
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Genet., 19 April 2023
Sec. Neurogenomics
Volume 14 - 2023 | https://doi.org/10.3389/fgene.2023.930692
 Peiwei Zhao1†
Peiwei Zhao1† Yanqiu Hu1†Juan Hu2†Cheng Li3†
Yanqiu Hu1†Juan Hu2†Cheng Li3† Yufeng Huang1
Yufeng Huang1 Lei Zhang1Sukun Luo1Hongmin Zhu2*Jun Jiang3*
Lei Zhang1Sukun Luo1Hongmin Zhu2*Jun Jiang3* Xuelian He1*
Xuelian He1*Introduction: Congenital disorders of glycosylation (CDGs) are a genetically heterogeneous group of metabolic disorders caused by abnormal protein or lpid glycosylation. DPM2 is one subunit of a heterotrimeric complex for dolichol-phosphatemannose synthase (DPMS), a key enzyme in glycosylation, and only four patients with DPM2-CDG have been reported.
Methods: Whole-exome sequencing (WES) was performed in a Chinese family having two siblings with a mild form of DPM2-CDG with developmental delay, mild intellectual disability, hypotonia, and increased serum creatine kinase. Sanger sequencing was used to validate the variants identified in the siblings and their parents. In vitro functional study was performed.
Results: A homozygous mutation, c.197G>A (p.Gly66Glu) in exon 4 of DPM2 (NM_003863) was identified by whole exome sequencing (WES). In vitro functional analysis demonstrated that this variant increased the expression level of DPM2 protein and western blot revealed a significant decrease in ICAM1, a universal biomarker for hypoglycosylation in patients with CDG, suggesting abnormal N-linked glycosylation. We also reviewed the 4 previously reported patients carrying homozygous or compound heterozygous variants of DMP2 gene, and found that patients with variants within the region encoding the first domain had more severe clinical symptoms than those with variants within the second domain. However, the actual genotype-phenotype relationship needs more study.
Discussion: Overall, our study broadens the variant spectrum of DPM2 gene, attempts to explain the different phenotypes in patients with different DPM2 variants, and emphasizes the need of further functional studies to understand the underlying pathophysiology of the phenotypic heterogeneity.
Congenital disorders of glycosylation (CDGs) are a group of inherited metabolic disorders caused by abnormal glycosylation of proteins or lipids (Al Teneiji et al., 2017). Glycosylation is an essential and the most common cellular process for post-translational modification of proteins and lipids, functioning in cell–cell and macromolecular interactions, protein folding, and protein signaling (Moremen et al., 2012; Abu Bakar et al., 2018). Patients with glycosylation defects often present with a broad spectrum of complications, usually involving the nervous system, liver, heart, muscular, eyes, and immune system (Freeze et al., 2012; Rymen and Jaeken, 2014; Sadat et al., 2014).
Dolichol-phosphate-mannose (DPM) synthase catalyzes the formation of DPM and plays an important role in N-glycosylation, as well as in C-mannosylation, O-mannosylation, and the formation of glycosyl-phosphatidylinositol (GPI) anchors (Maeda and Kinoshita, 2008). DPM2, a hydrophobic protein with 84 amino acids, is a subunit of the heterotrimeric DPM synthase complex and is required for DPM enzyme activity, possibly by stabilizing the DPM3 protein (Maeda et al., 2000; Lefeber et al., 2009).
DPM2-CDG (MIM:615042) is caused by variants in DPM2 gene and is an extremely rare CDG type. Until now, only four cases from three families have been reported, and three of them with the variant c.68A>G (p.Tyr23Cys) died before 3 years old due to severe epilepsy, muscular dystrophy, and acute respiratory infections (Barone et al., 2012; Radenkovic et al., 2021).
In this study, we reported two siblings from a Chinese family with dysmorphic features, developmental delay, mild intellectual disability, hypotonia, strabismus, and increased serum creatine kinase. A homozygous variant c.197G>A (p.Gly66Glu) was found in DPM2 gene in these two patients. In addition, we reviewed all reported patients with DPM2-CDG and analyzed the correlation between genotypes and clinical presentations.
This study has been approved by the Institutional Review Board of Wuhan Children’s Hospital, Tongji Medical College, Huazhong University of Science and Technology (No. 2021R060-E01). Two siblings with developmental delay and mild intellectual disability were recruited in this study. Upon obtaining informed consent, peripheral venous blood was withdrawn from the siblings (at the age of 11 years and 19 years, respectively) and both parents. Genomic DNA was extracted from leukocytes of whole blood samples using the QIAamp Blood DNA mini kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions. RNA was extracted using TRIzol reagent (Invitrogen) if the peripheral venous blood was adequate. In this study, RNA was obtained from peripheral venous blood of the younger sister. Three healthy controls age-matched with the younger sister were recruited, and informed consents were obtained from all subjects' parents before peripheral venous blood samples were drawn from them.
