
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Genet. , 22 January 2024
Sec. Genetics of Common and Rare Diseases
Volume 14 - 2023 | https://doi.org/10.3389/fgene.2023.1322462
This article is part of the Research Topic The Etiology and Pathogenesis of Craniomaxillofacial Birth Defects View all 8 articles
 Alexandra Topa1,2*
Alexandra Topa1,2* Anna Rohlin1,2
Anna Rohlin1,2 André Fehr1,3
André Fehr1,3 Lovisa Lovmar2Göran Stenman1,3Peter Tarnow4Giovanni Maltese4Madiha Bhatti-Søfteland4Lars Kölby4
Lovisa Lovmar2Göran Stenman1,3Peter Tarnow4Giovanni Maltese4Madiha Bhatti-Søfteland4Lars Kölby4Background: This study assessed the diagnostic yield of high-throughput sequencing methods in a cohort of craniosynostosis (CS) patients not presenting causal variants identified through previous targeted analysis.
Methods: Whole-genome or whole-exome sequencing (WGS/WES) was performed in a cohort of 59 patients (from 57 families) assessed by retrospective phenotyping as having syndromic or nonsyndromic CS.
Results: A syndromic form was identified in 51% of the unrelated cases. A genetic cause was identified in 38% of syndromic cases, with novel variants detected in FGFR2 (a rare Alu insertion), TWIST1, TCF12, KIAA0586, HDAC9, FOXP1, and NSD2. Additionally, we report two patients with rare recurrent variants in KAT6A and YY1 as well as two patients with structural genomic aberrations: one with a 22q13 duplication and one with a complex rearrangement involving chromosome 2 (2p25 duplication including SOX11 and deletion of 2q22). Moreover, we identified potentially relevant variants in 87% of the remaining families with no previously detected causal variants, including novel variants in ADAMTSL4, ASH1L, ATRX, C2CD3, CHD5, ERF, H4C5, IFT122, IFT140, KDM6B, KMT2D, LTBP1, MAP3K7, NOTCH2, NSD1, SOS1, SPRY1, POLR2A, PRRX1, RECQL4, TAB2, TAOK1, TET3, TGFBR1, TCF20, and ZBTB20.
Conclusion: These results confirm WGS/WES as a powerful diagnostic tool capable of either targeted in silico or broad genomic analysis depending on phenotypic presentation (e.g., classical or unusual forms of syndromic CS).
Craniosynostosis (CS) represents the premature closure of the skull sutures and occurs in ∼1 in 1,300 live births in the Swedish population (Tarnow et al., 2022). CS occurrence in the absence of other developmental anomalies and/or psychomotor delay is classified as nonsyndromic CS (NCS) and represents the most common presentation observed in 70%–75% of all patients with CS (Lattanzi et al., 2017; Armand et al., 2019).
Another classification is based on the type of suture(s) involved (sagittal, coronal, metopic, lambdoid) and can be restricted to a single suture or a multisuture form, such as pansynostosis, where all sutures are affected. Isolated sagittal synostosis is the most frequent form of CS, occurring in 45%–58% of all NCS cases (Lajeunie et al., 1996). Multisuture CS, specifically bicoronal synostosis, is frequently observed in syndromic forms of CS (SCS) (Twigg and Wilkie, 2015).
The etiology of CS is largely unknown. The interplay between genetic and environmental factors appears to play a role, especially in isolated NCS (Timberlake and Persing, 2018). However, genetic factors have a determinant role in the etiology of SCS, and there are several known core genes (FGFR1, FGFR2, FGFR3, TWIST1) associated with classical CS syndromes, such as Crouzon, Saethre–Chotzen, Pfeiffer, Muenke, and Apert (Muenke et al., 1994; Reardon et al., 1994; Wilkie et al., 1995; Bellus et al., 1996; Howard et al., 1997). Additional genes have been identified in other less frequently observed syndromes where CS is a main symptom, including craniofrontonasal (EFNB1), Antley–Bixler (POR), Carpenter types 1 (RAB23) and 2 (MEGF8), and CS and dental anomalies (IL11RA) (Flück et al., 2004; Twigg et al., 2004; Jenkins et al., 2007; Nieminen et al., 2011; Twigg et al., 2012). The development of whole-exome and whole-genome sequencing (WES/WGS) technologies has facilitated the discovery of new genes associated with CS syndromes, such as ERF (CS type 4), TCF12 (CS type 3), and P4HB and SEC24D (Cole–Carpenter types 1 and 2), as well as genes involved in syndromes where CS is a variable feature [e.g., Weiss–Kruszka (ZNF462) and Say–Barber–Biesecker–Young–Simpson (KAT6B) syndromes] (Clayton-Smith et al., 2011; Sharma et al., 2013; Twigg et al., 2013; Garbes et al., 2015; Rauch et al., 2015; Weiss et al., 2017). Moreover, mutations in SMAD6 have been detected by WES in subsets of patients with midline NCS (Timberlake et al., 2016). Notably, up to 14% of CS cases are caused by chromosomal aberrations (Wilkie et al., 2010). Low-penetrant mutations in CS core genes (FGFR2, FGFR3, TWIST1) have been detected in NCS presentations, underlining the existence of a phenotypic continuum between NSC and SCS (Heuzé et al., 2014).
Phenotypic presentation plays an important role in the genetic testing strategy for CS patients. Targeted screening covering CS core genes (FGFR2, FGFR3, TWIST1, FGFR1, EFNB1, TCF12, ERF) has a high diagnostic yield of up to 90% in SCS patients with the most recognizable classical phenotypes. In particular, bicoronal synostosis is an indicator of SCS (Wilkie et al., 2017). However, considering the significant variable expressivity and phenotypic heterogeneity of CS syndromes, recent studies demonstrate the advantage of next-generation sequencing (NGS) using gene panels with less-frequently mutated CS core genes, such as IL11RA, POR, MSX2, and CDC45. Furthermore, WES and WGS enable targeted in silico analysis of a custom-designed CS-related gene panel and performance of a broader exome/genome analysis in cases returning negative results. This is a cost-effective strategy that increases the diagnostic yield and can be adapted to the phenotypic presentation (Miller et al., 2017; Tønne et al., 2020; Hyder et al., 2021; Bukowska-Olech et al., 2022; Tønne et al., 2022).
The aim of the present study was to use WES and WGS to search for rare genetic variants that can explain CS within a cohort of clinically well-characterized SCS and NCS patients that had previously undergone targeted mutation screening (see 2.2 Methods) with negative results.
