 Abdallah Alhaj Sulaiman
Abdallah Alhaj Sulaiman Dana E. Al-Ansari1,2
Dana E. Al-Ansari1,2 Reem Ali
Reem Ali Mustapha Aouida
Mustapha Aouida Dindial Ramotar
Dindial Ramotar- 1Qatar Foundation, Division of Biological and Biomedical Sciences, College of Health and Life Sciences, Hamad Bin Khalifa University, Doha, Qatar
- 2National Heart and Lung Institute, Imperial College London, London, United Kingdom
Quinoxaline is a heterocyclic compound with a two-membered ring structure that undergoes redox cycling to produce toxic free radicals. It has antiviral, antibacterial, antifungal, and antitumor activities. However, the biological functions that are involved in mounting a response against the toxic effects of quinoxaline have not been investigated. Herein, we performed a genome-wide screen using the yeast haploid mutant collection and reported the identification of 12 mutants that displayed varying sensitivity towards quinoxaline. No mutant was recovered that showed resistance to quinoxaline. The quinoxaline-sensitive mutants were deleted for genes that encode cell cycle function, as well as genes that belong to other physiological pathways such as the vacuolar detoxification process. Three of the highly sensitive gene-deletion mutants lack the DDC1, DUN1, and MFT1 genes. While Ddc1 and Dun1 are known to perform roles in the cell cycle arrest pathway, the role of Mft1 remains unclear. We show that the mft1Δ mutant is as sensitive to quinoxaline as the ddc1Δ mutant. However, the double mutant ddc1Δ mft1Δ lacking the DDC1 and MFT1 genes, is extremely sensitive to quinoxaline, as compared to the ddc1Δ and mft1Δ single mutants. We further show that the mft1Δ mutant is unable to arrest in the G2/M phase in response to the drug. We conclude that Mft1 performs a unique function independent of Ddc1 in the cell cycle arrest pathway in response to quinoxaline exposure. This is the first demonstration that quinoxaline exerts its toxic effect likely by inducing oxidative DNA damage causing cell cycle arrest. We suggest that clinical applications of quinoxaline and its derivatives should entail targeting cancer cells with defective cell cycle arrest.
Introduction
Quinoxaline (QXN) is an organic heterocyclic compound from the class diazines, with the chemical formula (C8H6N2) (see structure Supplementary Figure S1A). QXN is prepared via a condensation reaction of 1,2-diaminobenzene with a 1,2-dicarbonyl compound (Pereira et al., 2015; Chauhan et al., 2020). The derivatives of QXN are produced through different reactions that include oxidation, reduction, and nitration, as well as catalytic and non-catalytic reactions (Sato and Katritzky, 2008; Buravchenko and Shchekotikhin, 2023). QXN and its derivatives have various biological activities such as anti-fungal, anti-viral and anti-bacterial (Chandrasekhar et al., 2010; Patel et al., 2016; Montana et al., 2020). These molecules also exhibit pharmacological applications in neurological disorders, cardiovascular diseases, diabetes and cancer (Heravi et al., 2009; El Newahie et al., 2019). QXN also exhibits anti-inflammatory characteristics as its derivatives can inhibit the production of pro-inflammatory cytokines and other molecules involved in the inflammatory response, making the QXN family of drugs potential candidates for the treatment of inflammatory diseases (Tariq et al., 2018). Despite the pharmacological potential and its wide applications for more than half a century, the exact underlying molecular mechanism of QXN biological activity is still unknown. Nonetheless, a very early study hinted that QXN derivatives can damage DNA (Suter et al., 1978).
It was several decades later that Cheng G. et al. (2015) used a derivative of QXN, quinoxaline 1, 4-di-N-oxides, to understand how this family of drugs induced its antibacterial effect on Escherichia coli (Cheng et al., 2015). The study revealed that the drug primarily induces the bacterial SOS response, that is, the induction of at least 20 genes encoding proteins involved in DNA damage and repair such as recA, uvrA, B, and umuD, C, as well as attenuating the cell division and cause oxidative stress (Cheng et al., 2015). The evidence revealed that upon entry the drug is redox activated and then diminished the viability of E. coli by causing oxidative DNA damage via the elevation of reactive oxygen species (Cheng et al., 2015). Thus, enhancing the effectiveness of these quinoxaline derivatives would imply targeting the DNA damage response systems such as preventing cell cycle arrest so that the cells are unable to effectively commit to repairing damaged DNA and hence accumulate unrepaired DNA lesions triggering cell death. Olaquindox, another derivative of QXN that is used as a livestock feed additive, has also been shown to induce the production of reactive oxygen species in human HepG2 cells causing oxidative DNA damage and arresting the cells in the S-phase (Li et al., 2017). Other QXN derivatives such as triazole-linked quinoxalines can recognize and bind to telomeric G-quadruplexes leading to the inhibition of breast cancer cell growth by arresting the cells at the G0/G1 phase (Hu and Lin, 2023).
