. C., Vevey, M., Genova, F., Scienski, K., et al. (2018). A copy number variant scan in the autochthonous Valdostana Red Pied cattle breed and comparison with specialized dairy populations. PLoS One 13, e0204669. doi:10.1371/journal.pone.0204669
Uffo, O., Acosta, A., Ribot, A., Ruiz, K., Ronda, R., and Martínez, S. (2013). Molecular characterization of the Siboney de Cuba cattle. Biotecnol. Apl. 30, 232–233.
Upadhyay, M., Bortoluzzi, C., Barbato, M., Ajmone-Marsan, P., Colli, L., Ginja, C., et al. (2019). Deciphering the patterns of genetic admixture and diversity in southern European cattle using genome-wide SNPs. Evol. Appl. 12, 951–963. doi:10.1111/eva.12770
Upadhyay, M. R., Chen, W., Lenstra, J. A., Goderie, C. R. J., MacHugh, D. E., Park, S. D. E., et al. (2017). Genetic origin, admixture and population history of aurochs (Bos primigenius) and primitive European cattle. Heredity 118, 169–176. doi:10.1038/hdy.2016.79
Vitorino, M., Silva, A. C., Inacio, J. M., Ramalho, J. S., Gur, M., Fainsod, A., et al. (2015). Xenopus Pkdcc1 and Pkdcc2 are two new tyrosine kinases involved in the regulation of JNK dependent Wnt/PCP signaling pathway. PloS One 10, e0135504. doi:10.1371/journal.pone.0135504
Wang, K., Li, M., Hadley, D., Liu, R., Glessner, J., Grant, S. F., et al. (2007). PennCNV: an integrated hidden Markov model designed for high-resolution copy number variation detection in whole-genome SNP genotyping data. Genome Res. 17, 1665–1674. doi:10.1101/gr.6861907
Wei, J., and Liang, B. S. (2012). PPM1B and P-IKKβ expression levels correlated inversely with rat gastrocnemius atrophy after denervation. Braz. J. Med. Biol. 45, 711–715. doi:10.1590/s0100-879x2012007500080
Yurchenko, A., Yudin, N., Aitnazarov, R., Plyusnina, A., Brukhin, V., Soloshenko, V., et al. (2018). Genome-wide genotyping uncovers genetic profiles and history of the Russian cattle breeds. Heredity 120, 125–137. doi:10.1038/s41437-017-0024-3
Zhou, J., Liu, L., Lopdell, T. J., Garrick, D. J., and Shi, Y. (2021). HandyCNV: standardized summary, annotation, comparison, and visualization of copy number variant, copy number variation region, and runs of homozygosity. Front. Genet. 12, 731355. doi:10.3389/fgene.2021.731355
Zhou, J., Liu, L., Reynolds, E., Huang, X., Garrick, D., and Shi, Y. (2022). Discovering copy number variation in dual-purpose XinJiang Brown cattle. Front. Genet. 12, 747431. doi:10.3389/fgene.2021.747431
Keywords: cattle, genetic diversity, composite breed, copy number variant, Siboney de Cuba
Citation: Cendron F, Ledesma-Rodríguez A, Mastrangelo S, Sardina MT, Díaz-Herrera DF, Uffo Reinosa O, Cassandro M and Penasa M (2024) Genome-wide analysis of the Siboney de Cuba cattle breed: genetic characterization and framing with cattle breeds worldwide. Front. Genet. 15:1302580. doi: 10.3389/fgene.2024.1302580
Received: 26 September 2023; Accepted: 08 January 2024;
Published: 26 January 2024.
Edited by:
Lingyang Xu, Chinese Academy of Agricultural Sciences, ChinaReviewed by:
Bharat Bhushan, Indian Veterinary Research Institute (IVRI), IndiaCopyright © 2024 Cendron, Ledesma-Rodríguez, Mastrangelo, Sardina, Díaz-Herrera, Uffo Reinosa, Cassandro and Penasa. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Filippo Cendron, ZmlsaXBwby5jZW5kcm9uQHVuaXBkLml0
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.