Whole-exome sequencing (WES) and subsequent data analysis were conducted with the help of a third party medical laboratory (Chigene Lab, Beijing China). For WES, the target DNA fragments were enriched by hybridization to construct an exome library. High-throughput sequencing was performed using an Illumina NovaSeq 6000 sequencer. Single-nucleotide and indel variants were identified using the Genome Analysis Toolkit (GATK). Paired-end alignment was performed using the Burrows–Wheeler aligner (BWA). SNPs and indels were filtered and screened according to sequence depth and mutation quality. The variants were annotated using the OMIM, ClinVar, and HGMD databases. Candidate gene variants were confirmed by Sanger sequencing using self-designed primers in the patients and their parents. A protein sequence conservation analysis of mutation sites was conducted using MEGA software. The protocol used for WES was the same as described previously (Zhao et al., 2021).
HCT116 cells were grown in DMEM supplemented with 10% fetal bovine serum (Gibco). The DPM2 coding sequence was amplified from HCT116 cells with the oligonucleotides 5′-ATG GCC ACG GGG ACA GAC-3′ and 5′-TCA CTG AGC CTT CTT GGT CAC TCTC-3′ using Pfu DNA polymerase (TransGen Biotech). The wildtype DPM2 (WT-DPM2) construct was obtained by inserting the amplified fragment into the pcDNA3.1(+) expression plasmid using BamHI and EcoR I restrictions sites.
To generate DPM2-mutated protein (G66E-DPM2), site-directed mutagenesis was performed with the oligonucleotides 5′- GAC GCT CCT GTT TGT GGA ACT GTT CAT CTC -3′ and 5′- TCC ACA AAC AGG AGC AGC AGG AGG CCTG -3′ using overlap PCR. In addition, Y23C-DPM2 was generated as a control. All the positive clones were verified for the correct sequence by Sanger sequencing. HCT116 cells cultured in a 6-well plate were transfected with 1 μg plasmids using Lip3000 (Invitrogen) according to the manufacturer’s instructions. Proteins were then resolved by SDS-PAGE on 12% gels (Invitrogen) and electrotransferred onto PVDF membranes.
Total RNA was extracted from HCT116 cells using TRIzol reagent (Invitrogen, United States). The first complementary DNA was synthesized from RNA using reverse transcriptase (Takara, Dalian). Quantitative primers used were as follows: DPM2-F CTT GCC ATT CAT CGA CAG TCAG; DPM2-R CTT GGT CAC TCT CTT GGT CTTC.
Real-time PCR was performed using SYBR Green PCR kit (Takara, Dalian) and GAPDH as an internal control; the primers used for GAPDH were as follows: F: GAG CGA GAT CCC TCC AAA ATC AAG and R: GGT TCA CAC CCA TGA CGA ACATG.
Cells were lysed in 1% NP-40 lysis buffer (50 mM Tris, pH 7.4, 150 mM NaCl, 1 mM EDTA, 1% NP-40, and 0.5% sodium deoxycholate) for 30 min on ice and then centrifuged at 12,000 rpm for 10 min. The protein concentration was determined by BCA assay (Thermo Fisher Scientific), and 20 ug total protein was separated by 12% SDS-PAGE and subsequently transferred to PVDF membranes. After blocking in 2% BSA (bovine serum albumin), membranes were probed with the following Abs: anti-Flag (200 ug/mL, Santa Cruz Biotechnology, Inc.), anti-ICAM1 (500 ug/mL, Proteintech, 60299-1-Ig), and anti-GAPDH (1000 ug/mL, Proteintech, 60004-1-Ig); all Abs were diluted (1:3000) during hybridization. Bound Abs were detected using secondary Abs (Cell Signaling Technology, Inc.) and enhanced chemiluminescence (ECL, Thermo Fisher Scientific).
To examine the localization of DPM2, a co-localization analysis of DPM2 with an endoplasmic reticulum (ER) marker (PDI) was performed. Cells adhered to poly-L-lysine-coated slides were fixed for 15 min with 3% paraformaldehyde and then permeabilized for 15 min in 0.1% Triton X-100/PBS (phosphate-buffered saline). After blocking for 60 min in 3% BSA/PBS, cell spots were incubated with the anti-DPM2 and anti-PDI antibody (Proteintech, 66422-1-Ig) for 60 min. Cells were washed in PBS three times and then incubated with goat anti-rabbit secondary Ab conjugated to Alexa Fluor 488 and Alexa Fluor 555 (Cell Signaling Technology, Inc.). Cells were then counterstained with DAPI, and fluorescent images were acquired on a confocal microscope (Leica Stellaris 5) using an oil immersion objective.