One approach to understanding the mechanism of action of QXN is to take advantage of the available tools in the budding yeast Saccharomyces cerevisiae (Sulaiman et al., 2022; Mohanty et al., 2023; Sulaiman et al., 2023). These include a collection of ∼4,800 haploid mutants, each deleted for a single non-essential gene. The haploid mutant collection can be used for different applications such as understanding biological processes, response to environmental stresses, as well as understanding molecular mechanisms of drug actions (Aouida et al., 2004). In this study, we used the haploid mutant collection to identify gene-deletion mutants that would cause either resistance or sensitivity to QXN. We first used the parent strain of the mutant collection to establish the concentration of QXN that would kill 50% of the cells (IC50) and used a range of QXN concentrations to identify mutants from the collection that are either resistant or sensitive to the drug. Herein, we reported the isolation of 12 mutants from the entire collection that exhibited varying sensitivities to QXN, and none displaying resistance to the drug. Two of the QXN-hypersensitive mutants, deleted for the DUN1 and the DDC1 gene, were defective in the DNA damage response and the cell cycle arrest pathways, respectively, suggesting that QXN acts by damaging the DNA and leading to cell cycle arrest. Another QXN-hypersensitive mutant is defective for the MTF1 gene. MTF1 encodes a protein that belongs to the THO complex involved in RNA transaction, but has not been previously shown to play a role in the cell cycle arrest pathway. We showed that the mft1Δ mutants are unable to properly arrest cells in the S phase and lack the G2/M arrest in response to QXN. Moreover, when deleted in the ddc1Δ caused the resulting double mutant, ddc1Δ mft1Δ to be sensitized to the drug. The data imply that Mft1 has a function that is distinct from the Ddc1 protein in the cell cycle arrest pathway in response to QXN exposure. This is the first report demonstrating that the toxic effects induced by QXN trigger cell cycle arrest. Thus, cancer cells with defective cell cycle arrest are expected to be hypersensitive to QXN.
Materials and methods
Yeast strains and media
BY4741 wild-type strain was the background of the gene deletion library, which was obtained from EUROSCARF (Frankfurt, Germany) (Giaever et al., 2002). Regular YPD (Yeast extract-peptone-dextrose) media from FORMEDIUM CCM0105 or SD minimal media were used to grow the strains. Quinoxaline 99% was purchased from Sigma-Aldrich with a catalogue number of Q1603, dissolved in sterile water, and kept as 1 M stock solution at–20°C. The 1 M stock solution of QXN can be stored for up to 1 year without losing efficacy. Hydroxyurea was purchased from Sigma-Aldrich, dissolved in sterile distilled water, filter sterilized and stored at–20°C as a 2 M stock.
Spot test analysis to establish the sensitivity of strain BY4741 to QXN
The parent strain BY4741 was grown overnight in YPD broth at 30°C, and then the OD600 was adjusted to 1.0. The stock was then serially diluted to 1:10, 1:20, 1:50, and 1:100 and then spotted onto YPD plates without and with different QXN concentrations of 9.5, 11.5, 13.5, and 15.5 mM. The plates were incubated at 30°C for 48 h, and the Image Lab Touch Software (BioRad) was used to document the results.
Survival assay
To assess the survival of S. cerevisiae in response to QXN and determine the lethal concentration at 50% (IC50). The parent strain BY4741 was grown overnight, and then the OD600 was adjusted to 0.6. Yeast cells were treated with increasing concentrations of QXN for a period of 4 h, then spotted onto YPD plate and incubated for 48 h at 30°C. The colonies were counted using ImageJ software, and the survival plot was prepared using GraphPad Prism.
High-throughput QXN screen
The yeast haploid mutant collection strains were arrayed as previously described (Aouida et al., 2004), except for using a 96-floating pin replicator operated by ROTOR HDA (Singer, United Kingdom). A total of 96 colonies were arrayed per solid YPD plate without or with the indicated concentration of QXN. The ROTOR HDA was set to pin each mutant once from the master plate onto the drug plate and ensure that the mutant was deposited onto the plates by using five pinnings on the same spot. The pin was changed between each drug concentration to prevent the drug from contaminating the master plate. The plates were incubated for 48 h at 30°C and photographed with a digital camera (Bio-Rad Chemi-Doc MP Imaging system 2000) to visually compare the growth of every mutant in the presence or absence of QXN (see illustration in Supplementary Figure S3).
Spot test analysis to verify the QXN hypersensitive strains
The QXN sensitive strains were precultured in YPD media for 24 h at 30°C. The OD600 was adjusted to 1.0 and serially diluted to 1:10, 1:50: 1:100: 1:500, and 1:1,000, and 5 µL of each dilution was spotted onto YPD agar plates without drug zero, and with 5.0, 6.35, 7.62, 9.0, and 10.0 mM of QXN. The plates were incubated at 30°C for 48 h, and results were documented using the Bio-Rad Chemi-Doc MP Imaging system 2000.
Mutant construction
The strains were constructed using the one-step gene targeting method as described previously by Baker Brachmann et al. (1998) using the following primers:
MFT1-F: 5′ATGCCTCTGTCACAAAAACAAATAGACCAAGTTAGAACCAAAGTGCAC TAAGATTGTACTGAGAGTGCAC 3′
MFT1-R:5′TCATTTTACTTCTTCAACAGAGGAAGACGCACTAAAATCGCTCTGTGCG GTATTTCACACCGC 3′
The verification primers are as follows:
MFT1 Verifications F: 5′ CAAGGCCGAGATACATAGTTCCGCGG 3′
MFT1 Verifications R: 5′ AAGGTGACGCACCATACAGAACTTGCTC 3′. Briefly, the MFT1-F and MFT1-R primers bearing the universal sequence were used for PCR amplification from the pRS306 plasmid the URA3 gene (Baker et al. #15). The resulting PCR fragment was introduced into the parent strain BY4741 and the isogenic ddc1Δ::KANMX strain by the standard lithium acetate method and selected for Ura + colonies. Ura + colonies carrying the mft1Δ::URA3 were verified using the verification primers.
QXN and HU sensitivities of the single and double mutant
For the sensitivities towards QXN, spot test (as described above) and growth curve assays were performed. For the growth curve, the cells were cultured overnight in YPD media then the OD600 was adjusted to ∼0.2 with fresh YPD media in a 96-well plate. Drugs were added to the cells at the indicated concentrations and growth was monitored for 24 h in a TECAN plate reader with shaking. The temperature was set to 30°C and the OD was taken every 2 h. The result was plotted as a graph of OD600 against the time (Sulaiman et al., 2022). For HU sensitivity, the spot test was performed as above. The experiments represent three independent repeats.
