 Huan Ye
Huan Ye Jiahui Fan1
Jiahui Fan1 Huamei Yue
Huamei Yue Rui Ruan
Rui Ruan Chuangju Li
Chuangju Li
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
DATA REPORT article
Front. Genet. , 01 November 2023
Sec. Livestock Genomics
Volume 14 - 2023 | https://doi.org/10.3389/fgene.2023.1297119
This article is part of the Research Topic Omics Applied to Livestock Genetics,volume II View all 16 articles
The largefin longbarbel catfish, Hemibagrus macropterus, is an economically important fish species in southwestern China, with males growing faster than females. This study presents a high-quality chromosome-level genome assembly of the largefin longbarbel catfish, generated by integrating Illumina short reads, PacBio HiFi long reads, and Hi-C data. The assembled genome size was 858.5 Mb, with a contig and scaffold N50 of 5.8 Mb and 28.4 Mb, respectively. A total of 656 contigs were successfully anchored to 30 pseudochromosomes with a BUSCO score of 97.7%, consistent with the number of chromosomes analyzed by karyotype. The genome contained 29.5% repeat sequences, and a predicted total of 26,613 protein-coding genes, of which 25,769 (96.8%) were functionally annotated in different databases. Evolutionary analysis showed that H. macropterus was most closely related to H. wyckioides, with a divergence time of approximately 16.3 million years. Chromosomal syntenic relationships among H. macropterus, H. wyckioides, and Pelteobagrus fulvidraco revealed a one-to-one relationship for most chromosomes, except for break, fission, and inversion of some chromosomes. The first high-quality reference genome will not only provide a valuable genetic resource for the study of sex determination mechanisms and genetic breeding of largefin longbarbel catfish, but also contribute to comparative analyses of genome and chromosome evolution within Siluriformes.
Catfish (order: Siluriformes) are a highly diverse and globally distributed group of actinopterygian fish, generally characterized by the whisker-like barbels, lack scales, and intramuscular spines (Gisbert et al., 2022). They comprise more than 4,500 species and account for nearly 12% of teleost fish (Fricke et al., 2022). Catfish are one of the most important aquaculture species worldwide (Gisbert et al., 2022). The number of chromosomes in catfish ranges from 2n = 24 to 100, with mainly continuous variation from 2n = 48 to 60 (Zhu and Pan, 2021). Consequently, catfish are considered suitable for studying genomic and chromosomal evolution in fish. With the rapid development of sequencing technologies, the chromosome-level genomes of more than 10 catfish species have been assembled, including Ictalurus punctatus (Liu et al., 2016), Pelteobagrus fulvidraco (Gong et al., 2018), Bagarius yarrelli (Jiang et al., 2019), Silurus meridionalis (Zheng et al., 2021), Leiocassis longirostris (He et al., 2021), Hemibagrus wyckioides (Shao et al., 2021), Pangasianodon hypophthalmus (Gao et al., 2021), Pseudobagrus ussuriensis (Zhu et al., 2022), Cranoglanis bouderius (Xu et al., 2022), Ictalurus furcatus (Wang et al., 2022), and Ancistrus triradiatus (Lemopoulos and Montoya-Burgos, 2022). These genomic resources facilitate studies of sex determination mechanisms (Bao et al., 2019; Gong et al., 2022), chromosomal and genome evolution (Zhu et al., 2022), ecological adaptation, and gene evolution and function (Liu et al., 2016; Zhou et al., 2023).
The largefin longbarbel catfish (Hemibagrus macropterus) (Figure 1A), belonging to the Bagridae family (Siluriformes), is an important commercial fish in southwestern China because of its high nutritional value (Zhang et al., 2009). It is a benthic dweller naturally distributed in the mainstream and tributaries of the Yangtze and Pearl River Basins (Zhu et al., 2007). Largefin longbarbel catfish exhibit sexual size dimorphism, with males growing faster than females. Two karyotypes (2n = 56 and 60) have been reported for this species among different populations (Hong and Zhou, 1984; Ma et al., 2013). However, the genetic resources of largefin longbarbel catfish are limited, which is unfavorable for understanding its genetic characteristics and developing breeding programs.