A total of 59 patients with CS from 57 families (one family comprises the mother and her two sons with similar phenotype) was included in the study. The following inclusion criteria were applied: patients with mostly coronal or multiple suture synostosis with either syndromic or nonsyndromic presentation, and without a detected pathogenic or likely pathogenic variant at previous targeted testing. Thirty-seven patients up to 2016 were retrieved from the Gothenburg Craniofacial Registry at the Sahlgrenska University Hospital, and the remaining 22 patients were retrieved from the clinical laboratory records for patients with negative outcomes following mutation screening between 2016 and 2020. Patient phenotypes were assessed retroactively by corroborating the medical records with registered photos and three-dimensional computed tomography reconstructions. A patient was considered to have a syndromic form of CS if one or several of the following features were present: 1) craniofacial changes involving the eyes (e.g., proptosis, hypertelorism), maxilla (e.g., midface retrusion with relative prognathism, micrognathia), nasal pyramid (e.g., short, small, beaked), and ears (e.g., low-set, dysplastic); 2) neurodevelopmental abnormalities (e.g., motor and/or speech delay, intellectual disability, seizures); and 3) other associated malformations (e.g., cleft palate, heart defect).
Thirty-seven patients were initially analyzed using a custom-designed NGS 63 CS-gene panel (Topa et al., 2020; Topa et al., 2022). Twenty-one patients were analyzed in a clinical setting using either an in silico CS-gene panel on a WES (6 patients)/WGS platform (15 patients) or a targeted NGS 12 CS-gene panel (1 patient) using the HaloPlex system (Agilent Technologies, Santa Clara, CA, United states) (Supplementary Methods S1). Complementary multiplex ligation-dependent probe amplification covering 10 genes (TWIST1, FGFR1, FGFR2, FGFR3, MSX2, ALX1, ALX3, ALX4, EFNB1, RUNX2) was performed for all 21 clinically screened cases.
Genomic DNA was extracted from blood samples. Parental samples were not systematically collected but obtained a posteriori in selected cases with phenotypically relevant variants for subsequent segregation analysis. The majority of patients were analyzed as singletons, except for three cases, where parental and sibling samples were available for Trio, Quatro, and Duo analysis, respectively. Overall, data from 52 WGS and five WES analyses of index family cases were processed for this study (Supplementary Methods S1).
All 57 index family cases were initially screened using an in silico panel that included 133 genes using Alissa Interpret (Agilent Technologies) for the variant-filtration pipeline (Supplementary Methods S1). The genes were selected according to their association with a CS phenotype and using PubMed and OMIM databases.
WGS and WES analyses were performed by filtration of variants from vc files and those containing copy number variations (CNVs) using the following software programs: Moon (Invitae Corp., San Francisco, CA, United States) comprising human phenotype ontology (HPO)-term-driven analysis; and Alissa Interpret (Agilent Technologies) using a classic variant-filtration pipeline, including primarily population frequency, inheritance pattern, molecular aspects, and CS-related HPO terms. Assessment of CNVs was performed on WGS data using the described variant-interpretation software and the Integrative Genomics Viewer (https://software.broadinstitute.org/software/igv/). Clinically relevant sequence variants and CNVs not fulfilling technical quality thresholds were confirmed by Sanger sequencing and multiplex ligation-dependent probe amplification or SNP array. mRNA/cDNA analysis was performed for novel variants with predicted splice effects.
Variant interpretation and classification were performed manually based on criteria, such as minimal allele/genotype frequency in gnomAD (https://gnomad.broadinstitute.org/), genotype–phenotype correlation, location at protein level, and evolutionary conservation. Additionally, computational data were evaluated using Alamut Visual Plus software (Sophia Genetics, Lausanne, Switzerland) with integrated in silico prediction tools, such as SIFT (https://bio.tools/sift), polyphen2 (http://genetics.bwh.harvard.edu/pph2/), and MutationTaster (https://www.mutationtaster.org/). The results were also compared with variant databases, such as HGMD professional (https://my.qiagendigitalinsights.com/bbp/) and ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/), in combination with in-house information. Variant classification was performed in accordance with American College of Medical Genetics and Genomics guidelines (Richards et al., 2015).
The gender proportion was 1:1.2 (27 males and 32 females). A syndromic form of CS was recognized in 51% of families. The distribution of the sutural pattern in the SCS and NCS groups is depicted in Supplementary Figure S1. In the multiple synostosis group (20 cases, including 16 SCS), the involvement of different sutures was as follows: coronal, 16 cases; sagittal, 13 cases; lambdoid, seven cases (5 combined with coronal); metopic, four cases (3 combined with coronal and 1 with sagittal); frontosphenoidal, two cases (combined with coronal); and pansynostosis, one case. Combined coronal and sagittal involvement were observed in 10 of 20 cases and predominantly in SCS. Mercedes synostosis (sagittal and bilateral lambdoid) was noted in two SCS cases. The phenotypic details, including the sutural pattern for each patient, are shown in Supplementary Table S1.
Molecular outcomes according to phenotypic presentation and suture pattern are depicted in Supplementary Figures S2, S3. We detected causal (pathogenic or likely pathogenic according to the ACMG criteria) variants in 19% of the families and 38% of unrelated patients with SCS (Table 1). Seven causal and novel variants were detected in FGFR2, TWIST1, TCF12, KIAA0586, HDAC9, FOXP1, and NSD2, and a de novo deletion in HDAC9 (P2605_132) detected using the in silico panel on WGS data was confirmed by multiplex ligation-dependent probe amplification. In this case, the breakpoints were located upstream of TWIST1, and the deletion included HDAC9, PRPS1L1, and SNX13 (Supplementary Figure S4), none of which had been clearly associated with disease in humans. The typical Apert phenotype in one patient (P_3) prompted a detailed examination of FGFR2, which revealed insertion of an Alu element 18-bp upstream of the boundary between intron 7 and exon 8 (NM_000141.5). This was subsequently confirmed by targeted NGS analysis at an external lab. Additionally, we identified the novel splice variant in TCF12 in our clinical screening and classified it as a variant of uncertain significance. The variant was subsequently upgraded to likely pathogenic after complementary parental segregation and mRNA analyses. cDNA sequencing revealed introduction of a cryptic acceptor splice site (TTTCTAG) at position −8 of intron 18, leading to a frameshift and an early stop codon. Furthermore, we detected larger de novo structural genomic rearrangements in two patients (P_2 and P2605_175) (Table 1). For patient P2605_175, we were unable to confirm the breakpoints in the complex rearrangement involving chromosome 2 by SNP array. Supplementary Table S2 outlines additional findings in CS-relevant genes with possible modulator effects, detected in patients with causal variants.
