 Bin Liu1†
Bin Liu1† Ziman Yang
Ziman Yang Qing Liu
Qing Liu Ying Zhang
Ying Zhang Hui Ding
Hui Ding Hongyan Lai
Hongyan Lai- 1Department of Anesthesiology, The Fourth People’s Hospital of Sichuan Province, Chengdu, Sichuan, China
- 2School of Life Science and Technology, University of Electronic Science and Technology of China, Chengdu, China
- 3Department of Pain, The Affiliated Traditional Chinese Medicine Hospital of Southwest Medical University, Luzhou, Sichuan, China
- 4Department of Anesthesiology, The Affiliated Traditional Chinese Medicine Hospital of Southwest Medical University, Luzhou, Sichuan, China
- 5Chongqing Key Laboratory of Big Data for Bio Intelligence, Chongqing University of Posts and Telecommunications, Chongqing, China
- 6Research Center of Integrated Traditional Chinese and Western Medicine, The Affiliated Traditional Chinese Medicine Hospital of Southwest Medical University, Luzhou, Sichuan, China
Allergy is an autoimmune disorder described as an undesirable response of the immune system to typically innocuous substance in the environment. Studies have shown that the ability of proteins to trigger allergic reactions in susceptible individuals can be evaluated by bioinformatics tools. However, developing computational methods to accurately identify new allergenic proteins remains a vital challenge. This work aims to propose a machine learning model based on multi-feature fusion for predicting allergenic proteins efficiently. Firstly, we prepared a benchmark dataset of allergenic and non-allergenic protein sequences and pretested on it with a machine-learning platform. Then, three preferable feature extraction methods, including amino acid composition (AAC), dipeptide composition (DPC) and composition of k-spaced amino acid pairs (CKSAAP) were chosen to extract protein sequence features. Subsequently, these features were fused and optimized by Pearson correlation coefficient (PCC) and principal component analysis (PCA). Finally, the most representative features were picked out to build the optimal predictor based on random forest (RF) algorithm. Performance evaluation results via 5-fold cross-validation showed that the final model, called iAller (https://github.com/laihongyan/iAller), could precisely distinguish allergenic proteins from non-allergenic proteins. The prediction accuracy and AUC value for validation dataset achieved 91.4% and 0.97%, respectively. This model will provide guide for users to identify more allergenic proteins.
1 Introduction
Allergic diseases are a group of immune-mediated inflammatory response diseases, including allergic asthma, allergic rhinitis, atopic dermatitis, food allergy. These diseases are caused by the hypersensitivity of body immune system to normally harmless environmental substances (Miescher and Vogel, 2002). With the change of worldwide environment, the incidence of allergic diseases has increased considerably in the past few years. Patients with allergic diseases often have complex clinical manifestations and a high risk of recurrence (Wang et al., 2023a). Biomedical researchers are increasingly concerned about these diseases.
Substances that can induce allergic reactions, typically proteins, are called allergens (Galli et al., 2008). Allergenic proteins for humans are often derived from aeroallergens, food allergens, personal care products and so on. Allergic reactions are generally grouped into two classes. The well-studied and common class is mediated by immunoglobulin E (IgE), which is one of the five primary human immunoglobulins. An IgE-mediated (type I hypersensitivity) allergy occurs when the body encounter allergenic proteins containing immunogenic and antigenic structures. The mechanism is that allergenic proteins enter body and drive immune cells to produce lots of allergenic protein-specific IgE antibodies. When the body re-exposure to the allergenic proteins, these IgEs will bind to them and lead to the activation of other immune cells as well as the initiation of inflammation response (Platts-Mills, 2001; Oseroff et al., 2012; Guo et al., 2023). The specific recognition and interaction for allergenic proteins is based on their sequences and structures.