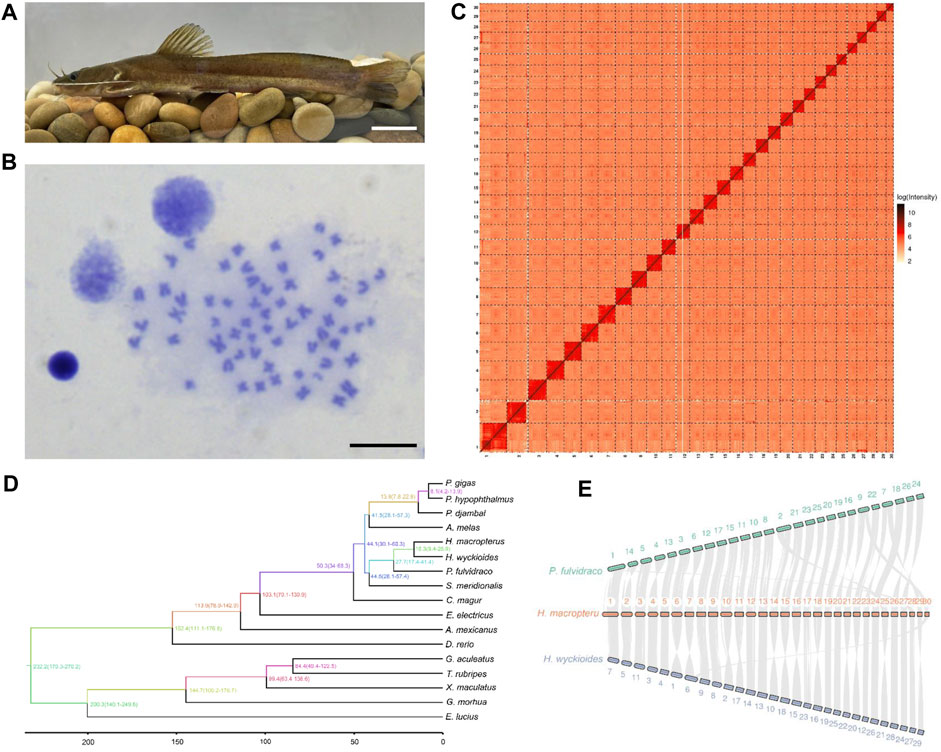
FIGURE 1. Chromosome-level genome assembly of Hemibagrus macropterus and comparative genomics analysis. (A) Representative image of H. macropterus. Scale bar represents 5 cm. (B) Karyotype of male H. macropterus. Scale bar represents 10 µm. (C) Heatmap of Hi-C interactions among 30 pseudochromosomes. Colour depth represents the density of the Hi-C interactions. (D) Phylogenetic relationships between H. macropterus and 16 other fish species based on 3,105 single-copy orthologous genes. (E) Chromosomal syntenic relationships among H. macropterus, H. wyckioides, and Pelteobagrus fulvidraco.
In this study, we present the first high-quality chromosome-level reference genome of H. macropterus. The assembled genome will be beneficial for exploring the genome evolution, sex determination mechanisms, and genetic breeding of largefin longbarbel catfish. Furthermore, this contribution to the genomic resources of Siluriformes will facilitate future comparative genomic studies among catfish.
A total of 31.4 Gb Illumina clean data were used to assess genome size and heterozygosity in H. macropterus. The predicted genome size was approximately 873.7 Mb and the estimated heterozygosity rate was 0.37%. For de novo genome assembly, 41.8 Gb PacBio HiFi reads were preliminarily assembled into 691 contigs with an N50 length of 5.8 Mb, covering 98.3% of the estimated genome. Using the Hi-C technique, a total of 656 contigs were successfully anchored to 30 pseudochromosomes (Figure 1C), consistent with the number of chromosomes analyzed by karyotype (Figure 1B) and reported in a previous study (Hong and Zhou, 1984). The assembled chromosome-level genome consisted of 35 contigs and 30 scaffolds, with a contig and scaffold N50 length of 5.8 and 28.4 Mb, respectively (Table 1), which represented 98.3% of the estimated genome. The guanine-cytosine (GC) content was 40%, similar with that of other Bagridae (Zhu et al., 2022). Benchmarking Universal Single-Copy Orthologs (BUSCO) analysis revealed 97.7% of BUSCO genes identified in the genome (Supplementary Table S1), indicating high completeness for the genome assembly.