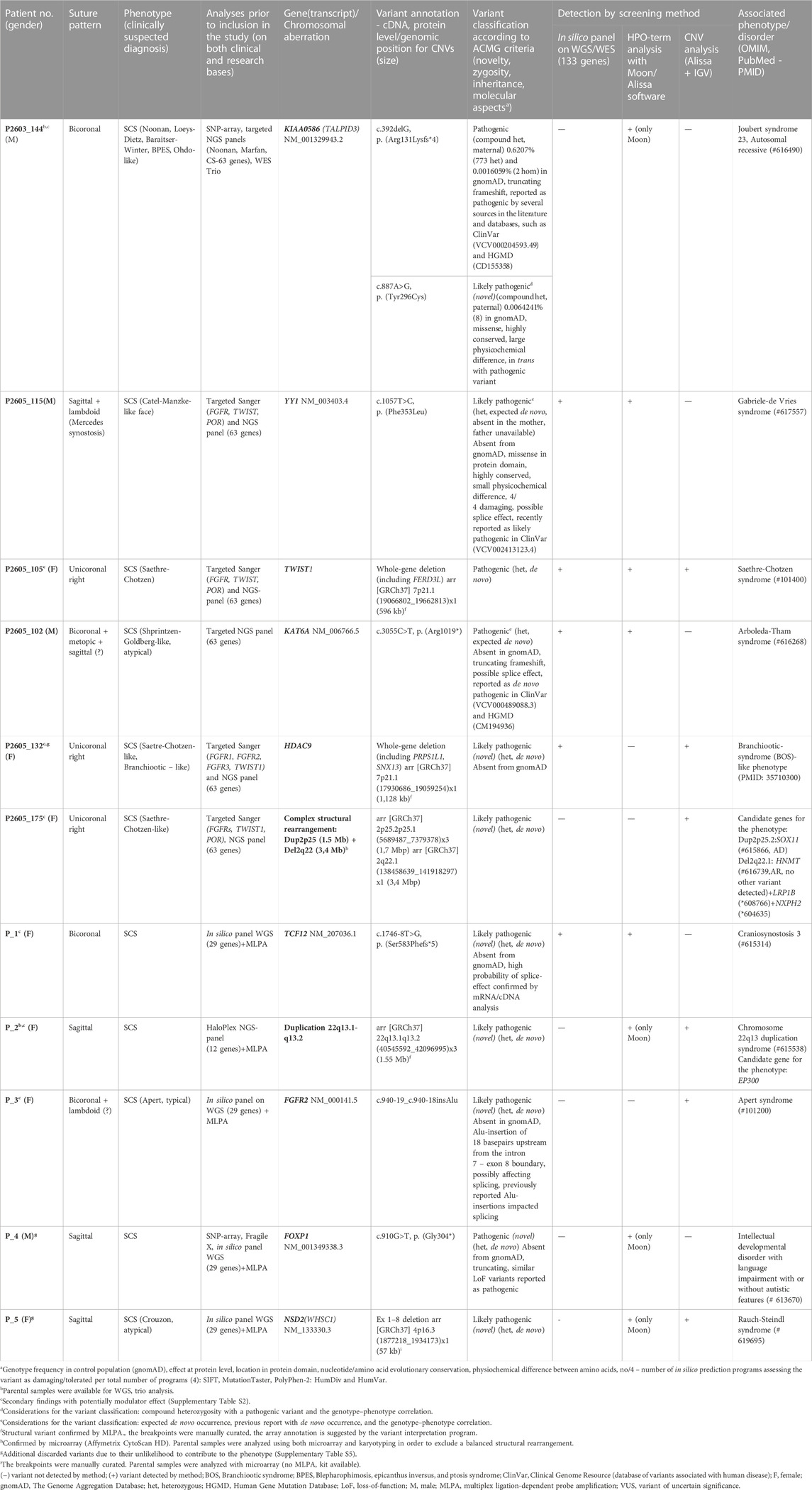
TABLE 1. Causal variants detected by WGS in patients with syndromic SCS.
Novel and possibly relevant variants were detected in 87% of families in which no causal variants were observed (Supplementary Tables S3, S4). When parental samples were available, segregation analysis was performed in cases harboring very low frequency variants in control populations and predicting damaging effects. Thirty-four novel and possibly relevant variants were detected in ADAMTSL4, ASH1L, ATRX, C2CD3, CHD5(2), DMP1, ERF, H4C5, IFT122 (2), IFT140, KDM6B, KMT2D, LTBP1(2), MAP3K7, NOTCH2, NSD1(2), SOS1, SPRY1, POLR2A, PRRX1, RECQL4, TAB2, TAOK1(3), TET3(2), TGFBR1, TCF20, and ZBTB20. New causal and possibly relevant variants were detected in 23 of 37 patients previously screened using the targeted 63-gene NGS panel. Eight variants that had been observed but not reported at previous targeted NGS analysis were confirmed with WGS and reassessed as possibly relevant (Supplementary Tables S3, S4). Additional variants, including novel ones considered unlikely to contribute to the phenotype and/or discarded after segregation analysis, are listed in Supplementary Table S5.
A total of 115 genetic variants were detected using a combination of the in silico panel along with HPO-driven and CNV analyses. The majority of variants (89%) were detected by HPO-driven filtration, of which 50% were identified exclusively by one software program (Moon). The in silico panel detected 41% of the variants, of which 8% were identified exclusively by this filtration method. Six of the causal variants (5.2%) were detected by CNV analysis combined with the in silico panel and HPO filtration in four cases (Table 1). Overall, causal and potentially significant variants were identified in 89% of the unrelated patients. The contribution of WGS/WES to the diagnostic yield (causal variants) was 16% for patients initially analyzed using the targeted 63-gene NGS panel and 25% for patients initially analyzed using the in silico clinical panel.
In this study we applied WES or WGS analyses of a cohort of patients with SCS and NCS and that previously tested negative in a targeted mutation screening program (Topa et al., 2020; Topa et al., 2022). The results revealed a diagnostic yield of 38% in patients with SCS and possibly relevant variants in 87% of the remaining patients (NCS included).
We report causal and likely causal variants (whereof eight novel) in the following genes (Table 1): 1) CS core genes FGFR2, TWIST1, and TCF12; 2) HDAC9, a gene situated in a candidate region with regulatory elements for TWIST1; 3) genes occasionally associated with CS (e.g., FOXP1, NSD2, and YY1); and 4) KIAA0586, which was not previously associated with CS. Furthermore, we report two de novo chromosomal aberrations (a microduplication of 22q13.1-q13.2 and a complex rearrangement of chromosome 2) and an additional case of a recurrent pathogenic variant in KAT6A.
The previously reported pathogenic variant in KIAA0586 (centrosomal protein homolog to chicken TALPID3) is the most frequently identified variant in Joubert syndrome type 23. This variant has been observed in a homozygous state in two individuals in the control population, suggesting a hypomorphic allele with pathogenic effect only in a compound heterozygous state with more severe variants (Pauli et al., 2019). The phenotype of the CS patient harboring this variant was not suggestive of Joubert syndrome, although some of the features, such as gross motor delay with impaired balance and coordination, were indicative of cerebellar abnormalities. To the best of our knowledge, this is the first case of CS among KIAA0586-related disorders, which are hybrid ciliopathies with a broad phenotypic spectrum ranging from severe skeletal abnormalities to Joubert syndrome. As such, patients may present a clinical picture that overlaps with these entities (Alby et al., 2015; Malicdan et al., 2015).
Recurrent de novo occurrence of the same pathogenic variant in KAT6A was reported by Kennedy et al. (2019) in two patients, one of which presented with sagittal CS. Microcephaly and CS are recurrent features in Arboleda−Tham syndrome, with several cases of midline synostosis and pansynostosis described in association with truncating variants in KAT6A (Tham et al., 2015; Kennedy et al., 2019; Timberlake et al., 2019; Marji et al., 2021; Korakavi et al., 2022). Interestingly, Clarke et al. (2018) reported two rare missense variants in KAT6A in three cases of isolated NCS (two metopic and one coronal).