The common methods used to determine protein allergenicity potential are traditional immunochemical, biochemical and immunological methods (Kimber et al., 2003; Ladics and Selgrade, 2009; Zhou et al., 2023). With the development of bioinformatics and machine learning algorithms, massive computational strategies for identifying allergenic proteins have emerged and evolved over time (Saha and Raghava, 2006; Gupta et al., 2013; Sharma et al., 2021a; Lathwal et al., 2021). Thereinto, the key idea of early reported methods is to seek sequence similarity, which is mainly based on the guidelines about evaluating the potential allergenicity of novel food proteins proposed by the United Nations Food and Agriculture Organization (FAO) and the World Health Organization (WHO). These methods, such as SDAP, Allermatch, AllerTool, AllerHunter, generally assess protein potential allergenicity by searching for similar sequences on the basis of local or global sequence alignment algorithms, such as BLAST, FASTA program, etc (Ivanciuc et al., 2003; Fiers et al., 2004; Zhang et al., 2007; Muh et al., 2009). Another class of technology involves the identification of allergen-related motifs by using motif search tool, such as MEME/MAST. Furthermore, ensemble approaches, such as proAP and AlgPred 2.0, have also been developed based on both sequence similarity and motif eliciting strategy (Soeria-Atmadja et al., 2006; Wang et al., 2013; Sharma et al., 2021b). In recent years, several feature vector-based approaches have been reported, including APPEL, AllerTOP, AllergenFP, AllerCatPro, ProAll-D (Cui et al., 2007; Dimitrov et al., 2013; Dimitrov et al., 2014; Nguyen et al., 2022; Shanthappa and Kumar, 2022). In general, they take sequence-derived compositional, evolutionary, structural and physicochemical information into consideration and achieve allergenic protein classification by using machine learning or deep learning models (Wang et al., 2021; Ao et al., 2022; Wu et al., 2023). For example, random forest (RF), support vector machine (SVM), decision tree (DT), k-nearest neighbors (KNN) and multilayer perceptron (MLP) were employed to establish AlgPred 2.0 on the basis of composition/evolutionary information-based features (Zhang et al., 2007). Different classification models, including Gaussian Naive Bayes, Radius Neighbour’s Classifier, Bagging Classifier, ADA Boost, Linear Discriminant Analysis, Quadratic Discriminant Analysis, Extra Tree Classifier and Long Short-Term Memory (LSTM), have been considered in the study of ProAll-D (Cui et al., 2007).
Although there are presently a number of computational methods for detecting allergenic proteins, due to the limitations of prediction performance, it is still need to train more effective and robust allergenic protein classifiers. In this work, we focused on allergenic proteins for human beings and developed iAller to distinguish them from non-allergenic proteins. The major implement procedures have been shown in Figure 1, which includes 1) constructing a benchmark dataset consisting of 2,210 positive and 2,210 negative sample sequences; 2) conducting pre-analysis on the whole dataset with iLearnPlus by combining nine feature descriptors with four machine learning algorithms; 3) selecting AAC, DPC and CKSAAP feature extraction methods with excellent performance to encode sequence samples, as well as RF algorithm to build classifier; 4) integrating these three types of features and performing feature selection and dimensionality reduction by using PCC and PCA; 5) training and determining the optimal classifier on training dataset through 5-fold cross-validation; 6) assessing the prediction performance of the optimal RF model on validation dataset. The high accuracy and AUC value of 91.4% and 0.97 suggest that this model should be an excellent choice for identifying allergenic proteins.
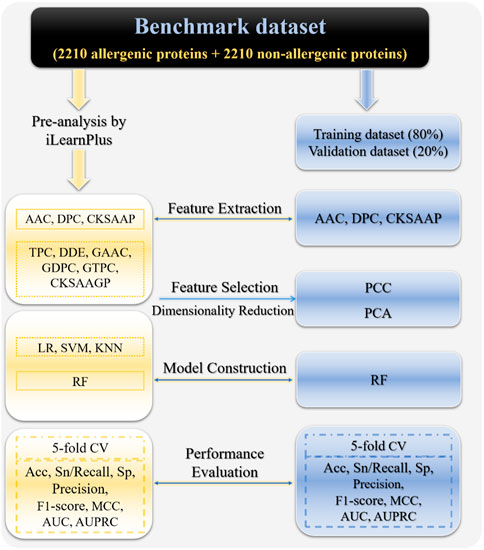
FIGURE 1. The frame of establishing the allergenic protein classifier in this study. Abbreviations: AAC, amino acid composition; DPC, dipeptide composition; TPC, tripeptides composition; DDE, dipeptide deviation from expected mean, CKSAAP, composition of k-spaced amino acid pairs; GAAC, grouped amino acid composition; GDPC, grouped dipeptide composition; GTPC, grouped tripeptide composition; CKSAAGP, composition of k-spaced amino acid group pairs; RF, random forest; LR, logistic regression; SVM, support vector machine; KNN, k-nearest neighbors, Acc, accuracy; Sn, sensitivity; Sp, specificity; MCC, Matthews correlation coefficient; AUC, area under receiver operating characteristic (ROC) curve; AUPRC, area under precision-recall curve.
2 Materials and methods
2.1 Protein sequence benchmark dataset
The main significance of this work is to provide a theoretical basis for the study of human allergic reactions. Hence, this work primarily focused on proteins causing allergic reactions in human beings, excluding other species. The sequence benchmark dataset was composed of 2,210 non-allergenic proteins and 2,210 allergenic proteins. These allergenic proteins were originated from various human allergens, mainly including wheat, rice, seafood product, pollen, dust and so on. This dataset had been studied by ProAll-D project and could be freely accessible from https://doi.org/10.17632/tjmt97xpjf.1. Proteins with high homology had been removed by CD-HIT program for avoiding sequence redundancy and ensuring the objectivity of experimental results (Shanthappa and Kumar, 2022).