The patient was admitted to the Rehabilitation Department in Wuhan Children’s Hospital due to developmental delay, mild intellectual disability, hypertonia, and strabismus at 11 years old. She was the younger daughter of healthy non-consanguineous parents without relevant family medical history. She was born at 40 weeks of gestation by caesarean section (on the request of her family) with a birth weight of 3.40 kg (50–75th) and a birth length of 50 cm (50–75th). Head circumference and Apgar scores were not available. She showed delayed motor milestones with head control at 4 months, turning over at 7 months, and unassisted walking at 3 years old. Even when she was 11 years old, she walked unsteadily, on tiptoe, with the center of gravity of the body in the sacrococcygeal region, and she had severe strephenopodia on the right. She had undergone Achilles tendon lengthening procedure. Now her walking posture improved, but she still needed the help of hands to get up or down stairs. Her upper limbs were normal and could freely move. Language development was also considerably delayed, and she spoke the first words at 4 years old. At present, she still had a poor expressive vocabulary with scant sentences and inadequate words and mild dysarthria. She also showed intellectual disability and was not able to count to 10. She had no obvious facial abnormalities other than strabismus which had not been corrected by surgery so far. In addition, she had recurrent infections at the age of preschool.
Blood biochemistry examinations showed normal parameters of liver function, including alanine transaminase (ALT), asparagine transaminase (AST), and gamma glutamyl transpeptidase (γ-GGT) but abnormal levels of creatine kinase (CK) (2097 U/L, reference: 20–250 U/L) and CK-MB (Muscle/Brain) (58 U/L, reference 0–25 U/L). There was no obvious abnormality in blood amino acid or urine organic acid analyzed by mass spectrometry. Electroencephalogram examination showed that occipital background activity was slightly slower. Electrophysiological evaluation showed slowed motor conduction velocity, longer motor latency, and decreased amplitude on the left median and ulnar nerves, but no sensory nerve action potential was detected in the median, ulnar, and sural nerves in the right lower limb, suggesting multiple peripheral nerve injury (Figures 1A, B). She did not undergo echocardiogram evaluation. The brain MRI scan showed slightly longer patchy signals on T1 and T2 and high intensity signals in the FLAIR (fluid-attenuated inversion recovery) sequence in the white matter of bilateral parietal lobes, an indication of demyelinating lesions (Figure 1C).
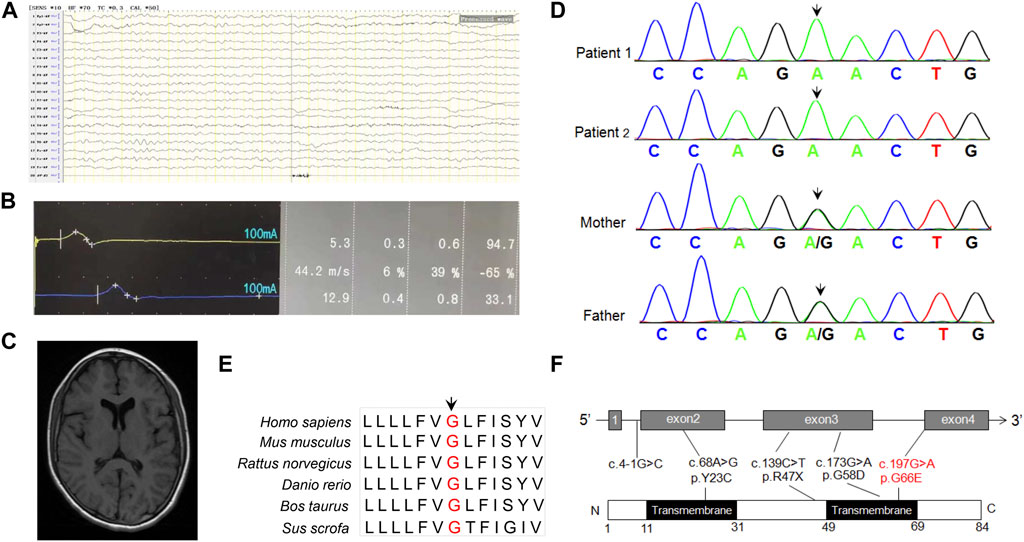
FIGURE 1. (A) EEG prompts slow background activity. (B) The motor nerve conduction amplitude of the right nervus peroneus communis was decreased, the terminal latency was prolonged, and the speed was normal, indicating that the axon and demyelination were damaged. (C) MRI shows abnormal signal foci in the white matter of the brain, and demyelination change in the younger sister. (D) Sanger sequencing of DPM2 variants in the family of this study. (E) Conservation analysis of DPM2 protein among different species. The position of the variants at amino acid 66 is indicated in red and highly conserved throughout all indicated species. (F) Scheme of the distribution of DPM2 variants. The variants in red were reported in the present study.
The elder sister had language and motor delay during childhood, and the developmental milestones were delayed. However, her conditions were better than those of her younger sister. She was able to control her head at 3 months, sat independently at 10 months, and walked unassisted at about 2 years old. Like her sister, she had the problem of recurrent infections at the age of preschool.
She had mild intellectual disability and exercise intolerance. Laboratory investigations showed increased creatine kinase (2022 U/L) and CK-MB (34 U/L) but normal liver and renal function parameters.