There is only one report of CS in Gabriele-de-Vries syndrome (Gabriele et al., 2017), with that describing a likely pathogenic de novo missense variant in YY1 encoding a transcription factor [c.1015A>C, p. (Lys339Gln)]. However, facial asymmetry and a broad head have been described in other patients, suggesting that mild CS may be underdiagnosed. Furthermore, the variant in our patient [c.1057T>C, p. (Phe353Leu)] is expected to have occurred sporadically and is located in the same zinc-finger protein domain as the variant reported by Gabriele et al. (2017), thus supporting its potential pathogenic role.
To the best of our knowledge, this study includes the second reported case of CS associated with a de novo truncating variant in FOXP1. Urreizti et al. (2018) reported a patient with metopic synostosis and an Opitz C-trigonocephaly-like phenotype. The sagittal synostosis in our patient may have occurred independent of the FOXP1 variant, explaining only the neurodevelopmental symptoms. However, recent transcriptome studies show that neural crest-derived cells from the sagittal suture of human embryonic calvaria are highly enriched for the FOXP1/2 transcriptional network (He et al., 2021). Moreover, a truncating variant in FOXP2 inherited from an affected parent was recently reported in a patient with sagittal SCS (Tønne et al., 2022). This further supports the involvement of FOXP1/2 in midline synostosis.
The deletion identified in NSD2 is to the best of our knowledge the first limited to exons 1 through 8. Larger structural variants (SVs) involving NSD2 are associated with Wolf–Hirschhorn 4p16.3 deletion syndrome, and loss-of-function (LoF) and missense variants in NSD2 were recently associated with the developmental disorder Rauch–Steindl syndrome (Zanoni et al., 2021). In that study, CS was diagnosed in one of the patients with a de novo missense mutation in NSD2, but the authors attributed this to the co-occurrence of a second de novo missense mutation in AGO2, which is associated with skull deformities, such as scaphocephaly. However, the microcephaly, dolichocephaly, and craniofacial asymmetry described in Rauch–Steindl syndrome suggest that CS is underdiagnosed. The present findings support that CS may occasionally be part of Rauch–Steindl syndrome.
We report the fourth case of CS and deletion involving HDAC9 without disrupting the TWIST1-coding region. Recently, Hirsch et al. (2022) showed that SVs involving HDAC9 disrupt TWIST1-regulatory elements within HDAC9 in patients with CS. Our patient had coronal synostosis that was also described in two of the previously reported patients with deletion and one with a translocation breakpoint disrupting the HDAC9–TWIST1 locus (Hirsch et al., 2022). Interestingly, the craniofacial features of our patient during infancy were not suggestive of Sathre–Chotzen syndrome. No information about possible syndactyly was available, and the presence of dysplastic helices, hearing loss, and a branchial cyst suggested branchiootic syndrome.
CS has not been documented in previous cases of 22q13.1-q13.2 duplications, although prominent forehead with brachycephaly are recurrent features (Samanich et al., 2012; Rahikkala et al., 2013). Notably, de novo mutations in EP300 (included in the 22q13 duplication) are suggested to result in gain-of-function and are associated with a phenotype distinct from Rubinstein–Taybi syndrome caused by LoF variants (Menke et al., 2018). The facial appearance with prominent forehead and short up-slanting palpebral fissures are reminiscent of features observed in association with 22q13.1-q13.2 duplications, suggesting EP300 as a candidate gene for the phenotype. However, the sagittal synostosis in our patient could be related to the paternally inherited MSX2 variant with incomplete penetrance.
Another interesting finding was the de novo complex genomic rearrangement, including a 2p25 duplication and 2q22 deletion in a patient with a Saethre–Chotzen-like phenotype. The duplication involves SOX11, in which a de novo missense variant was reported in a patient with lambdoid synostosis and Coffin–Siris features (Timberlake et al., 2019). We could not determine a clear correlation between the phenotype and the genes included in the 2q22 deletion (containing the known protein-coding genes HNMT, LRP1B, and NXPH2). Only HNMT is disease-associated, specifically with a recessive type of intellectual disability (OMIM # 616739). Both LRP1B and NXPH2 are dosage-sensitive and likely intolerant to haploinsufficiency.
Alu insertions represent a very rare mutational mechanism in Apert syndrome. To the best of our knowledge, there are only three previously reported cases caused by similar Alu insertions (Oldridge et al., 1999; Bochukova et al., 2009). We recommend undertaking specific searches for this type of mutation in patients with typical Apert syndrome and without coding variants in FGFR2.
Overall, we observed enrichment of causal variants in developmental genes, including those encoding transcription factors and chromatin modifiers (e.g., FOXP1, KAT6A, NSD2, YY1) associated with several disorders with pleiotropic phenotypes. CS is a variable feature in these pathologies, which may depend on the widespread downstream transcriptional and epigenetic effects of these genes (Zollino et al., 2017).
The variant in ATRX (Supplementary Table S3) may be related to the Carpenter-like phenotype observed in patient P2605_136. A recent report identified a missense variant (NM_000489: c.6511A>G p. Met2171Val) in the C-terminal helicase domain of ATRX in two brothers with a Carpenter-like phenotype (patient photos not provided) accompanied by coronal and sagittal synostosis (Sáenz et al., 2021). However, the same variant was reported in a patient with ATR-X syndrome in the absence of noticeable skeletal abnormalities (Wada et al., 2013). The missense variant in our patient (c.690T>G, p. (Ile230Met)) is located in a zinc-finger protein domain in which neighboring mutations have previously been associated with ATR-X syndrome (Gibbons et al., 2008; Arvio and Lähdetie, 2021).
The novel TET3 (Supplementary Table S3) variants in the patient with multisuture synostosis are of particular interest for CS. This case and previous reports of patients with TET3-related craniofacial involvement including brachycephaly and asymmetric skull shapes point out towards a possible role of TET3-variants in the dysregulation of gene expression during suture development (Beck et al., 2020; Seyama et al., 2022).
Additionally, we found novel variants in POLR2A and PRRX1 (Supplementary Table S3) genes that have recently been associated with metopic and lambdoid synostosis, respectively (Tønne et al., 2022; Timberlake et al., 2023). Interestingly, previous studies demonstrated that PRRX1 is expressed in calvarial sutural mesenchymal stem cells (Wilk et al., 2017).
Notably, we observed two patients with pathogenic and likely pathogenic variants in GSK and ABCC8 (Supplementary Table S3), respectively, in which mutations lead to familial hyperinsulinemic hypoglycemia. The GSK variant explained the recurrent hypoglycemia in the patient with bicoronal synostosis. Because activation of the IGF1-signaling pathway is involved in CS pathogenesis (Al-Rekabi et al., 2016; Gustafson et al., 2019), we cannot rule out a possible role for the GSK and ABCC8 variants in CS occurrence in these patients.