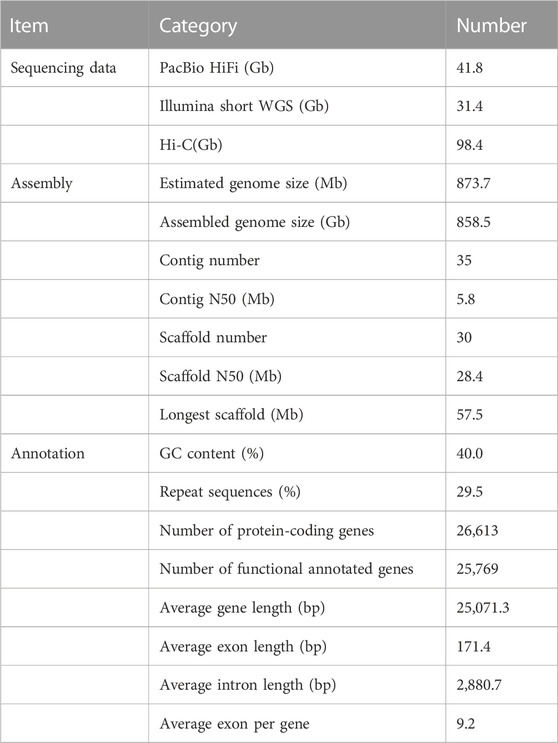
TABLE 1. Statistics of Hemibagrus macropterus genome assembly and annotation.
The genome of H. macropterus contained 29.5% repetitive sequences (Supplementary Table S2), with transposable element (TE) accounting for 18.06% of the assembled genome. The largest proportion of TE was terminal inverted repeats (9.29%), followed by long terminal repeat retrotransposons (7.43%). Together with homology, de novo, and RNA-seq prediction methods, 26,613 protein-coding genes were annotated (Supplementary Table S3). The average length of gene, exon, and intron was 25,071, 171, and 2,881 bp, respectively (Table 1). BUSCO assessments showed that 98.2% complete BUSCO genes were predicted, including 96.1% single copy and 2.1% duplicated genes (Supplementary Table S4). These results indicated high-quality genome assembly and annotation of H. macropterus.
The evolutionary relationships between H. macropterus and other teleosts were determined based on the analysis of 3,105 single-copy orthologous genes from of 17 fish genomes (Supplementary Figure S1). H. wyckioides was most closely related to H. macropterus, consistent with their taxonomic relationship (Fricke et al., 2022), and clustered with P. fulvidraco (Figure 1D). The nine species of Siluriformes formed a monophyletic clade, and then together with Electrophorus electricus (Gymnotiformes), Astyanax mexicanus (Characiformes), and Danio rerio (Cypriniformes), formed the clade of Ostariophysan. According to the fossil calibration times, the estimated divergence time between H. macropterus and H. wyckioides was approximately 16.3 Mya, and the divergence time between M. macropterus and P. fulvidraco was around 27.7 Mya.
Through comparative genomic analysis, we identified 60 and 28 gene families, respectively, that underwent significant expansion and contraction in H. macropterus (Supplementary Figure S2). Enrichment analysis revealed that the expanded and contracted genes were enriched in 18 and 7 Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways, respectively (Supplementary Tables S5, S6), with most involved in the immunity, metabolism, and hormone biosynthesis. These results provide valuable preliminary information on the biological properties of the species.
We compared the chromosome syntenies between H. macropterus and two other catfish species. The karyotype of these three species (P. fulvidraco, H. macropterus, and H. wyckioides) was 2n = 52, 2n = 60, and 2n = 59, respectively. Most of the chromosomes between P. fulvidraco and H. macropterus exhibited a one-to-one relationship (Figure 1E), whereas the chromosomes (Chr) 1, 2, 7, and 9 of P. fulvidraco broke into two chromosomes in H. macropterus. H. macropterus and H. wyckioides displayed a strong one-to-one correspondence among their chromosomes, except for the fission of H. wyckioides Chr6 into Chr7 and 30, and inversion of some chromosomes (Chr6, 7, 10, 11, 14, 15, 16, 18, 21, 22, 25, and 29) in H. macropterus. It was recently reported that the sex-determining region of P. fulvidraco and H. wyckioides was located on the Chr 2 and 26, respectively (Gong et al., 2022), whereas the correspondent chromosomes in H. macropterus were Chr14 and 15 and 24, respectively, indicating the complexity of the sex-determining region or chromosome in catfish.