Cell cycle analysis
The cell cycle analysis was performed following the protocol described by Haase and Reed, 2002; Haase (2003). Briefly, strains were grown overnight. The following day, cultures were adjusted to an optical density (OD 600) of ∼1.0 and treated with QXN (9 mM) for 4 h or left untreated. The cells were washed and left to recover for 0, 30, 60, and 120 min. Post-treatment cells were washed with distilled H2O twice and centrifuged. Pellets were resuspended in 70% ethanol for 30 min for fixation. The cells were washed and treated with 100 μg/mL RNase at 37°C for 16 h. The following day, the cells were washed and resuspended in 1 μg/μL proteinase K at 50°C for 30 min. The cells were washed with PBS and stained with 2.5 μg/mL propidium iodide for 20 min and analyzed on BD Fortessa X-20 flow cytometer. The data were analyzed by FlowJo software.
Results
Determination of QXN drug concentration that inhibits the growth of the wild-type yeast strain BY4741
We first determined the effective concentration of QXN that would kill 50% (IC50) of the parental strain BY4741 that was used for creating the haploid gene-deletion mutant collection. Briefly, the BY4741 strain was grown overnight, OD 600 was adjusted to 1.0 followed by acute treatment for 4 h with increasing concentrations of QXN in liquid culture, and allowed to grow overnight in fresh media. The final OD 600 was measured and plotted against QXN concentrations. The result indicated that the growth of the cells was severely impeded by increasing concentrations of QXN, showing a significant decrease at 11.5 mM and more drastically at 13.5 mM (Supplementary Figure S1B). The QXN IC50 for the BY4741 strain was determined to be ∼12 mM. In a separate experiment, the cells were adjusted to OD 600 of ∼0.2 in liquid YPD and incubated in the absence and presence of increasing concentrations of QXN. The final OD 600 was determined after 24 h of growth. Under this chronic drug treatment, the growth of the cells was significantly delayed (Supplementary Figure S1C). At least, 50% growth was observed at 16 h when the cells were exposed to 12.5 mM of QXN as compared to the control (Supplementary Figure S1C). Only slight growth of BY4741 was observed at 16 h with 15 mM QXN (Supplementary Figure S1C). Thus, the strain BY4741 is unable to tolerate the toxic effects of QXN concentrations greater than 15 mM.
We next determined the QXN concentrations that would effectively prevent the growth of strain BY4741 on solid YPD media to establish a suitable condition to perform the drug screening analysis of the haploid mutant collection. Briefly, four independent cultures of the BY4741 strain were grown overnight and the next day the OD 600 was adjusted to 1.0 before serially diluted and spotted onto plates with increasing concentrations of QXN (Supplementary Figure S2A). A 100-fold dilution of each culture grew on plates with 9.5 mM QXN, but very poorly on plates with 11.5 mM QXN, suggesting that this later dose is lethal to the strain, and mutants that are sensitive to the drug are not expected to grow at 9.5 mM QXN. Similarly, when cells of OD 600–1.0 were diluted 1000-fold and plated directly onto plates with increasing concentrations of QXN, the viability of the cells sharply decreased (Supplementary Figure S2B; and the quantification shown in Supplementary Figure S2C).
Isolation of QXN-sensitive strains from the yeast haploid mutant collection
The yeast haploid mutant collection consisted of 4,852 strains representing single gene deletion for nearly 4,000 genes (Aouida et al., 2004). Briefly, the mutant collection was allowed to grow overnight in YPD liquid culture in 96-well plates, and the strains were pinned onto YPD solid plates without and with increasing concentrations of QXN up to 19.2 mM, followed by growth for 48 h at 30°C before scoring for the most sensitive strains to the drug (see illustration in Supplementary Figure S3, and Materials and Methods). The entire haploid mutant collection was screened independently three times and from all the analyses, 12 mutants were found to be very sensitive to QXN at concentrations lower than 9.5 mM (Figure 1 and Supplementary Table S1). These mutants were retested and three gene-deletion mutants dun1Δ, ddc1Δ, and mft1Δ showed severe sensitivity to QXN. The DUN1 and DDC1 genes have been shown to encode proteins involved in cell cycle function, while MFT1 encodes a protein that belongs to the THO complex involved in transcription (Supplementary Table S1). Two of the sensitive strains were deleted for genes with no known biological function (Figure 1 and Supplementary Table S1), and the other mutants are defective in physiological pathways that affect, for example, vacuolar function (Cohen et al., 2003). The sensitivities of these 12 mutants were independently confirmed using spot test analysis (Figure 2), and the data were consistent with the initial screen performed by the robot.

FIGURE 1. Identification of the QXN-sensitive yeast mutants from the haploid mutant collection. Briefly, frozen stocks of the haploid collection were allowed to grow overnight in YPD media and subjected to the robotic screen as outlined in the illustration shown in Supplementary Figure S3. Each spot represents a mutant from the collection that was spotted on solid YPD plates without and with 5 mM of QXN. Following 2 days of growth at 30°C, the plates were photographed. The opened square highlighted the mutants (e.g., ddc1Δ and mft1Δ) that are sensitive to QXN and the arrows point to the assigned gene name from the Saccharomyces Genome Database. The data are representative of three independent High-Throughput screens.