We found an inherited novel variant in ERF in a patient with unicoronal synostosis, and two variants in SPRY1 in two patients with multiple synostosis and involvement of the sagittal suture (Supplementary Table S4). The variants were inherited from an a priori unaffected parent. A truncating variant in SPRY1 was reported by Timberlake et al. (2016) in a woman with mild cranial dysmorphism and her two children with sagittal synostosis. Tooze et al. (2023) also reported a case of syndromic sagittal synostosis with complete LoF of SPRY1 due to bi-allelic inheritance of a truncating variant from healthy parents. These findings suggest that heterozygous SPRY1 variants may play a modifier role in the occurrence of sagittal NCS, whereas bi-allelic LoF variants lead to a more severe phenotype with SCS.
Additionally, we observed enrichment of possibly relevant variants in genes involved in the TGF-β pathway (ADAMTSL4, LTBP1, LTBP4 and TGFBR1 - Supplementary Table S4) with a role in the morphogenesis of the skull sutures (Rawlins and Opperman, 2008). Furthermore, CS was recently reported in patients with bi-allelic mutations in ADAMTSL4 and LTBP1 (Pottie et al., 2021; Gustafson et al., 2022). Also, we observed enrichment of heterozygous carriers of variants in CS-related autosomal recessive genes from the ciliopathy spectrum (e.g., IFT122, IFT140, and WDR19 - Supplementary Table S4). These findings, together with results from previous studies showing enrichment of damaging variants in genes associated with SCS in patients with NCS, suggest an oligogenic involvement with incomplete penetrance in the occurrence of single-suture NCS (Clarke et al., 2018; Gustafson et al., 2019; Tiberio et al., 2021).
This study has limitations in its retrospective nature, reduced number of patients and partly limited access to phenotypic details. An additional limitation is the lack of parental samples, which complicated the interpretation of variants of uncertain significance. Also, the cost of high-throughput sequencing methods limited the access to WGS/WES Trio in those cases where parental samples were obtained a posteriori for targeted segregation analysis of detected variants in singleton WGS/WES analyses. Another limitation resides in the capacity of the bioinformatic pipeline to detect CNVs/SVs from genomic data, which results in the possibility that certain smaller SVs, indels, or intragenic deletions may have been missed. Furthermore, CNV analysis was unavailable for WES data from five patients. Also, the access to functional genomic studies such as transcriptomics in order to verify the effect of potentially causal variants was limited. However, we have ongoing projects using long-read sequencing and RNA-sequencing to study complex genomic variants such as the structural rearrangement in patient P2605_175 and the Alu-insertion in FGFR2 in patient P_3, respectively.
In summary, our findings demonstrate the power of WGS/WES as a diagnostic tool capable of generating an increased diagnostic yield in patients with unusual syndromic presentations. This is evident in view of the heterogeneity of CS and the detection of new candidate genes with pleiotropic effects. In particular, attention should be given to the phenotypic assessment of the patients and their parents, which enables a reliable interpretation of the genetic variants. Additionally, the results demonstrate the advantage of using HPO-driven variant filtration for detecting new candidate genes or variants in genes rarely associated with CS. Furthermore, the results presented a high detection rate of possibly relevant variants in patients with NCS, thereby emphasizing the need for more extensive studies, including transcriptome analysis of larger patient cohorts. Such studies will promote a broader understanding of the molecular pathogenesis of SCS as well as NCS with suggested polygenic contribution.
The causal variants were submitted to ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/) under the following accession numbers: 1) SUB13964571: SCV004174841 - SCV004174846; 2) SUB14012407: SCV004174877 - SCV004174882.
The study was approved by the Regional Ethical Review Board (No 303-15). Informed consent to participate in the study was obtained from all patients or their parents.
AT: Conceptualization, Data curation, Formal Analysis, Funding acquisition, Investigation, Methodology, Validation, Writing–original draft, Writing–review and editing, Visualization. AR: Data curation, Formal Analysis, Funding acquisition, Investigation, Methodology, Resources, Supervision, Validation, Writing–original draft, Writing–review and editing. AF: Investigation, Resources, Validation, Writing–review and editing. LL: Funding acquisition, Project administration, Writing–review and editing. GS: Funding acquisition, Investigation, Resources, Supervision, Validation, Writing–original draft, Writing–review and editing. PT: Resources, Writing–review and editing. GM: Resources, Writing–review and editing. MB-S: Resources, Writing–review and editing. LK: Funding acquisition, Investigation, Methodology, Project administration, Resources, Supervision, Validation, Writing–original draft, Writing–review and editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This study was funded by the ALF agreement, The Swedish Research Council (Grant/Award No: ALFGBG-716621), Gothenburg Medical Society and the Ågrenska Foundation.
We want to thank the Clinical Genomics infrastructure offered by SciLifeLab, Stockholm, Sweden, for assistance with genome sequencing, as well as SciLifeLab, Gothenburg, Sweden, for their assistance with both sequencing and bioinformatics analyses of the data.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2023.1322462/full#supplementary-material
Alby, C., Piquand, K., Huber, C., Megarbané, A., Ichkou, A., Legendre, M., et al. (2015). Mutations in KIAA0586 cause lethal ciliopathies ranging from a hydrolethalus phenotype to short-rib polydactyly syndrome. Am. J. Hum. Genet. 97, 311–318. doi:10.1016/j.ajhg.2015.06.003