A male H. macropterus was collected for genome sequencing from the Wuhan section of the Yangtze River. After anesthesia with 3-aminobenzoic acid ethyl ester methanesulfonate-222 (MS-222) (Sigma-Aldrich, St. Louis, MO, United States), muscle tissue was collected, frozen in liquid nitrogen, and stored at −80°C, while other tissues including brain, gills, heart, intestine, kidney, liver, spleen, and testis were stored in RNAlater solution (Sigma-Aldrich). All experiments involving in the handling and treatment of fish were conducted in accordance with the Guidelines for the Care and Use of Laboratory Animals of the Yangtze River Fisheries Research Institute, Chinese Academy of Fishery Sciences.
Total genomic DNA was isolated from frozen muscle tissue using a Blood & Cell Culture DNA Kit (Qiagen, Hilden, Germany). DNA quality and purity were determined using a Nanodrop 2000 (Thermo Fisher Scientific, Waltham, MA, United States) and agarose gel electrophoresis. A 350 bp paired-end library was constructed using an Illumina TruSeq DNA Nano Preparation Kit (Illumina, San Diego, CA, United States) and sequenced on an Illumina HiSeq 2500 platform (Illumina). Approximately 5 μg of genomic DNA was used to construct a PacBio SMRTbell library. The library was sequenced using a PacBio Circular Consensus Sequencing (CCS) Platform (PacBio, Menlo Park, CA, United States). A Hi-C library was prepared using a GrandOmics Hi-C kit according to the manufacturer’s instructions and sequenced on an Illumina NovaSeq platform (Illumina). Total RNA was extracted from different tissues of H. macropterus using an RNeasy Plus Mini Kit (Qiagen). A complementary DNA library was constructed using a TruSeq Stranded mRNA-Seq kit on the Illumina HiSeq 2500 platform (Illumina).
Three male H. macropterus juveniles were injected twice with phytohemagglutinin (PHA) at 10 μg/g body weight with a 12 h interval, injected with colchicine at 10 μg/g body weight for 3 h, and anaesthetised using MS-222. Kidney cells were collected by hypotonic and fixation treatments as previously described (Zhu and Gui, 2007) and the number of mitotic metaphase chromosomes was counted in 100 cells.
Illumina clean short reads were used to estimate genome size and heterozygosity based on k-mer frequency distribution analysis using Jellyfish (Marçais and Kingsford, 2011). The PacBio long reads were assembled de novo into contigs using Hifiasm (Cheng et al., 2021), and then polished using Illumina short reads and NextPolish (Hu et al., 2019). For chromosome-level assembly of the H. macropterus genome, clean Hi-C reads were mapped to the primary genome using Bowtie2 (Langmead and Salzberg, 2012). HiC-Pro was used to validate interacting paired reads (Servant et al., 2015). Primary assembly scaffolds were oriented, ordered, and clustered on pseudochromosomes using LACHESIS (Korbel and Lee, 2013). JuiceBox was used to adjust the placement and orientation errors (Durand et al., 2016), and a Hi-C heat map was constructed.
Two strategies were used to assess genome completeness. The BUSCO completeness score of the assembled genome was evaluated using the Actinopterygii database (Simão et al., 2015), and RNA-seq data were mapped back to the genome using HISAT2 with default settings (Kim et al., 2015).
Repetitive elements of the H. macropterus genome were annotated using both homology and de novo strategies. According to the structural features, tandem and simple sequence repeats were predicted using TRF (Benson, 1999) and MISA (Beier et al., 2017), respectively, with default parameters. The transposable elements were identified using LTR_Finder (Ou and Jiang, 2019), LTRharverst (Ellinghaus et al., 2008), and LTR_retriver (Ou and Jiang, 2017). De novo annotation of other repeat sequences was performed using RepeatModeler (Price et al., 2005), followed by genome-scale detection using RepeatMasker (Chen, 2004). The combined results of these two predictions provided the final annotation of the non-redundant repeat elements in the genome.
We combined de novo, homology, and transcriptome-based methods to predict protein-coding genes. For de novo prediction, we used Augustus (Keller et al., 2011), GlimmerHMM (Majoros et al., 2004), and Geneid (Blanco et al., 2007) with their default parameters. Protein sequences of D. rerio, E. electricus, Esox lucius, Gadus morhua, H. wyckioides, Silurus meridionalis, P. fulvidraco, and Takifugu rubripes were aligned to the genome of H. macropterus using TBLASTN (Camacho et al., 2009). GeneWise (Birney et al., 2004) was used to predict the gene structure according to homology alignments. For transcriptome-based prediction, protein-coding regions were identified by aligning the transcripts with the assembled genome using PASA (Haas et al., 2003). Transposons were removed using TransposonPSI, and the final non-redundant reference gene set was obtained using EVidenceModeler (Haas et al., 2008).