In order to identify the pathogenic gene for a molecular diagnosis and prognosis, trio-WES (including the younger sister and parents) was conducted. Bioinformatics analysis was performed to identify candidate variants according to a filtering strategy based on population frequency, variant classification, and variant functional damaging prediction. A homozygous variant c.197G>A (p.Gly66Glu) in exon 4 of DPM2 (NM_003863) was found in the younger sister, inherited from her heterozygous parents. This trio-WES result was confirmed by Sanger sequencing, which also identified the same homozygous variant in the elder sister (Figure 1D). The variant c.197G>A (p.Gly66Glu) was not found in any variant database (i.e., gnomAD, ClinVar, and 1000 Genomes). Bioinformatics analysis showed that this site is conserved among different species (Figure 1E), and the amino acid substitution is probably damaging, with a high PolyPhen-2 score of 1, a SIFT tolerance index of 0.006, a deleterious REVEL score of 0.837, and a damaging M-CAP score of 0.386. According to the standards and guidelines recommended by the American College of Medical Genetics and Genomics (ACMG), this variant should be classified as VUS (variant with uncertain significance). However, the grade could be upgraded to be likely pathogenic as more sources of evidence from the functional study were obtained (see the following paragraph) (PS3, in vitro or in vivo functional study supportive of a damaging effect on the gene or gene product; PM2, absent from controls or at extremely low frequency if recessive in the Exome Sequencing Project, 1000 Genomes Project, or Exome Aggregation Consortium; PP3, multiple lines of computational evidence support a deleterious effect on the gene or gene product). All germline variants (Tyr23Cys, c.4-1G>C, Arg47Ter, Gly58Asp) reported before, including the novel one identified in our study were shown in Figure 1F.
After performing quantitative real-time PCR by using RNA extracted from PBMCs of the younger sister and three age-matched normal controls, we observed that the in vivo level of DPM2 mRNA was slightly but significantly higher (p < 0.05) in the younger sister than that of controls (Figure 2A), which could be due to individual variations. We then investigated the expression levels of Gly66Glu (G66E) variant and wildtype (WT), as well as a reported pathogenic variant Tyr23Cys (Y23C) in an in vitro system, by introducing plasmid expressing Flag-tagged G66E-DPM2, WT-DPM2, and Y23C-DPM2, into HCT116 cells, respectively. As shown in Figures 2B, C, expression of G66E-DPM2 dramatically increased at both mRNA and protein levels (p < 0.01 and p < 0.001, respectively), compared to WT-DPM2, whereas Y23C-DPM2 showed no significant changes, which could be due to the effect of the Gly66Glu variant. However, the efficiency of transfection was also an important factor that might be responsible for the observed changes.
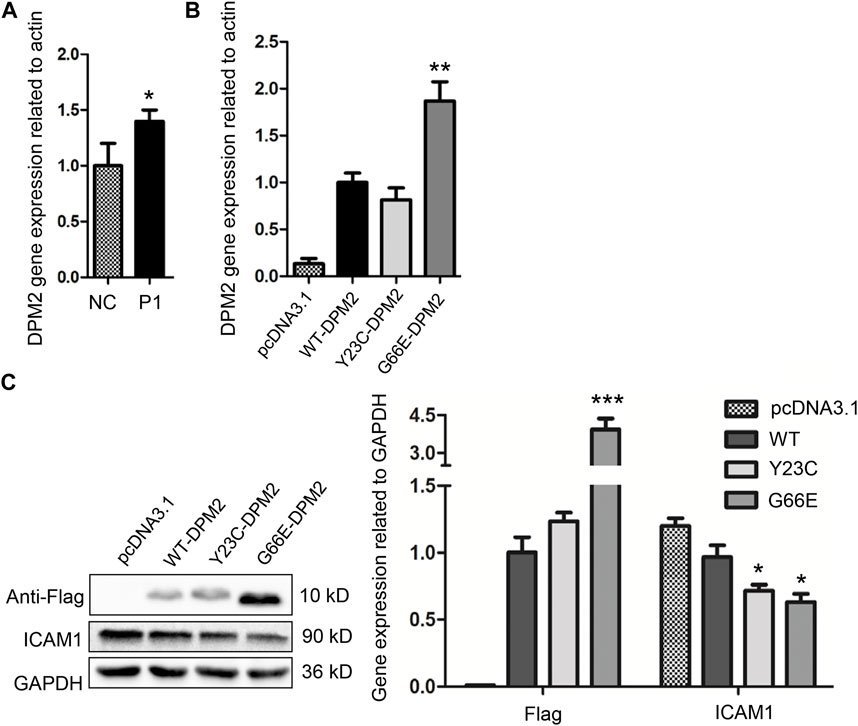
FIGURE 2. Expression level of DPM2 in PBMCs of patient 1 and normal control (A). (B) Expression level of DPM2 mRNA in HCT116 cells transfected with wildtype or mutant DPM2. (C) Protein expression level of DPM2 and ICAM1 in HCT116 cells. NC, normal control; P1, patient 1 (*, p < 0.05; **, p < 0.01; ***, p < 0.001).