Al-Rekabi, Z., Wheeler, M. M., Leonard, A., Fura, A. M., Juhlin, I., Frazar, C., et al. (2016). Activation of the IGF1 pathway mediates changes in cellular contractility and motility in single-suture craniosynostosis. J. Cell Sci. 29, 483–491. doi:10.1242/jcs.175976
Armand, T., Schaefer, E., Di Rocco, F., Edery, P., Collet, C., and Rossi, M. (2019). Genetic bases of craniosynostoses: an update. Neurochirurgie 65, 196–201. doi:10.1016/j.neuchi.2019.10.003
Arvio, M., and Lähdetie, J. (2021). Natural history of alpha-thalassemia X-linked intellectual disability syndrome: a case report of a 45-year-old man. Am. J. Med. Genet. A 185, 2164–2167. doi:10.1002/ajmg.a.62213
Beck, D. B., Petracovici, A., He, C., Moore, H. W., Louie, R. J., Ansar, M., et al. (2020). Delineation of a human Mendelian disorder of the DNA demethylation machinery: TET3 deficiency. Am. J. Hum. Genet. 106, 234–245. doi:10.1016/j.ajhg.2019.12.007
Bellus, G. A., Gaudenz, K., Zackai, E. H., Clarke, L. A., Szabo, J., Francomano, C. A., et al. (1996). Identical mutations in three different fibroblast growth factor receptor genes in autosomal dominant craniosynostosis syndromes. Nat. Genet. 14, 174–176. doi:10.1038/ng1096-174
Bochukova, E. G., Roscioli, T., Hedges, D. J., Taylor, I. B., Johnson, D., David, D. J., et al. (2009). Rare mutations of FGFR2 causing apert syndrome: identification of the first partial gene deletion, and an Alu element insertion from a new subfamily. Hum. Mutat. 30, 204–211. doi:10.1002/humu.20825
Bukowska-Olech, E., Sowińska-Seidler, A., Larysz, D., Gawliński, P., Koczyk, G., Popiel, D., et al. (2022). Results from genetic studies in patients affected with craniosynostosis: clinical and molecular aspects. Front. Mol. Biosci. 9, 865494. doi:10.3389/fmolb.2022.865494
Clarke, C. M., Fok, V. T., Gustafson, J. A., Smyth, M. D., Timms, A. E., Frazar, C. D., et al. (2018). Single suture craniosynostosis: identification of rare variants in genes associated with syndromic forms. Am. J. Med. Genet. A 176, 290–300. doi:10.1002/ajmg.a.38540
Clayton-Smith, J., O'Sullivan, J., Daly, S., Bhaskar, S., Day, R., Anderson, B., et al. (2011). Whole-exome-sequencing identifies mutations in histone acetyltransferase gene KAT6B in individuals with the Say-Barber-Biesecker variant of Ohdo syndrome. Am. J. Hum. Genet. 89, 675–681. doi:10.1016/j.ajhg.2011.10.008
Flück, C. E., Tajima, T., Pandey, A. V., Arlt, W., Okuhara, K., Verge, C. F., et al. (2004). Mutant P450 oxidoreductase causes disordered steroidogenesis with and without Antley-Bixler syndrome. Nat. Genet. 36, 228–230. doi:10.1038/ng1300
Gabriele, M., Vulto-van Silfhout, A. T., Germain, P. L., Vitriolo, A., Kumar, R., Douglas, E., et al. (2017). YY1 haploinsufficiency causes an intellectual disability syndrome featuring transcriptional and chromatin dysfunction. Am. J. Hum. Genet. 100, 907–925. doi:10.1016/j.ajhg.2017.05.006
Garbes, L., Kim, K., Rieß, A., Hoyer-Kuhn, H., Beleggia, F., Bevot, A., et al. (2015). Mutations in SEC24D, encoding a component of the COPII machinery, cause a syndromic form of osteogenesis imperfecta. Am. J. Hum. Genet. 96, 432–439. doi:10.1016/j.ajhg.2015.01.002
Gibbons, R. J., Wada, T., Fisher, C. A., Malik, N., Mitson, M. J., Steensma, D. P., et al. (2008). Mutations in the chromatin-associated protein ATRX. Hum. Mutat. 29, 796–802. doi:10.1002/humu.20734
Gustafson, J., Bjork, M., van Ravenswaaij-Arts, C. M. A., and Cunningham, M. L. (2022). Mechanism of disease: recessive ADAMTSL4 mutations and craniosynostosis with ectopia lentis. Case Rep. Genet. 26, 3239260. doi:10.1155/2022/3239260
Gustafson, J. A., Park, S. S., and Cunningham, M. L. (2019). Calvarial osteoblast gene expression in patients with craniosynostosis leads to novel polygenic mouse model. PLoS One 14, e0221402. doi:10.1371/journal.pone.0221402
He, J., Yan, J., Wang, J., Zhao, L., Xin, Q., Zeng, Y., et al. (2021). Dissecting human embryonic skeletal stem cell ontogeny by single-cell transcriptomic and functional analyses. Cell Res. 31, 742–757. doi:10.1038/s41422-021-00467-z
Heuzé, Y., Holmes, G., Peter, I., Richtsmeier, J. T., and Jabs, E. W. (2014). Closing the gap: genetic and genomic continuum from syndromic to nonsyndromic craniosynostoses. Curr. Genet. Med. Rep. 2, 135–145. doi:10.1007/s40142-014-0042-x
Hirsch, N., Dahan, I., D'Haene, E., Avni, M., Vergult, S., Vidal-García, M., et al. (2022). HDAC9 structural variants disrupting TWIST1 transcriptional regulation lead to craniofacial and limb malformations. Genome Res. 32, 1242–1253. doi:10.1101/gr.276196.121
Howard, T. D., Paznekas, W. A., Green, E. D., Chiang, L. C., Ma, N., Ortiz de Luna, R. I., et al. (1997). Mutations in TWIST, a basic helix-loop-helix transcription factor, in Saethre-Chotzen syndrome. Nat. Genet. 15, 36–41. doi:10.1038/ng0197-36
Hyder, Z., Calpena, E., Pei, Y., Tooze, R. S., Brittain, H., Twigg, S. R. F., et al. (2021). Evaluating the performance of a clinical genome sequencing program for diagnosis of rare genetic disease, seen through the lens of craniosynostosis. Genet. Med. 23, 2360–2368. doi:10.1038/s41436-021-01297-5
Jenkins, D., Seelow, D., Jehee, F. S., Perlyn, C. A., Alonso, L. G., Bueno, D. F., et al. (2007). RAB23 mutations in Carpenter syndrome imply an unexpected role for hedgehog signaling in cranial-suture development and obesity. Am. J. Hum. Genet. 80, 1162–1170. doi:10.1086/518047
Kennedy, J., Goudie, D., Blair, E., Chandler, K., Joss, S., McKay, V., et al. (2019). KAT6A Syndrome: genotype-phenotype correlation in 76 patients with pathogenic KAT6A variants. Genet. Med. 21, 850–860. doi:10.1038/s41436-018-0259-2
Korakavi, N., Bupp, C., Grysko, B., Juusola, J., Borta, C., and Madura, C. (2022). First case of pan-suture craniosynostosis due to de novo mosaic KAT6A mutation. Childs Nerv. Syst. 38, 173–177. doi:10.1007/s00381-021-05111-0