FIGURE 2. Spot test analysis confirms the sensitivity of the mutants to QXN. Cells were grown overnight, serially diluted and spotted onto plates with the indicated concentrations of QXN. The plates shown in panels (A–C) were photographed after 48 h incubation at 30°C. The QXN-sensitive strains are compared to the parent strain BY4741. The experiment was independently repeated at least three times.
ddc1Δ and mft1Δ function in separate pathways
Since the dun1Δ, ddc1Δ, and mft1Δ mutants were the most sensitive to QXN, besides the two mutants with unknown functions, and Dun1 and Ddc1 are involved in cell cycle arrest, we examined whether Mft1 would be implicated in the cell cycle process, as such role has not been assigned to this protein. We first independently examined whether recreating the mft1Δ mutant would result in sensitivity to QXN. Indeed, deletion of the MFT1 gene in the parent strain BY4741, using one-step gene targeting and confirmed by PCR analysis, resulted in new mft1Δ mutants, which showed nearly the same level of sensitivity to QXN as the screened mutant (Figure 3A). We next checked whether Mft1 and Ddc1 belong to the same pathway. As such, the MFT1 gene was deleted in the single ddc1Δ mutant to produce the double deletion ddc1Δ mft1Δ mutant. We tested the sensitivity of the single mutant against the double mutant and the result showed that the single mft1Δ and the single ddc1Δ mutants were as sensitive to QXN as compared to the parent strain BY4741 (Figure 3A). In contrast, the ddc1Δ mft1Δ double mutant was extremely sensitive to QXN (Figure 3A). Consistent with this observation, the mft1Δ and the ddc1Δ single mutants showed the same extent of growth inhibition in liquid culture in the presence of 5 mM QXN, while the double mutant did not grow, as compared to the parent and under no drug treatment (Figure 3D vs.Figure 3C). These findings strongly suggest that both Ddc1 and Mft1 function in separate pathways.
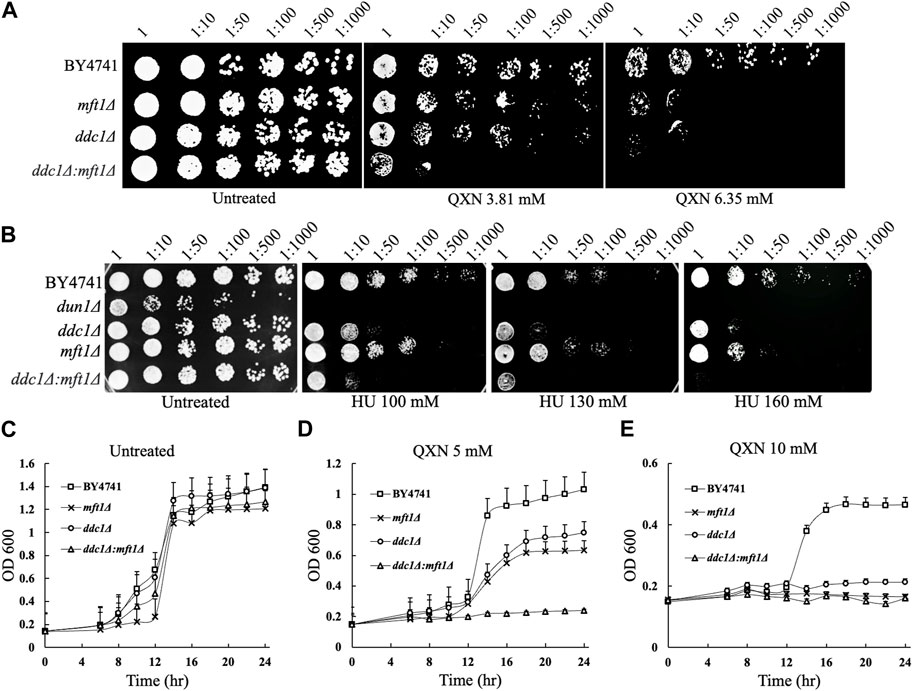
FIGURE 3. Comparison of the sensitivity of the parent, the single mutants mft1Δ and ddc1Δ and the double mutant ddc1Δ mft1Δ towards QXN and HU. (A,B), The indicated strains were grown overnight, serially diluted and spotted onto plates containing QXN and HU, respectively. The data is representative of two independent experiments. The plates were photographed after 48 h incubation at 30°C. BY4741 is the parent. (C–E), Cells from overnight cultures were adjusted to OD600 of ∼0.15 and allowed to grow in the absence (panel C) and presence (panels D and E) of the indicated QXN concentrations. The OD600 was taken at the indicated time and plotted. The results shown are the averages of three independent experiments and the error bars indicate the standard deviation.
The dun1Δ mutant is known to be defective in S phase arrest and this mutant is sensitive to hydroxyurea (HU) (Palou et al., 2010), an agent that inhibits the ribonucleotide reductase needed to maintain the pool of dNTP for DNA synthesis. Since the ddc1Δ and mft1Δ mutants showed nearly the same level of sensitivity to QXN as the dun1Δ single mutant (Figure 3A), we checked whether the ddc1Δ and mft1Δ mutants would be as sensitive to HU. While the dun1Δ mutant displayed extreme sensitivity to HU, the ddc1Δ and the mft1Δ single mutants were also sensitive to HU, but to a lesser extent (Figure 3B). However, the ddc1Δ mft1Δ double mutant was more sensitive to HU than either the ddc1Δ or the mft1Δ single mutant, suggesting that both Ddc1 and Mft1 contribute to the S phase arrest.