For the functional annotation, the gene set was aligned to proteins deposited in the SwissProt and NCBI non-redundant protein databases using BLASTP. KEGG pathways were annotated by the KEGG Automatic Annotation Server (Moriya et al., 2007). Gene Ontology and protein domains were identified using InterProScan (Jones et al., 2014) with default parameters.
Orthologous gene families were identified by comparing the predicted protein sequences of H. macropterus with those of 16 other fish using OrthoMCL (Li et al., 2003). Single-copy gene orthogroups among these species were selected and aligned using MAFFT V7 (Yamada et al., 2016). A maximum likelihood phylogenetic tree was constructed using RAXML7 (Stamatakis, 2015) with 1,000 bootstrap replicates. Divergence time was estimated using MCMCTREE in PAML4 (Yang, 2007), and calibrated using fossil divergence times from the TimeTree database (http://www.timetree.org/). Based on the results of OrthoMCL, expanded and contracted gene families were analyzed via CAFE (De Bie et al., 2006), and functional enrichment analysis was performed by alignment homologues against the KEGG pathway database.
To investigate the chromosomal syntenic relationships among H. macropterus, H. wyckioides, and P. fulvidraco, MCscan (Tang et al., 2008) was applied to determine the syntenic blocks. Proteomes were compared between the pairs of species using BLASTP with an e-value of 1e-5, and a minimum of four genes in each block were used for synteny calling.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
The animal studies were approved by the Animal Experimental Ethical Inspection of Laboratory Animal Centre, Yangtze River Fisheries Research Institute, Chinese Academy of Fishery Sciences. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent was obtained from the owners for the participation of their animals in this study.
HYe: Conceptualization, Data curation, Investigation, Methodology, Resources, Software, Validation, Visualization, Writing–original draft, Writing–review and editing. JF: Data curation, Investigation, Methodology, Validation, Writing–original draft. YH: Data curation, Investigation, Validation, Visualization, Writing–original draft. HYu: Investigation, Methodology, Writing–original draft. RR: Writing–original draft. SL: Data curation, Resources, Writing–original draft. CH: Resources, Writing–original draft. YX: Conceptualization, Funding acquisition, Resources, Supervision, Writing–original draft. CL: Conceptualization, Writing–original draft, Writing–review and editing.
The authors declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by the National Key R&D Program of China (2022YFD2400101); Chongqing Science and Technology Bureau (cstc2021jxjl80023); and the Science and Technology Project of Guizhou Province, China [(2020)4Y027].
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2023.1297119/full#supplementary-material
SUPPLEMENTARY FIGURE S1 | Comparison of the number of homologous genes present in the genomes of Hemibagrus macropterus and 16 other fish species.
SUPPLEMENTARY FIGURE S2 | Expansion and contraction of Hemibagrus macropterus gene families. The number and sector with green, red, and blue represent the specific values of the gene families of expansion, contraction, and stability for each species, respectively. MRCA, most recent common ancestor.