Lajeunie, E., Le Merrer, M., Bonaiti-Pellie, C., Marchac, D., and Renier, D. (1996). Genetic study of scaphocephaly. Am. J. Med. Genet. 62, 282–285. doi:10.1002/(sici)1096-8628(19960329)62:3<282::aid-ajmg15>3.0.co;2-g
Lattanzi, W., Barba, M., Di Pietro, L., and Boyadjiev, S. A. (2017). Genetic advances in craniosynostosis. Am. J. Med. Genet. A 173, 1406–1429. doi:10.1002/ajmg.a.38159
Malicdan, M. C., Vilboux, T., Stephen, J., Maglic, D., Mian, L., Konzman, D., et al. (2015). Mutations in human homologue of chicken talpid3 gene (KIAA0586) cause a hybrid ciliopathy with overlapping features of Jeune and Joubert syndromes. J. Med. Genet. 52, 830–839. doi:10.1136/jmedgenet-2015-103316
Marji, F. P., Hall, J. A., Anstadt, E., Madan-Khetarpal, S., Goldstein, J. A., and Losee, J. E. (2021). A novel frameshift mutation in KAT6A is associated with pancraniosynostosis. J. Pediatr. Genet. 10, 81–84. doi:10.1055/s-0040-1710330
Menke, L. A., Gardeitchik, T., Hammond, P., Heimdal, K. R., Houge, G., Hufnagel, S. B., et al. (2018). Further delineation of an entity caused by CREBBP and EP300 mutations but not resembling Rubinstein-Taybi syndrome. Am. J. Med. Genet. A 176, 862–876. doi:10.1002/ajmg.a.38626
Miller, K. A., Twigg, S. R., McGowan, S. J., Phipps, J. M., Fenwick, A. L., Johnson, D., et al. (2017). Diagnostic value of exome and whole genome sequencing in craniosynostosis. J. Med. Genet. 54, 260–268. doi:10.1136/jmedgenet-2016-104215
Muenke, M., Schell, U., Hehr, A., Robin, N. H., Losken, H. W., Schinzel, A., et al. (1994). A common mutation in the fibroblast growth factor receptor 1 gene in Pfeiffer syndrome. Nat. Genet. 8, 269–274. doi:10.1038/ng1194-269
Nieminen, P., Morgan, N. V., Fenwick, A. L., Parmanen, S., Veistinen, L., Mikkola, M. L., et al. (2011). Inactivation of IL11 signaling causes craniosynostosis, delayed tooth eruption, and supernumerary teeth. Am. J. Hum. Genet. 89, 67–81. doi:10.1016/j.ajhg.2011.05.024
Oldridge, M., Zackai, E. H., McDonald-McGinn, D. M., Iseki, S., Morriss-Kay, G. M., Twigg, S. R., et al. (1999). De novo alu-element insertions in FGFR2 identify a distinct pathological basis for Apert syndrome. Am. J. Hum. Genet. 64, 446–461. doi:10.1086/302245
Pauli, S., Altmüller, J., Schröder, S., Ohlenbusch, A., Dreha-Kulaczewski, S., Bergmann, C., et al. (2019). Homozygosity for the c.428delG variant in KIAA0586 in a healthy individual: implications for molecular testing in patients with Joubert syndrome. J. Med. Genet. 56, 261–264. doi:10.1136/jmedgenet-2018-105470
Pottie, L., Adamo, C. S., Beyens, A., Lütke, S., Tapaneeyaphan, P., De Clercq, A., et al. (2021). Bi-allelic premature truncating variants in LTBP1 cause cutis laxa syndrome. Am. J. Hum. Genet. 108, 1095–1114. doi:10.1016/j.ajhg.2021.04.016
Rahikkala, E., Forsström, L. M., Kokkonen, H., Knuutila, S., Mustonen, A., and Ignatius, J. (2013). Report of interstitial 22q13.1q13.2 microduplication in two siblings with distinctive dysmorphic features, heart defect and mental retardation. Eur. J. Med. Genet. 56, 389–396. doi:10.1016/j.ejmg.2013.05.004
Rauch, F., Fahiminiya, S., Majewski, J., Carrot-Zhang, J., Boudko, S., Glorieux, F., et al. (2015). Cole-Carpenter syndrome is caused by a heterozygous missense mutation in P4HB. Am. J. Hum. Genet. 96, 425–431. doi:10.1016/j.ajhg.2014.12.027
Rawlins, J. T., and Opperman, L. A. (2008). Tgf-beta regulation of suture morphogenesis and growth. Front. Oral Biol. 12, 178–196. doi:10.1159/000115038
Reardon, W., Winter, R. M., Rutland, P., Pulleyn, L. J., Jones, B. M., and Malcolm, S. (1994). Mutations in the fibroblast growth factor receptor 2 gene cause Crouzon syndrome. Nat. Genet. 8, 98–103. doi:10.1038/ng0994-98
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of medical genetics and genomics and the association for molecular pathology. Genet. Med. 17, 405–424. doi:10.1038/gim.2015.30
Sáenz, S. S., Arias, B., Hosomichi, K., and Romero, V. I. (2021). The limits of clinical findings in similar phenotypes, from Carpenter to ATRX syndrome using a whole exome sequencing approach: a case review. Hum. Genomics 15, 49. doi:10.1186/s40246-021-00348-x
Samanich, J., Montagna, C., Morrow, B. E., and Babcock, M. (2012). Interstitial duplication of 22q13.2 in a girl with short stature, impaired speech and language, and dysmorphism. J. Pediatr. Genet. 1, 47–53. doi:10.3233/pge-2012-009
Seyama, R., Tsuchida, N., Okada, Y., Sakata, S., Hamada, K., Azuma, Y., et al. (2022). Two families with TET3-related disorder showing neurodevelopmental delay with craniofacial dysmorphisms. J. Hum. Genet. 67, 157–164. doi:10.1038/s10038-021-00986-y
Sharma, V. P., Fenwick, A. L., Brockop, M. S., McGowan, S. J., Goos, J. A., Hoogeboom, A. J., et al. (2013). Mutations in TCF12, encoding a basic helix-loop-helix partner of TWIST1, are a frequent cause of coronal craniosynostosis. Nat. Genet. 45, 304–307. doi:10.1038/ng.2531
Tarnow, P., Kölby, L., Maltese, G., Söfteland, M. B., Lewén, A., Nilsson, P., et al. (2022). Incidence of non-syndromic and syndromic craniosynostosis in Sweden. J. Craniofac Surg. 33, 1517–1520. doi:10.1097/scs.0000000000008457
Tham, E., Lindstrand, A., Santani, A., Malmgren, H., Nesbitt, A., Dubbs, H. A., et al. (2015). Dominant mutations in KAT6A cause intellectual disability with recognizable syndromic features. Am. J. Hum. Genet. 96, 507–513. doi:10.1016/j.ajhg.2015.01.016
Tiberio, F., Parolini, O., and Lattanzi, W. (2021). Ciliary signalling and mechanotransduction in the pathophysiology of craniosynostosis. Genes (Basel) 12, 1073. doi:10.3390/genes12071073
Timberlake, A. T., Choi, J., Zaidi, S., Lu, Q., Nelson-Williams, C., Brooks, E. D., et al. (2016). Two locus inheritance of non-syndromic midline craniosynostosis via rare SMAD6 and common BMP2 alleles. Elife 5, e20125. doi:10.7554/eLife.20125
Timberlake, A. T., Jin, S. C., Nelson-Williams, C., Wu, R., Furey, C. G., Islam, B., et al. (2019). Mutations in TFAP2B and previously unimplicated genes of the BMP, Wnt, and Hedgehog pathways in syndromic craniosynostosis. Proc. Natl. Acad. Sci. U. S. A. 116, 15116–15121. doi:10.1073/pnas.1902041116