The mft1Δ mutant is defective in G2/M arrest in response to QXN exposure
We examined whether QXN would trigger cell cycle arrest in a specific phase in the parent cells and whether this arrest would be disrupted by the loss of Ddc1 or Mft1 or both. Briefly, the asynchronous population of the parent, the single mutants ddc1Δ and mft1Δ, and the double mutant ddc1Δ mft1Δ were untreated and treated for 4 h with QXN (9.5 mM). The cells were washed and allowed to recover in fresh media without QXN. Samples were taken at 0, 30, 60 and 120 min, and processed for cell cycle analysis using the Sytox Green dye (Haase, 2003). In response to QXN, and at 0 mins recovery time, the parent showed an increase in the number of cells in the G2/M phase as compared to the asynchronous population of untreated cells (Figure 4A). Following 30 min of recovery, the parent displayed an increase in the number of cells in S phase arrest and by 120 min resumed normal cycling (Figure 4A). These observations suggest that the dose of QXN is capable of inducing toxic DNA lesions thereby triggering cell cycle arrest in both the S and G2/M phases. The ddc1Δ mutant showed the same extent of G2/M arrest as the parent following treatment with QXN, and displayed no S phase arrest during the 30-min or 60-min recovery, but retained the G2/M arrest and by 120 min remedied the QXN-induced lesions (Figure 4C). Thus, it would appear that the ddc1Δ mutant lacks the S phase arrest required to process QXN-induced DNA lesions. In contrast, the mft1Δ mutant showed no major G2/M arrest upon treatment with QXN, as compared to the parent or the ddc1Δ mutant, but showed a delayed S phase arrest that appeared during the 60-min recovery phase (Figure 4B). The mft1Δ mutant failed to recover the normal cell cycle progression after 120 min, as compared to the parent (Figure 4B vs. Figure 4A). We interpret this observation to suggest that the mft1Δ mutant is defective in activating the G2/M phase arrest in response to QXN treatment. In the case of the ddc1Δ mft1Δ double mutant, it showed a synergistic effect in response to the QXN treatment with no S phase arrest at 30 min and a weakened G2/M arrest at 60 min, with most of the cells accumulating in the G1 phase (Figure 4D). Based on the above findings, it would appear that Mft1 plays a role in triggering the G2/M phase arrest upon exposure to QXN.
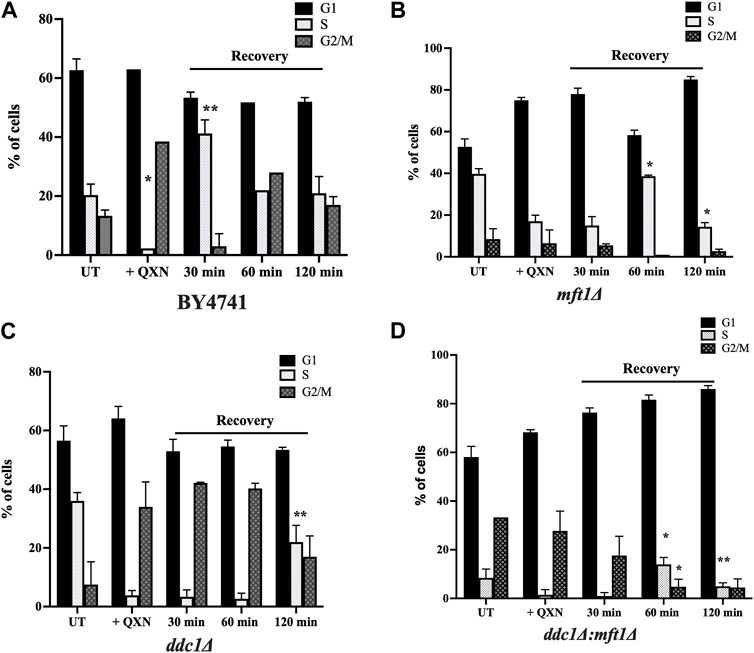
FIGURE 4. Cell cycle analysis of the parent and the indicated mutants in response to QXN and following recovery. The exponentially growing parent and mutant strains were untreated and treated with QXN (9 mM for 4 h), and the cells were washed and allowed to grow in fresh media without QXN. Samples were taken at 0, 30, 60 and 120 min as recovery time for FACS analysis as described in the Materials and Methods. (A–D), The percentage of cells in the indicated phases for the untreated, treated, and post-treatment (recovery) for the parent and the mutants. Error bars represent mean ± SD. *p < 0.05, **p < 0.01. See Supplementary Figure S4 for the graphic representation of the cell cycle profiles for the indicated strains.
Discussion
In this study, we identified 12 gene-deletion mutants from the S. cerevisiae haploid mutant collection that displayed varying sensitivity to QXN. However, we found none of the mutants in the entire collection to be resistant to the drug. It is possible that more than one gene might be involved in contributing to the QXN-resistance phenotype or such a resistant gene is essential for viability. Nonetheless, of the QXN-sensitive mutants, three of these (dun1Δ, ddc1Δ, and mft1Δ) displayed hypersensitivity to QXN, suggesting that the Dun1, Ddc1, and Mft1 proteins perform a major role in the response to the drug. Dun1 is a serine/threonine protein kinase that operates in the S phase checkpoint, for example, when the replication fork is obstructed. Dun1 also has a well-defined role in upregulating the levels of the dNTPs pool (Gershon and Kupiec, 2021). It induces transcription of the ribonucleotide reductase genes RNR2, RNR3 and RNR4 and triggers the cytoplasmic-nuclear redistribution of the ribonucleotide reductase Rnr2-Rnr4 upon genotoxic stress. Dun1 can phosphorylate several proteins such as the DNA repair protein Rad55, the superoxide dismutase Sod1, and the repressor Crt1 that inhibits ribonucleotide reductase gene expression as well as the inhibitor, Sml1, of Rnr1. In addition, Dun1 is required to regulate telomere length, as well as late firing of origins of replication (Gershon and Kupiec, 2021). Thus, the sensitivity of the dun1Δ mutant to QXN can be explained by a defect in the S-phase checkpoint, causing damaged DNA such as incomplete replicated DNA to enter the G2/M phase.