Bao, L., Tian, C., Liu, S., Zhang, Y., Elaswad, A., Yuan, Z., et al. (2019). The Y chromosome sequence of the channel catfish suggests novel sex determination mechanisms in teleost fish. BMC Biol. 17 (1), 6. doi:10.1186/s12915-019-0627-7
Beier, S., Thiel, T., Münch, T., Scholz, U., and Mascher, M. (2017). MISA-web: a web server for microsatellite prediction. Bioinformatics 33 (16), 2583–2585. doi:10.1093/bioinformatics/btx198
Benson, G. (1999). Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27 (2), 573–580. doi:10.1093/nar/27.2.573
Birney, E., Clamp, M., and Durbin, R. (2004). GeneWise and genomewise. Genome Res. 14 (5), 988–995. doi:10.1101/gr.1865504
Blanco, E., Parra, G., and Guigó, R. (2007). Using geneid to identify genes. Curr. Protoc. Bioinform. 18 (1), Unit 4.3–4.3.28. doi:10.1002/0471250953.bi0403s18
Camacho, C., Coulouris, G., Avagyan, V., Ma, N., Papadopoulos, J., Bealer, K., et al. (2009). BLAST+: architecture and applications. BMC Bioinforma. 10 (1), 421. doi:10.1186/1471-2105-10-421
Chen, N. (2004). Using RepeatMasker to identify repetitive elements in genomic sequences. Curr. Protoc. Bioinform. 5 (1), Unit 4.10–14.10.14. doi:10.1002/0471250953.bi0410s05
Cheng, H., Concepcion, G. T., Feng, X., Zhang, H., and Li, H. (2021). Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat. Methods 18 (2), 170–175. doi:10.1038/s41592-020-01056-5
De Bie, T., Cristianini, N., Demuth, J. P., and Hahn, M. W. (2006). CAFE: a computational tool for the study of gene family evolution. Bioinformatics 22 (10), 1269–1271. doi:10.1093/bioinformatics/btl097
Durand, N. C., Robinson, J. T., Shamim, M. S., Machol, I., Mesirov, J. P., Lander, E. S., et al. (2016). Juicebox provides a visualization system for Hi-C contact maps with unlimited zoom. Cell Sys 3 (1), 99–101. doi:10.1016/j.cels.2015.07.012
Ellinghaus, D., Kurtz, S., and Willhoeft, U. (2008). LTRharvest, an efficient and flexible software for de novo detection of LTR retrotransposons. BMC Bioinforma. 9 (1), 18. doi:10.1186/1471-2105-9-18
Fricke, R., Eschmeyer, W. N., and Van der Laan, R. (2022). Eschmeyer’s catalog of fishes: genera, species, references. Available at: http://researcharchive.calacademy.org/research/ichthyology/catalog/fishcatmain.asp.Electronic version (Accessed January 23, 2023).
Gao, Z., You, X., Zhang, X., Chen, J., Xu, T., Huang, Y., et al. (2021). A chromosome-level genome assembly of the striped catfish (Pangasianodon hypophthalmus). Genomics 113 (5), 3349–3356. doi:10.1016/j.ygeno.2021.07.026
Gisbert, E., Luz, R. K., Fernández, I., Pradhan, P. K., Salhi, M., Mozanzadeh, M. T., et al. (2022). Development, nutrition, and rearing practices of relevant catfish species (Siluriformes) at early stages. Rev. Aquac. 14 (1), 73–105. doi:10.1111/raq.12586
Gong, G., Dan, C., Xiao, S., Guo, W., Huang, P., Xiong, Y., et al. (2018). Chromosomal-level assembly of yellow catfish genome using third-generation DNA sequencing and Hi-C analysis. GigaScience 7 (11), giy120. doi:10.1093/gigascience/giy120
Gong, G., Xiong, Y., Xiao, S., Li, X.-Y., Huang, P., Liao, Q., et al. (2022). Origin and chromatin remodeling of young X/Y sex chromosomes in catfish with sexual plasticity. Natl. Sci. Rev. 10 (2), nwac239. doi:10.1093/nsr/nwac239
Haas, B. J., Delcher, A. L., Mount, S. M., Wortman, J. R., Smith, R. K., Hannick, L. I., et al. (2003). Improving the Arabidopsis genome annotation using maximal transcript alignment assemblies. Nucleic Acids Res. 31 (19), 5654–5666. doi:10.1093/nar/gkg770
Haas, B. J., Salzberg, S. L., Zhu, W., Pertea, M., Allen, J. E., Orvis, J., et al. (2008). Automated eukaryotic gene structure annotation using EVidenceModeler and the program to assemble spliced alignments. Genome Biol. 9 (1), R7. doi:10.1186/gb-2008-9-1-r7
He, W. P., Zhou, J., Li, Z., Jing, T. S., Li, C. H., Yang, Y. J., et al. (2021). Chromosome-level genome assembly of the Chinese longsnout catfish Leiocassis longirostris. Zool. Res. 42 (4), 417–422. doi:10.24272/j.issn.2095-8137.2020.327
Hong, Y. H., and Zhou, T. (1984). Karyotype of nine species of Chinese catfishes (Bagridae). Zool. Res. 5, 21–28. (in Chinese).