As reported previously, patients with Tyr23Cys DPM2 had severe clinical symptoms and died before 3 years old (Zhao et al., 2021), but this variant did not change the levels of mRNA or protein in our in vitro expression system (Figures 2B, C). In order to further figure out the possible pathogenic mechanism, immunofluorescence assay was used to examine the localization of mutated DPM2. However, neither Tyr23Cys nor Gly66Glu altered the cellular localization of DPM2 protein (Figure 3). Although the protein level and localization of Y23C-DPM2 were unchanged, ICAM1, a universal biomarker for hypoglycosylation in patients with CDG (He et al., 2012), was decreased in HCT116 cells (Figure 2C), which indicates that DPM2 may regulate glycosylation through a mechanism other than the protein level, consistent with reported involvement of Tyr23 in binding between DPM1 and DPM2 (Maeda and Kinoshita, 2008).
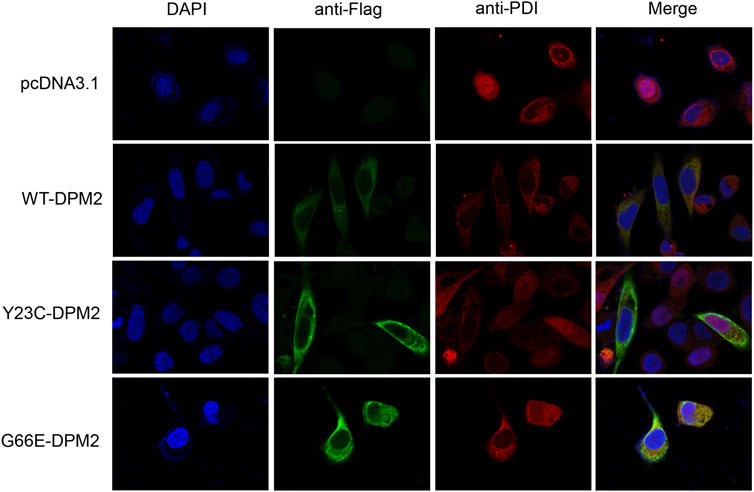
FIGURE 3. DPM2 protein expression and subcellular localization in HCT116 cells.
Literature review of DPM2-CDG was conducted by searching for all cases reported since 2012, the year in which DPM2-CDG was first reported (Zhao et al., 2021), using the keywords “DPM2 gene” and “congenital disorders of glycosylation.” The searched databases included PubMed, Medline, and ClinVar. We reviewed two articles including four cases with DPM2-CDG (He et al., 2012; Zhao et al., 2021), three of which were reported by Rita Barone et al. in 2012. The demographic characteristics and clinical features of these patients (P3–P6), as well as the two siblings (P1–P2) we reported, are summarized in Table 1.
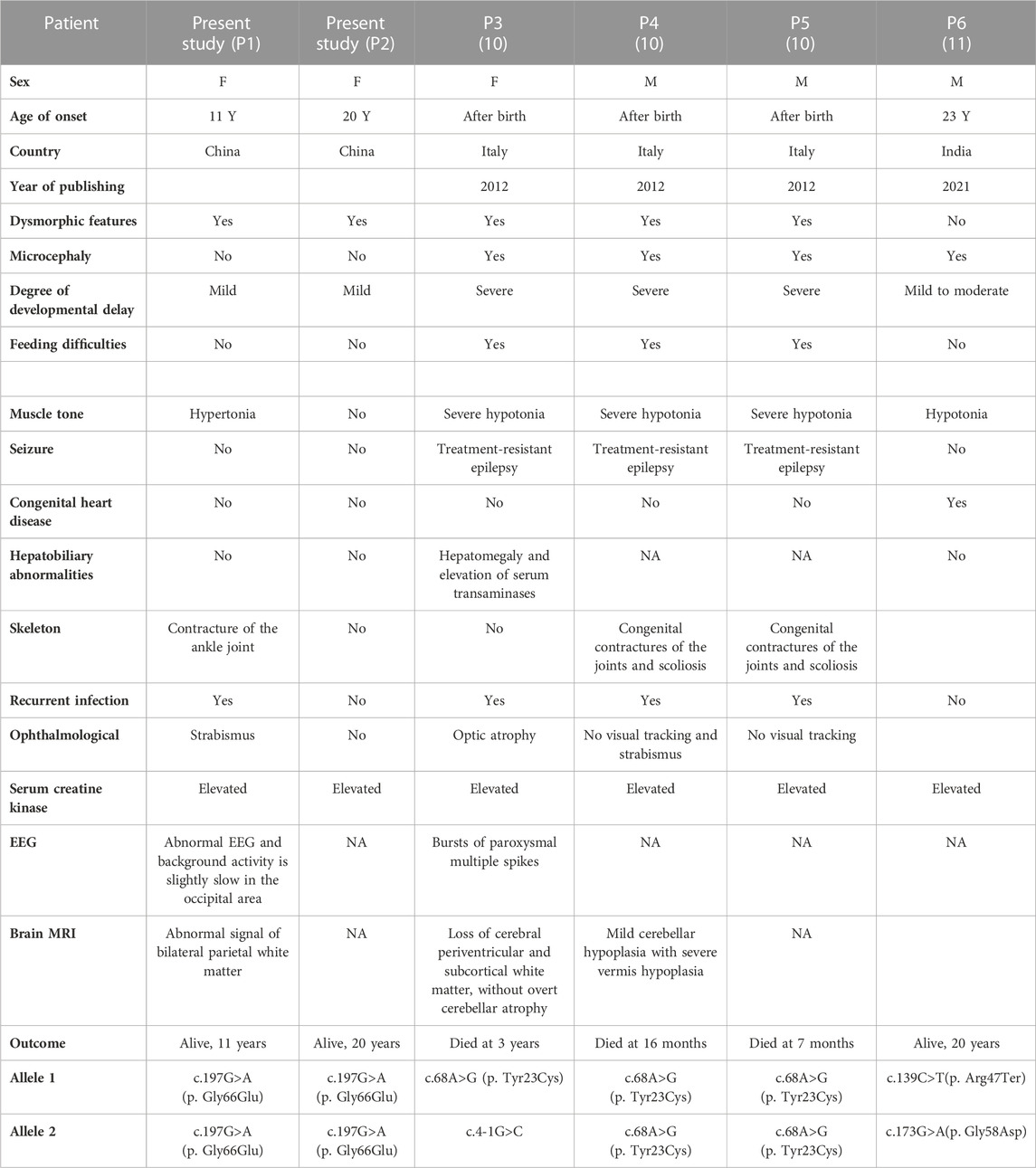
TABLE 1. Clinical characteristics of patients with DPM2-CDG.
As shown in Table 1, there were three male patients and three female patients, aged from 2 months to 23 years. The main clinical manifestations of the patients were progressive microcephaly, severe visual defect, intractable epilepsy, muscle damage, liver involvement, and decreased coagulation factors. These three patients reported by Rita Barone et al. in 2012 died at a very early age due to acute respiratory infections. The fourth patient was a 23-year-old adult male with truncal hypotonia, hypertonicity, congenital heart defects, intellectual disability, and generalized muscle wasting (He et al., 2012). In this study, we reported two cases of DPM2-CDG with much milder symptoms which broadened the phenotypic spectrum of the disease.
There are four germline variants (Tyr23Cys, c.4-1G>C, Arg47Ter, and Gly58Asp) reported previously. Three patients with Tyr23Cys and c.4-1G>C variants had severe symptoms, and one patient with compound heterozygous variants (Arg47Ter and Gly58Asp) had relatively mild symptoms. Two patients in this study carried the Gly66Glu variant, and their clinical presentations were much less severe than those of the previously described patients.