Ddc1 is a DNA damage checkpoint protein that inhibits the cell cycle in response to unrepaired DNA lesions (Hong and Roeder, 2002). The observation of the S phase arrest in the parent strain in response to QXN treatment, and not in the ddc1Δ mutant, is consistent with QXN causing replication intermediates requiring the function of Ddc1 to trigger S phase arrest. In the absence of Ddc1, the QXN-induced DNA lesions would be expected to activate the G2/M phase arrest as observed (Figure 4). The Ddc1 protein forms a heterotrimeric complex with Rad17 and Mec3 (Rad17-Mec3-Ddc1), also referred to as the yeast counterpart of the human 9-1-1 complex. The Rad17-Mec3-Ddc1 complex forms a ring structure, similar to the PCNA-like checkpoint complex, and plays a role in ensuring the integrity of the DNA before DNA synthesis or separation of the daughter chromosomes. This complex is loaded onto the site of DNA damage to form nuclear foci, aided by its interaction with the RAD24-RFC checkpoint clamp loader, which then activates the Mec1 kinase for cell cycle arrest during the G1 and G2/M phases (Liu, 2019). The activation of the Mec1 kinase requires the function of Ddc1, which has a C-terminal tail that is heavily phosphorylated, and implicated in the recruitment of various DNA damage response proteins. Thus, the defective S phase arrest observed by the ddc1Δ mutant in response to QXN could be explained by the failure of the mutant to activate the Mec1 kinase (Liu, 2019). Interestingly, while Ddc1 is involved in the interaction with Rad17, our screen did not detect the rad17Δ mutant as one that is sensitive to QXN or any member of the RAD24-RFC complex, suggesting that Ddc1 may perform a specific function in response to QXN. Based on our findings, we anticipate that the human counterpart, the 9-1-1 complex, of the yeast Rad17-Mec3-Ddc1 complex would play a pivotal role in QXN response.
In the case of Mft1, it has not been previously shown to be involved in cell cycle arrest, although little is known regarding its exact function. Mft1 belongs to a complex called THO that is involved in transcription elongation and mRNA export (Chávez et al., 2000). The THO complex consists of four proteins Hpr1, Mft1, Tho2 and Thp2. A defect in this THO complex impedes transcription such that it generates DNA-RNA hybrid (R loops), that is, displaced complementary single-stranded DNA, which can stimulate lethal transcription-associated recombination leading to genomic instability (Gómez-González et al., 2009). These R loops can perform specific functions such as DNA replication. However, R loops can cause DNA breaks leading to recombination and chromosomal rearrangements (García-Muse and Aguilera, 2019). Since the additional members of the THO complex were not recovered as QXN-sensitive mutants, it is reasonable to propose that Mft1 could have a distinct role in the response to QXN. The observation that the mft1Δ mutant is unable to efficiently arrest the cells in the S phase and showed no G2/M phase arrest, as compared to the parental strain in response to QXN, suggests that Mft1 performs a distinct role from Ddc1. We believe that Mft1 is required to trigger a G2/M phase arrest to allow the cells to process R-loop intermediates that might be created by QXN leading to DNA double-strand breaks. We conclude that Mft1 plays a major role in triggering G2/M phase arrest, while Ddc1 is required for activating the S phase arrest in response to QXN exposure. Thus, cells devoid of both Mft1 and Ddc1 are expected to be profoundly more sensitive to QXN, as opposed to the individual mutant (Figure 3D). So far, a counterpart of Mft1 does not appear to exist in human cells, although it is conserved in other yeast species indicating that Mft1 role may be specific for these fungi.
It is noteworthy that although our genome-wide screen identified other defective gene functions that are related to Golgi trafficking, vacuolar detoxification, and proteolysis, the major effects caused by QXN appear to involve processes that maintain the integrity of the DNA. As such, we believed that QXN could be an effective drug to target tumours that are defective in cell cycle arrest.
Scope statement
The drug quinoxaline and its derivatives have broad applications, in particular controlling bacterial, fungal and viral infections, as well as possessing antitumor activities. Quinoxaline can induce reactive oxygen species and is believed to damage various macromolecules including the DNA. However, the exact mechanism by which this drug causes toxicity is not known. We set out to search for molecular factors that would interfere with the responses of the budding yeast S. cerevisiae to quinoxaline. We exploited the library of yeast haploid gene-deletion mutants and identified at least 12 with varying sensitivity to quinoxaline. We showed that one of the hypersensitive mutants lacks the MFT1 gene with no previous role in quinoxaline response. We report for the first time that quinoxaline exerts its toxic effects by inducing cell cycle arrest and that Mft1 is required to mediate this process. The genetic characterization presented herein paves the way to determine precisely whether oxidative DNA lesions generated by reactive oxygen species produced by quinoxaline are responsible for cell cycle arrest. As such, any clinical application of quinoxaline and its derivatives should entail targeting cancer cells with defective cell cycle arrest.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding authors.
Author contributions
AAS: Data curation, Formal Analysis, Investigation, Methodology, Resources, Software, Validation, Visualization, Writing–original draft, Writing–review and editing. DA-A: Data curation, Formal Analysis, Funding acquisition, Investigation, Methodology, Resources, Validation, Visualization, Writing–review and editing. RA: Data curation, Formal Analysis, Investigation, Methodology, Resources, Software, Validation, Visualization, Writing–review and editing. MA: Conceptualization, Data curation, Formal Analysis, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing–original draft, Writing–review and editing. DR: Conceptualization, Data curation, Formal Analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Supervision, Validation, Visualization, Writing–original draft, Writing–review and editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. Qatar Foundation for the research funding to the College of Health and Life Sciences. Qatar National Research Fund for the Graduate Scholarship Research Award provided to DA-A (GSRA6-2-0603-19065).
Acknowledgments
We thank the College of Health and Life Sciences, Hamad Bin Khalifa University, for providing scholarships to AAS.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2023.1296383/full#supplementary-material
SUPPLEMENTARY FIGURE S1 | Growth rate analysis of strain BY4741 in response to acute and chronic exposure to quinoxaline (QXN). (A), Chemical Structure of quinoxaline. (B), Cells were grown overnight and the next day the OD600 was adjusted to OD 1.0 followed by acute treatment for 4 h with increasing concentrations of QXN in liquid culture. The cells were washed, diluted to OD600 of ∼0.2, and allowed to grow overnight in fresh media. The final OD600 after overnight growth was measured and plotted against QXN concentrations. (C), Cells were adjusted to OD600 of ∼0.2 in liquid YPD and incubated in the absence and presence of increasing concentrations of QXN for overnight treatment. The final OD600 was determined after 24 h of growth. The results shown in panels B and C are the averages of three independent experiments and the error bars indicate the standard deviation.