Hu, J., Fan, J., Sun, Z., and Liu, S. (2019). NextPolish: a fast and efficient genome polishing tool for long-read assembly. Bioinformatics 36 (7), 2253–2255. doi:10.1093/bioinformatics/btz891
Jiang, W., Lv, Y., Cheng, L., Yang, K., Bian, C., Wang, X., et al. (2019). Whole-genome sequencing of the giant devil catfish, Bagarius yarrelli. Genome Biol. Evol. 11 (8), 2071–2077. doi:10.1093/gbe/evz143
Jones, P., Binns, D., Chang, H. Y., Fraser, M., Li, W., McAnulla, C., et al. (2014). InterProScan 5: genome-scale protein function classification. Bioinformatics 30 (9), 1236–1240. doi:10.1093/bioinformatics/btu031
Keller, O., Kollmar, M., Stanke, M., and Waack, S. (2011). A novel hybrid gene prediction method employing protein multiple sequence alignments. Bioinformatics 27 (6), 757–763. doi:10.1093/bioinformatics/btr010
Kim, D., Langmead, B., and Salzberg, S. L. (2015). HISAT: a fast spliced aligner with low memory requirements. Nat. Methods 12 (4), 357–360. doi:10.1038/nmeth.3317
Korbel, J. O., and Lee, C. (2013). Genome assembly and haplotyping with Hi-C. Nat. Biotechnol. 31 (12), 1099–1101. doi:10.1038/nbt.2764
Langmead, B., and Salzberg, S. L. (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9 (4), 357–359. doi:10.1038/nmeth.1923
Lemopoulos, A., and Montoya-Burgos, J. I. (2022). Whole genome assembly of the armored loricariid catfish Ancistrus triradiatus highlights herbivory signatures. Mol. Genet. Genomics 297 (6), 1627–1642. doi:10.1007/s00438-022-01947-6
Li, L., Stoeckert, C. J., and Roos, D. S. (2003). OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 13 (9), 2178–2189. doi:10.1101/gr.1224503
Liu, Z., Liu, S., Yao, J., Bao, L., Zhang, J., Li, Y., et al. (2016). The channel catfish genome sequence provides insights into the evolution of scale formation in teleosts. Nat. Commun. 7 (1), 11757. doi:10.1038/ncomms11757
Ma, Y. G., Zhang, F., and Yuan, W. A. (2013). Analysis the karotype of Mystus macropterus bleeker in the jialin river. Mod. Agr. Sci. Technol. 2, 260–261. (in Chinese).
Majoros, W. H., Pertea, M., and Salzberg, S. L. (2004). TigrScan and GlimmerHMM: two open source ab initio eukaryotic gene-finders. Bioinformatics 20 (16), 2878–2879. doi:10.1093/bioinformatics/bth315
Marçais, G., and Kingsford, C. (2011). A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 27 (6), 764–770. doi:10.1093/bioinformatics/btr011
Moriya, Y., Itoh, M., Okuda, S., Yoshizawa, A. C., and Kanehisa, M. (2007). KAAS: an automatic genome annotation and pathway reconstruction server. Nucleic Acids Res. 35 (2), W182–W185. doi:10.1093/nar/gkm321
Ou, S., and Jiang, N. (2017). LTR_retriever: a highly accurate and sensitive program for identification of long terminal repeat retrotransposons. Plant Physiol. 176 (2), 1410–1422. doi:10.1104/pp.17.01310
Ou, S., and Jiang, N. (2019). LTR_FINDER_parallel: parallelization of LTR_FINDER enabling rapid identification of long terminal repeat retrotransposons. Mob. DNA 10 (1), 48. doi:10.1186/s13100-019-0193-0
Price, A. L., Jones, N. C., and Pevzner, P. A. (2005). De novo identification of repeat families in large genomes. Bioinformatics 21 (1), i351–i358. doi:10.1093/bioinformatics/bti1018
Servant, N., Varoquaux, N., Lajoie, B. R., Viara, E., Chen, C. J., Vert, J. P., et al. (2015). HiC-Pro: an optimized and flexible pipeline for Hi-C data processing. Genome Biol. 16 (1), 259. doi:10.1186/s13059-015-0831-x
Shao, F., Pan, H., Li, P., Ni, L., Xu, Y., and Peng, Z. (2021). Chromosome-level genome assembly of the Asian red-tail catfish (Hemibagrus wyckioides). Front. Genet. 12, 747684. doi:10.3389/fgene.2021.747684