Glycosylation is an essential biological process for various protein or lipid modifications. Many human disorders of glycosylation pathways have now been identified, including defects in synthetic pathways for N-linked oligosaccharides, O-linked oligosaccharides, glycophosphatidylinositol (GPI) anchors, and dolichols (Moremen et al., 2012; Al Teneiji et al., 2017). In this study, we identified a novel variant in DPM2 gene by WES in two Chinese siblings with mild developmental delay, hypotonia, and increased creatine kinase. Bioinformatics analysis and further in vitro experiment suggested that this variant was likely pathogenic.
DPM2 is one subunit of the heterotrimeric dolichol-phosphate-mannose (DPM) synthase complex (DPM1, DPM2, and DPM3), which catalyzes the synthesis of dolichol-P-mannose and plays a critical role in N-glycosylation as well as O-mannosylation (Barone et al., 2012). Deficiency in DPM1 has been associated with CDG syndrome due to deficient protein N-glycosylation, whereas defect in DPM3 causes a muscular dystrophy associated with abnormal O-mannosylation of dystroglycan (Waetzig et al., 2010; Barone et al., 2012; Yang et al., 2013; Bursle et al., 2017). DPM2 stabilizes the synthase complex (Watanabe et al., 2000). Although our study showed different expression levels between Gly66Glu and Tyr23Cys, the effect of transfection efficiency on protein expression should be considered. Furthermore, the tyrosine amino acid at 23 in the first transmembrane domain is highly conserved during evolution and is essential for proper function of DPM2 (Maeda et al., 1998). Thus, the protein expression level could not be a critical factor in maintaining the proper function of DPM2 in the process of glycosylation. Further study is needed to understand the detailed mechanism of DPM2 in modulating glycosylation and the effects of variants in DPM2 gene on glycosylation.
DPM2-CDG is extremely rare, and only six cases from four families with five variants (c.139C>T, c.173G>A, c.197G>A, c.4-1G>C, and c.68A>G) have been identified (Table 1). DPM2 is composed of 84 amino acids and contains two putative transmembrane domains (amino acid residues 11–31 and 49–69, Figure 1F). One previous study showed that the introduction of two amino acid substitutions (Phe21 and Tyr23) to the first transmembrane domain of DPM2 abolished its ability to associate with DPM1, suggesting that the first domain of DPM2 is involved in association with DPM1 (Maeda et al., 1998). We also noticed that three patients with Tyr23Cys (homozygous or compound heterozygous) from two families had more severe phenotypes, including profound developmental delay, intractable epilepsy, severe hypotonia, and early fatal outcome (Barone et al., 2012) compared to other three patients with variants in the second transmembrane domain. However, more cases are needed to establish a genotype–phenotype correlation.