SUPPLEMENTARY FIGURE S2 | Determination of the QXN concentrations needed to perform the genome-wide screen. (A), Four independent colonies of strain BY4741 were grown overnight, serially diluted and spotted onto solid YPD agar plates containing the indicated concentrations of QXN ranging from 0 to 15.5 mM. The plates were incubated for 48 hrs at 30°C and the images were taken using Bio-Rad Chemi-Doc MP Imaging system. (B), Cells adjusted to OD 600 of ∼1.0, treated for 4 h with the indicated concentrations of QXN, diluted 1000-fold and plated directly onto plates without QXN. The surviving colonies were scored after 48 h of growth at 30°C. The colonies of the plated yeast cells were counted using Image J. Data are mean SEM of n=3, and significance is assumed at p* < 0.05. (C), Plot of the surviving fraction of the cells following treatment with QXN.
SUPPLEMENTARY FIGURE S3 | Illustration of the screening approach to identify QXN-sensitive mutants. Frozen stocks of the haploid yeast mutant collection were cultured overnight into 96-well plates in YPD liquid media. The following day the cells were pinned by the aid of a ROTOR HDA onto plates without and with increasing concentrations of QXN. Plates were incubated at 30°C for 48 to 72 h and photographed using the Bio-Rad Chemi-Doc MP Imaging system. A representative plate is shown and a fading spot is indicative of a QXN-sensitive mutant.
SUPPLEMENTARY FIGURE S4 | Cell cycle profiles of the parent and the indicated mutants in response to QXN and post-treatment recovery. The cell cycle analysis was performed as described in Figure 4, and the percentage of cells in the indicated phases for the untreated, treated, and post-treatment (recovery) were derived from the graphic profiles. The representative profiles correspond to the untreated cells, treated cells without recovery (0 min), and treated cells recovered for 120 min. The G1 phase is shown in purple, the S phase in yellow, and G2/M in green.
References
Aouida, M., Pagé, N., Leduc, A., Peter, M., and Ramotar, D. (2004). A genome-wide screen in Saccharomyces cerevisiae reveals altered transport as a mechanism of resistance to the anticancer drug bleomycin. Cancer Res. 64 (3), 1102–1109. doi:10.1158/0008-5472.can-03-2729
Baker Brachmann, C., Davies, A., Cost, G. J., Caputo, E., Li, J., Hieter, P., et al. (1998). Designer deletion strains derived from Saccharomyces cerevisiae S288C: a useful set of strains and plasmids for PCR-mediated gene disruption and other applications. Yeast 14 (2), 115–132. doi:10.1002/(SICI)1097-0061(19980130)14:2<115::AID-YEA204>3.0.CO;2-2
Buravchenko, G. I., and Shchekotikhin, A. E. (2023). Quinoxaline 1, 4-dioxides: advances in chemistry and chemotherapeutic drug development. Pharmaceuticals 16 (8), 1174. doi:10.3390/ph16081174
Chandrasekhar, S., Reddy, N. K., and Kumar, V. P. (2010). Oxidation of alkynes using PdCl2/CuCl2 in PEG as a recyclable catalytic system: one-pot synthesis of quinoxalines. Tetrahedron Lett. 51 (28), 3623–3625. doi:10.1016/j.tetlet.2010.05.006
Chauhan, D. S., Singh, P., and Quraishi, M. (2020). Quinoxaline derivatives as efficient corrosion inhibitors: current status, challenges and future perspectives. J. Mol. Liq. 320, 114387. doi:10.1016/j.molliq.2020.114387
Chávez, S., Beilharz, T., Rondón, A. G., Erdjument-Bromage, H., Tempst, P., Svejstrup, J. Q., et al. (2000). A protein complex containing Tho2, Hpr1, Mft1 and a novel protein, Thp2, connects transcription elongation with mitotic recombination in Saccharomyces cerevisiae. EMBO J. 19 (21), 5824–5834. doi:10.1093/emboj/19.21.5824
Cheng, G., Li, B., Wang, C., Zhang, H., Liang, G., Weng, Z., et al. (2015). Systematic and molecular basis of the antibacterial action of quinoxaline 1, 4-di-N-oxides against Escherichia coli. Plos one 10 (8), e0136450. doi:10.1371/journal.pone.0136450
Cohen, M., Stutz, F., Belgareh, N., Haguenauer-Tsapis, R., and Dargemont, C. (2003). Ubp3 requires a cofactor, Bre5, to specifically de-ubiquitinate the COPII protein, Sec23. Nat. Cell Biol. 5 (7), 661–667. doi:10.1038/ncb1003
El Newahie, A. M., Nissan, Y. M., Ismail, N. S. M., Abou El Ella, D. A., Khojah, S. M., and Abouzid, K. A. M. (2019). Design and synthesis of new quinoxaline derivatives as anticancer agents and apoptotic inducers. Molecules 24 (6), 1175. doi:10.3390/molecules24061175
García-Muse, T., and Aguilera, A. (2019). R loops: from physiological to pathological roles. Cell 179 (3), 604–618. doi:10.1016/j.cell.2019.08.055
Gershon, L., and Kupiec, M. (2021). A novel role for Dun1 in the regulation of origin firing upon hyper-acetylation of H3K56. PLoS Genet. 17 (2), e1009391. doi:10.1371/journal.pgen.1009391