Timberlake, A. T., Kiziltug, E., Jin, S. C., Nelson-Williams, C., Loring, E., Allocco, A., et al. (2023). De novo mutations in the BMP signaling pathway in lambdoid craniosynostosis. Hum. Genet. 1, 21–32. doi:10.1007/s00439-022-02477-2
Timberlake, A. T., and Persing, J. A. (2018). Genetics of nonsyndromic craniosynostosis. Plast. Reconstr. Surg. 141, 1508–1516. doi:10.1097/PRS.0000000000004374
Tønne, E., Due-Tønnessen, B. J., Mero, I. L., Wiig, U. S., Kulseth, M. A., Vigeland, M. D., et al. (2020). Benefits of clinical criteria and high-throughput sequencing for diagnosing children with syndromic craniosynostosis. Eur. J. Hum. Genet. 29, 920–929. doi:10.1038/s41431-020-00788-4
Tønne, E., Due-Tønnessen, B. J., Vigeland, M. D., Amundsen, S. S., Ribarska, T., Åsten, P. M., et al. (2022). Whole-exome sequencing in syndromic craniosynostosis increases diagnostic yield and identifies candidate genes in osteogenic signaling pathways. Am. J. Med. Genet. A 188, 14641475. doi:10.1002/ajmg.a.62663
Tooze, R. S., Calpena, E., Twigg, S. R., D'Arco, F., Wakeling, E. L., Wilkie, A. O., et al. (2023). Craniosynostosis, inner ear, and renal anomalies in a child with complete loss of SPRY1 (sprouty homolog 1) function. J. Med. Genet. 60, 712–716. doi:10.1136/jmg-2022-108946
Topa, A., Rohlin, A., Andersson, M. K., Fehr, A., Lovmar, L., Stenman, G., et al. (2020). NGS targeted screening of 100 Scandinavian patients with coronal synostosis. Am. J. Med. Genet. A 182, 348–356. doi:10.1002/ajmg.a.61427
Topa, A., Rohlin, A., Andersson, M. K., Fehr, A., Lovmar, L., Stenman, G., et al. (2022). The outcome of targeted NGS screening in patients with syndromic forms of sagittal and pansynostosis - IL11RA is an emerging core-gene for pansynostosis. Eur. J. Med. Genet. 65, 104476. doi:10.1016/j.ejmg.2022.104476
Twigg, S. R., Kan, R., Babbs, C., Bochukova, E. G., Robertson, S. P., Wall, S. A., et al. (2004). Mutations of ephrin-B1 (EFNB1), a marker of tissue boundary formation, cause craniofrontonasal syndrome. Proc. Natl. Acad. Sci. U. S. A. 101, 8652–8657. doi:10.1073/pnas.0402819101
Twigg, S. R., Lloyd, D., Jenkins, D., Elçioglu, N. E., Cooper, C. D., Al-Sannaa, N., et al. (2012). Mutations in multidomain protein MEGF8 identify a Carpenter syndrome subtype associated with defective lateralization. Am. J. Hum. Genet. 91, 897–905. doi:10.1016/j.ajhg.2012.08.027
Twigg, S. R., Vorgia, E., McGowan, S. J., Peraki, I., Fenwick, A. L., Sharma, V. P., et al. (2013). Reduced dosage of ERF causes complex craniosynostosis in humans and mice and links ERK1/2 signaling to regulation of osteogenesis. Nat. Genet. 45, 308–313. doi:10.1038/ng.2539
Twigg, S. R., and Wilkie, A. O. (2015). A genetic-pathophysiological framework for craniosynostosis. Am. J. Hum. Genet. 97, 359–377. doi:10.1016/j.ajhg.2015.07.006
Urreizti, R., Damanti, S., Esteve, C., Franco-Valls, H., Castilla-Vallmanya, L., Tonda, R., et al. (2018). A de novo FOXP1 truncating mutation in a patient originally diagnosed as C syndrome. Sci. Rep. 8, 694. doi:10.1038/s41598-017-19109-9
Wada, T., Ban, H., Matsufuji, M., Okamoto, N., Enomoto, K., Kurosawa, K., et al. (2013). Neuroradiologic features in X-linked α-thalassemia/mental retardation syndrome. AJNR Am. J. Neuroradiol. 34, 2034–2038. doi:10.3174/ajnr.A3560
Weiss, K., Wigby, K., Fannemel, M., Henderson, L. B., Beck, N., Ghali, N., et al. (2017). Haploinsufficiency of ZNF462 is associated with craniofacial anomalies, corpus callosum dysgenesis, ptosis, and developmental delay. Eur. J. Hum. Genet. 25, 946–951. doi:10.1038/ejhg.2017.86
Wilk, K., Yeh, S. A., Mortensen, L. J., Ghaffarigarakani, S., Lombardo, C. M., Bassir, S. H., et al. (2017). Postnatal calvarial skeletal stem cells expressing PRX1 reside exclusively in the calvarial sutures and are required for bone regeneration. Stem Cell Rep. 8, 933–946. doi:10.1016/j.stemcr.2017.03.002
Wilkie, A. O., Byren, J. C., Hurst, J. A., Jayamohan, J., Johnson, D., Knight, S. J., et al. (2010). Prevalence and complications of single-gene and chromosomal disorders in craniosynostosis. Pediatrics 126, e391–e400. doi:10.1542/peds.2009-3491
Wilkie, A. O., Slaney, S. F., Oldridge, M., Poole, M. D., Ashworth, G. J., Hockley, A. D., et al. (1995). Apert syndrome results from localized mutations of FGFR2 and is allelic with Crouzon syndrome. Nat. Genet. 9, 165–172. doi:10.1038/ng0295-165
Wilkie, A. O. M., Johnson, D., and Wall, S. A. (2017). Clinical genetics of craniosynostosis. Curr. Opin. Pediatr. 29, 622–628. doi:10.1097/MOP.0000000000000542
Zanoni, P., Steindl, K., Sengupta, D., Joset, P., Bahr, A., Sticht, H., et al. (2021). Loss-of-function and missense variants in NSD2 cause decreased methylation activity and are associated with a distinct developmental phenotype. Genet. Med. 23, 1474–1483. doi:10.1038/s41436-021-01158-1
Zollino, M., Lattante, S., Orteschi, D., Frangella, S., Doronzio, P. N., Contaldo, I., et al. (2017). Syndromic craniosynostosis can define new candidate genes for suture development or result from the non-specifc effects of pleiotropic genes: rasopathies and chromatinopathies as examples. Front. Neurosci. 11, 587. doi:10.3389/fnins.2017.00587
Keywords: genetic, gene, suture, syndrome, skull, craniofacial, synostosis
Citation: Topa A, Rohlin A, Fehr A, Lovmar L, Stenman G, Tarnow P, Maltese G, Bhatti-Søfteland M and Kölby L (2024) The value of genome-wide analysis in craniosynostosis. Front. Genet. 14:1322462. doi: 10.3389/fgene.2023.1322462
Received: 16 October 2023; Accepted: 19 December 2023;
Published: 22 January 2024.
Edited by:
Yongchu Pan, Nanjing Medical University, ChinaReviewed by:
Ewelina Bukowska-Olech, Poznan University of Medical Sciences, PolandCopyright © 2024 Topa, Rohlin, Fehr, Lovmar, Stenman, Tarnow, Maltese, Bhatti-Søfteland and Kölby. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alexandra Topa, YWxleGFuZHJhLnRvcGFAdmdyZWdpb24uc2U=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.