In addition, although electrophysiological evaluation did not find any significant muscle damage and muscle biopsy was not available, the younger sister had difficulties in movement, especially in going up and down stairs, the elder sister had exercise intolerance, and both had elevated creatine kinase. This evidence suggested that the siblings suffered from muscle damage. As mentioned previously, in addition to N-glycosylation, DPM synthase plays a critical role in O-mannosylation, the defect in which contributes to dystroglycanopathies, a subgroup of the congenital muscular dystrophies (van Tol et al., 2019). Therefore, it is possible that Gly66Glu affects the O-mannosylation by interfering with the function of DPM2, and more evidence is needed to support this hypothesis.
In conclusion, we identified and characterized a novel DPM2 variant, c.197G>A (Gly66Glu) in two Chinese siblings with a milder form of CDG compared to patients with the Tyr23Cys variant. However, whether the phenotypes are linked to genotypes needs more study. Our study broadens the genetic spectrum of DPM2 variants, emphasizing the importance of WES in assisting the diagnosis of rare diseases. Our study also raises questions with regard to the possible pathogenic mechanism which needs further study.
The datasets for this article are not publicly available due to concerns regarding participant/patient anonymity. Requests to access the datasets should be directed to the corresponding authors.
The studies involving human participants were reviewed and approved by the Institutional Review Board of Wuhan Children’s Hospital, Tongji Medical College, Huazhong University of Science and Technology. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin. Written informed consent was obtained from the individual(s) and minor(s)’ legal guardian/next of kin for the publication of any potentially identifiable images or data included in this article.
Study conceptualization: XH and HZ. Study design: PZ, YaH, and XH. Literature research: PZ, YaH, and YH. Clinical information collection: JH, CL, and JJ. Data acquisition: PZ, YaH, HZ, and CL. Data analysis/interpretation: PZ, YaH, HZ, and LZ. Manuscript preparation: XH, PZ, and SL. Manuscript editing: XH and JJ. Manuscript final version approval: HZ, XH, and JJ.
This work was supported by the grants of the Wuhan Municipal Health Commission (Nos WX19C19, WX14A06, WX17Z12, and WX16C17), Youth Program of National Natural Science Foundation of China (No. 81700302), Natural Science Foundation of Hubei Province (2017CFB322), Construction Project of Research Division of Children’s Medical Imaging of Wuhan Children’s Hospital (2022FEYJS002), Construction Project of Wuhan Clinical Research Center for Children’s Medical Imaging (WSTB 2022-38), Wuhan Children’s Neurological Disease Clinical Medical Research Center (WK 2014-160), and Construction Project of Clinical Medical Research Center for Neurodevelopmental Disorders in Children in Hubei Province (HST 2020-19).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abu Bakar, N., Lefeber, D. J., and van Scherpenzeel, M. (2018). Clinical glycomics for the diagnosis of congenital disorders of glycosylation. J. Inherit. Metab. Dis. 41 (3), 499–513. doi:10.1007/s10545-018-0144-9
Al Teneiji, A., Bruun, T. U., Sidky, S., Cordeiro, D., Cohn, R. D., Mendoza-Londono, R., et al. (2017). Phenotypic and genotypic spectrum of congenital disorders of glycosylation type I and type II. Mol. Genet. Metab. 120 (3), 235–242. doi:10.1016/j.ymgme.2016.12.014
Barone, R., Aiello, C., Race, V., Morava, E., Foulquier, F., Riemersma, M., et al. (2012). DPM2-CDG: A muscular dystrophy-dystroglycanopathy syndrome with severe epilepsy. Ann. Neurol. 72 (4), 550–558. doi:10.1002/ana.23632
Bursle, C., Brown, D., Cardinal, J., Connor, F., Calvert, S., and Coman, D. (2017). DMP1-CDG (CDG1e) with Significant Gastrointestinal Manifestations; Phenotype and Genotype Expansion. JIMD Rep. 34, 27–32. doi:10.1007/8904_2016_7
Freeze, H. H., Eklund, E. A., Ng, B. G., and Patterson, M. C. (2012). Neurology of inherited glycosylation disorders. Lancet Neurol. 11 (5), 453–466. doi:10.1016/S1474-4422(12)70040-6