Giaever, G., Chu, A. M., Ni, L., Connelly, C., Riles, L., Véronneau, S., et al. (2002). Functional profiling of the Saccharomyces cerevisiae genome. nature 418 (6896), 387–391. doi:10.1038/nature00935
Gómez-González, B., Felipe-Abrio, I., and Aguilera, A. (2009). The S-phase checkpoint is required to respond to R-loops accumulated in THO mutants. Mol. Cell. Biol. 29 (19), 5203–5213. doi:10.1128/MCB.00402-09
Haase, S. B. (2003). Cell cycle analysis of budding yeast using SYTOX Green. Curr. Protoc. Cytom. 26 (1), Unit 7.23. doi:10.1002/0471142956.cy0723s26
Haase, S. B., and Reed, S. I. (2002). Improved flow cytometric analysis of the budding yeast cell cycle. Cell cycle 1 (2), 117–121. doi:10.4161/cc.1.2.114
Heravi, M. M., Baghernejad, B., and Oskooie, H. A. (2009). A novel three-component reaction for the synthesis of N-cyclohexyl-3-aryl-quinoxaline-2-amines. Tetrahedron Lett. 50 (7), 767–769. doi:10.1016/j.tetlet.2008.11.123
Hong, E. J., and Roeder, G. S. (2002). A role for Ddc1 in signaling meiotic double-strand breaks at the pachytene checkpoint. Genes Dev. 16 (3), 363–376. doi:10.1101/gad.938102
Hu, M.-H., and Lin, J.-H. (2023). New triazole-attached quinoxalines selectively recognize the telomeric multimeric G-quadruplexes and inhibit breast cancer cell growth. Int. J. Biol. Macromol. 241, 124548. doi:10.1016/j.ijbiomac.2023.124548
Li, D., Dai, C., Yang, X., Li, B., Xiao, X., and Tang, S. (2017). GADD45a regulates olaquindox-induced DNA damage and S-phase arrest in human hepatoma G2 cells via JNK/p38 pathways. Molecules 22 (1), 124. doi:10.3390/molecules22010124
Liu, W. (2019). The structure of the checkpoint clamp 9-1-1 complex and clamp loader Rad24-RFC in Saccharomyces cerevisiae. Biochem. Biophys. Res. Commun. 515 (4), 688–692. doi:10.1016/j.bbrc.2019.05.138
Mohanty, A., Alhaj Sulaiman, A., Moovarkumudalvan, B., Ali, R., Aouida, M., and Ramotar, D. (2023). The yeast permease Agp2 senses cycloheximide and undergoes degradation that requires the small protein brp1-cellular fate of Agp2 in response to cycloheximide. Int. J. Mol. Sci. 24 (8), 6975. doi:10.3390/ijms24086975
Montana, M., Montero, V., Khoumeri, O., and Vanelle, P. (2020). Quinoxaline derivatives as antiviral agents: a systematic review. Molecules 25 (12), 2784. doi:10.3390/molecules25122784
Palou, G., Palou, R., Guerra-Moreno, A., Duch, A., Travesa, A., and Quintana, D. G. (2010). Cyclin regulation by the s phase checkpoint. J. Biol. Chem. 285 (34), 26431–26440. doi:10.1074/jbc.M110.138669
Patel, S. B., Patel, B. D., Pannecouque, C., and Bhatt, H. G. (2016). Design, synthesis and anti-HIV activity of novel quinoxaline derivatives. Eur. J. Med. Chem. 117, 230–240. doi:10.1016/j.ejmech.2016.04.019
Pereira, J. A., Pessoa, A. M., Cordeiro, M. N. D. S., Fernandes, R., Prudêncio, C., Noronha, J. P., et al. (2015). Quinoxaline, its derivatives and applications: a State of the Art review. Eur. J. Med. Chem. 97, 664–672. doi:10.1016/j.ejmech.2014.06.058
Sato, N. (2008). “8.03 - pyrazines and their benzo derivatives,” in Comprehensive heterocyclic chemistry III. Editor A. R. Katritzky, (Oxford: Elsevier), 273–331.
Sulaiman, A. A., Ali, R., Aouida, M., Moovarkumudalvan, B., and Ramotar, D. (2022). The histone H2B Arg95 residue links the pheromone response pathway to rapamycin-induced G1 arrest in yeast. Sci. Rep. 12 (1), 10023. doi:10.1038/s41598-022-14053-9
Sulaiman, A. A., Ali, R., and Ramotar, D. (2023). The histone H2B Arg95 residue efficiently recruits the transcription factor Spt16 to mediate Ste5 expression of the pheromone response pathway. Sci. Rep. 13 (1), 10189. doi:10.1038/s41598-023-37339-y
Suter, W., Rosselet, A., and Knüsel, F. (1978). Mode of action of quindoxin and substituted quinoxaline-di-N-oxides on Escherichia coli. Antimicrob. Agents Chemother. 13 (5), 770–783. doi:10.1128/aac.13.5.770
Keywords: Saccharomyces cerevisiae, quinoxaline sensitive mutants, cell cycle arrest, genome-wide screening, drug resistance, antibacterial, antifungal, and antitumor activities
Citation: Sulaiman AA, Al-Ansari DE, Ali R, Aouida M and Ramotar D (2024) Mft1, identified from a genome-wide screen of the yeast haploid mutants, mediates cell cycle arrest to counteract quinoxaline-induced toxicity. Front. Genet. 14:1296383. doi: 10.3389/fgene.2023.1296383
Received: 18 September 2023; Accepted: 31 December 2023;
Published: 12 January 2024.
Edited by:
Quaiser Saquib, King Saud University, Saudi ArabiaReviewed by:
Devyani Haldar, Centre for DNA Fingerprinting and Diagnostics (CDFD), IndiaTabish H. Khan, Washington University in St. Louis, United States
Copyright © 2024 Sulaiman, Al-Ansari, Ali, Aouida and Ramotar. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mustapha Aouida, bWFvdWlkYUBoYmt1LmVkdS5xYQ==; Dindial Ramotar, ZHJhbW90YXJAaGJrdS5lZHUucWE=
†These authors share senior authorship