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V., and Zdobnov, E. M. (2015). BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31 (19), 3210–3212. doi:10.1093/bioinformatics/btv351
Stamatakis, A. (2015). Using RAxML to infer phylogenies. Curr. Protoc. Bioinform. 51 (1), 6.14.11–6. doi:10.1002/0471250953.bi0614s51
Tang, H., Wang, X., Bowers, J. E., Ming, R., Alam, M., and Paterson, A. H. (2008). Unraveling ancient hexaploidy through multiply-aligned angiosperm gene maps. Genome Res. 18 (12), 1944–1954. doi:10.1101/gr.080978.108
Wang, H., Su, B., Butts, I. A. E., Dunham, R. A., and Wang, X. (2022). Chromosome-level assembly and annotation of the blue catfish Ictalurus furcatus, an aquaculture species for hybrid catfish reproduction, epigenetics, and heterosis studies. GigaScience 11, giac070. doi:10.1093/gigascience/giac070
Xu, Y., Shao, F., Chen, W., Ni, L., and Peng, Z. (2022). A chromosome-level genome of the helmet catfish (Cranoglanis bouderius). Front. Genet. 13, 962406. doi:10.3389/fgene.2022.962406
Yamada, K. D., Tomii, K., and Katoh, K. (2016). Application of the MAFFT sequence alignment program to large data-reexamination of the usefulness of chained guide trees. Bioinformatics 32 (21), 3246–3251. doi:10.1093/bioinformatics/btw412
Yang, Z. (2007). PAML 4: phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 24 (8), 1586–1591. doi:10.1093/molbev/msm088
Zhang, M., Liu, W., and Li, G. (2009). Isolation and characterisation of collagens from the skin of largefin longbarbel catfish (Mystus macropterus). Food Chem. 115 (3), 826–831. doi:10.1016/j.foodchem.2009.01.006
Zheng, S., Shao, F., Tao, W., Liu, Z., Long, J., Wang, X., et al. (2021). Chromosome-level assembly of southern catfish (silurus meridionalis) provides insights into visual adaptation to nocturnal and benthic lifestyles. Mol. Ecol. Resour. 21 (5), 1575–1592. doi:10.1111/1755-0998.13338
Zhou, Y. L., Wu, J. J., Gong, G. R., Liu, M., Li, Z., Guo, X. F., et al. (2023). Barbel regeneration and function divergence in red-tail catfish (Hemibagrus wyckioides) based on the chromosome-level genomes and comparative transcriptomes. Int. J. Biol. Macromol. 232, 123374. doi:10.1016/j.ijbiomac.2023.123374
Zhu, H. P., and Gui, J. F. (2007). Identification of genome organization in the unusual allotetraploid form of Carassius auratus gibelio. Aquaculture 265 (1), 109–117. doi:10.1016/j.aquaculture.2006.10.026
Zhu, C., Pan, Z., Yao, Y., Wu, D., Yuan, M., Gan, L., et al. (2021). Progress of karyotype studies on Siluriformes in China. J. Fish. China 46 (4), 657–663. (in Chinese). doi:10.12122/j.issn.1673-4254.2021.05.04
Zhu, C. K., Zheng, Y. H., Gao, Y. Y., and Li, Y. J. (2007). Biological characteristics and prospects for captive aquaculture of the Mystus macropterus. Reserv. Fish. 153, 86–89. (in Chinese).
Keywords: Hemibagrus macropterus, genome assembly, genome annotation, comparative genomics, Hi-C, PacBio
Citation: Ye H, Fan J, Hou Y, Yue H, Ruan R, Li S, Hu C, Xie Y and Li C (2023) Chromosome-level genome assembly of the largefin longbarbel catfish (Hemibagrus macropterus). Front. Genet. 14:1297119. doi: 10.3389/fgene.2023.1297119
Received: 19 September 2023; Accepted: 23 October 2023;
Published: 01 November 2023.
Edited by:
Lucas Lima Verardo, Universidade Federal dos Vales do Jequitinhonha e Mucuri (UFVJM), BrazilReviewed by:
Mingzheng Duan, Zhaotong University, ChinaCopyright © 2023 Ye, Fan, Hou, Yue, Ruan, Li, Hu, Xie and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yong Xie, Nzk5MDkxMTQyQHFxLmNvbQ==; Chuangju Li, bGNqQHlmaS5hYy5jbg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.