He, P., Ng, B. G., Losfeld, M. E., Zhu, W., and Freeze, H. H. (2012). Identification of intercellular cell adhesion molecule 1 (ICAM-1) as a hypoglycosylation marker in congenital disorders of glycosylation cells. J. Biol. Chem. 287 (22), 18210–18217. doi:10.1074/jbc.M112.355677
Lefeber, D. J., Schönberger, J., Morava, E., Guillard, M., Huyben, K. M., Verrijp, K., et al. (2009). Deficiency of Dol-P-Man synthase subunit DPM3 bridges the congenital disorders of glycosylation with the dystroglycanopathies. Am. J. Hum. Genet. 85 (1), 76–86. doi:10.1016/j.ajhg.2009.06.006
Maeda, Y., and Kinoshita, T. (2008). Dolichol-phosphate mannose synthase: Structure, function and regulation. Biochim. Biophys. Acta 1780 (6), 861–868. doi:10.1016/j.bbagen.2008.03.005
Maeda, Y., Tanaka, S., Hino, J., Kangawa, K., and Kinoshita, T. (2000). Human dolichol-phosphate-mannose synthase consists of three subunits, DPM1, DPM2 and DPM3. EMBO J. 19 (11), 2475–2482. doi:10.1093/emboj/19.11.2475
Maeda, Y., Tomita, S., Watanabe, R., Ohishi, K., and Kinoshita, T. (1998). DPM2 regulates biosynthesis of dolichol phosphate-mannose in mammalian cells: Correct subcellular localization and stabilization of DPM1, and binding of dolichol phosphate. EMBO J. 17, 4920. doi:10.1093/emboj/17.17.4920
Moremen, K. W., Tiemeyer, M., and Nairn, A. V. (2012). Vertebrate protein glycosylation: Diversity, synthesis and function. Nat. Rev. Mol. Cell Biol. 13 (7), 448–462. doi:10.1038/nrm3383
Radenkovic, S., Fitzpatrick-Schmidt, T., Byeon, S. K., Madugundu, A. K., Saraswat, M., Lichty, A., et al. (2021). Expanding the clinical and metabolic phenotype of DPM2 deficient congenital disorders of glycosylation. Mol. Genet. Metab. 132 (1), 27–37. doi:10.1016/j.ymgme.2020.10.007
Rymen, D., and Jaeken, J. (2014). Skin manifestations in CDG. J. Inherit. Metab. Dis. 37 (5), 699–708. doi:10.1007/s10545-014-9678-7
Sadat, M. A., Moir, S., Chun, T. W., Lusso, P., Kaplan, G., Wolfe, L., et al. (2014). Glycosylation, hypogammaglobulinemia, and resistance to viral infections. Curr. Opin. Cell Biol. 370 (17), 1615–1625. doi:10.1056/nejmoa1302846
van Tol, W., Ashikov, A., Korsch, E., Bakar, N. A., Willemsen, M. A., Thiel, C., et al. (2019). A mutation in mannose-phosphate-dolichol utilization defect 1 reveals clinical symptoms of congenital disorders of glycosylation type I and dystroglycanopathy. JIMD Rep. 50, 31. doi:10.1002/jmd2.12060
Waetzig, G. H., Chalaris, A., Rosenstiel, P., Suthaus, J., Holland, C., Karl, N., et al. (2010). N-linked glycosylation is essential for the stability but not the signaling function of the interleukin-6 signal transducer glycoprotein 130. J. Biol. Chem. 285 (3), 1781–1789. doi:10.1074/jbc.M109.075952
Watanabe, R., Murakami, Y., Marmor, M. D., Inoue, N., Maeda, Y., Hino, J., et al. (2000). Initial enzyme for glycosylphosphatidylinositol biosynthesis requires PIG-P and is regulated by DPM2. EMBO J. 19 (16), 4402–4411. doi:10.1093/emboj/19.16.4402
Yang, A. C., Ng, B. G., Moore, S. A., Rush, J., Waechter, C. J., Raymond, K. M., et al. (2013). Congenital disorder of glycosylation due to DPM1 mutations presenting with dystroglycanopathy-type congenital muscular dystrophy. Mol. Genet. Metab. 110 (3), 345–351. doi:10.1016/j.ymgme.2013.06.016
Keywords: congenital disorders of glycosylation, DPM2, whole-exome sequencing, mutation, correlation of genotype–phenotype
Citation: Zhao P, Hu Y, Hu J, Li C, Huang Y, Zhang L, Luo S, Zhu H, Jiang J and He X (2023) Identification and characterization of a new variation in DPM2 gene in two Chinese siblings with mild intellectual impairment. Front. Genet. 14:930692. doi: 10.3389/fgene.2023.930692
Received: 28 April 2022; Accepted: 04 April 2023;
Published: 19 April 2023.
Edited by:
Jie Yuan, Sun Yat-sen University, ChinaReviewed by:
Rodrigo Starosta, Washington University in St. Louis, United StatesCopyright © 2023 Zhao, Hu, Hu, Li, Huang, Zhang, Luo, Zhu, Jiang and He. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hongmin Zhu, aHVqdWFuMTAzQDE2My5jb20=; Jun Jiang, amlhbmdqdW56bUAxNjMuY29t; Xuelian He, aGV4dWVsaWFuMjAxM0Bob3RtYWlsLmNvbQ==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.