2.2 Preliminary selection of analysis methods
For building a machine learning classifier for protein sequences, it is necessary to convert biological sequence information into feature vector information that can be processed by computers (Lu et al., 2022; Wang et al., 2023b; Dao et al., 2023; Le, 2023; Zhu et al., 2023). Therefore, it is very imortant to select appropriate feature extraction methods. For the above protein sequence datasets, we firstly performed pre-experiment by using iLearnPlus (Chen et al., 2021), a comprehensive and automated machine-learning platform. This online server could automatically generate and save evaluation metrics of the selected algorithms according to input data and parameter settings. Nine feature descriptors, including AAC, DPC, tripeptides composition (TPC), dipeptide deviation from expected mean (DDE), CKSAAP, grouped amino acid composition (GAAC), grouped dipeptide composition (GDPC), grouped tripeptide composition (GTPC) and composition of k-spaced amino acid group pairs (CKSAAGP), were applied to extract sample information of allergenic and non-allergenic proteins in our work. By comparing the performance of RF, logistic regression (LR), SVM and KNN classification models using these features, we finally chose the AAC, DPC, CKSAAP methods combined with RF algorithm for further detailed analysis.
2.3 Protein sequence features
2.3.1 Amino acid composition (AAC) feature
Amino acids are the basic units of proteins. Twenty types of amino acids are involved in protein composition, namely, Alanine (A), Cysteine (C), Aspartic acid (D), Glutamic (E), Phenylalanine (F), Glycine (G), Histidine (H), Isoleucine (I), Lysine (K), Leucine (L), Methionine (M), Asparagine (N), Proline (P), Glutamine (Q), Arginine (R), Serine (S), Threonine (T), Valine (V), Tryptophan (W), Tyrosine (Y). Among the numerous computational methods for transforming protein sequences into feature vectors, AAC coding method is the simplest and most intuitive one. The principle is to calculate the frequencies of twenty types of amino acids in protein sequence (Bhasin and Raghava, 2004; Sahoo et al., 2021). Based on AAC, every allergenic/non-allergenic protein sequence can be represented with a 20-dimension feature vector, as Formula (1),
where
2.3.2 Dipeptide composition (DPC) feature
Feature encoding method based on k-mer composition is to divide protein sequences into fragments with fixed length of k, and calculate the frequency of each type k-mer fragment. Such method can capture information about amino acid composition as well as local sequence order (Ahmad et al., 2016). When k = 2, namely, DPC, there are 20 × 20 = 400 kinds of 2-mers. Each protein sequence will be transformed into a numerical vector with 400 features. The calculation formula is as following,
the meaning of
2.3.3 Composition of k-spaced amino acid pairs (CKSAAP) feature
Another popular binary encoding strategy similar to DPC is CKSAAP. The encoding scheme is to count the occurrence times of 400 amino acid pairs separated by any k-mer in a given protein sequence (Ju and Wang, 2020). For example, when k = 1, a protein will be encoded to a 400-dimensional numerical vector with each feature factor being the frequency at which any one 1-spaced amino acid pair appears. In this work, we set k-spaced amino acid pair to k = 1, 2, 3 to encode allergenic and non-allergenic protein sequences, taking into account its prediction accuracy, computational time and complexity. A total of 1200 CKSAAP features were produced, as follow:
where
2.4 Feature fusion, selection and shrinkage
In the field of biomolecule sequence analysis, extracting features from a single perspective often leads to incomplete sequence information and low prediction performance of classification models. In order to improve this problem, the above AAC, DPC and CKSAAP three type features were fused together for cc. Every protein sequence of the benchmark dataset was represented as:
Feature vectors produced by multi-feature fusion methods were usually high-dimensional and redundant (Han et al., 2022; Yan et al., 2022; Zhao-Yue ZHANG et al., 2022; Ao et al., 2023). We further utilized two approaches, PCC combined with incremental feature selection (IFS) strategy and PCA, to select more informative features and reduce dimensionality (Karl Pearson, 1901; Stigler, 1989; Dao et al., 2018). PCC is often used to measure the strength and direction of a linear relationship between two variables. It is defined as the quotient of the covariance and standard deviation between two variables. A larger absolute value of the Pearson coefficient indicates a stronger linear relationship between the two variables. PCA is another common feature extraction and dimensionality reduction method. Its purpose is to transform a series of influence factors with correlations into a new set of mutually independent comprehensive indicators, while retaining as much information as possible on the original variables during the transformation. The core idea is to map the original high-dimensional data into a new low-dimensional space and to obtain a set of orthogonal basis vectors. PCA enables the map of raw data on this set vector to be with maximum variance and preserve the major characteristics. For example, a raw dataset with p variables will be converted q comprehensive principal components by a linear combination of optimally weighted original variables, where q is less than p. The detailed computation procedures of these methods are described in iLearnPlus.
2.5 Classifier construction with random forest (RF)
RF is an ensemble learning algorithm that combines several base learners into a strong learner by voting or averaging to improve the robustness and generalization performance (Breiman, 2001; Wei et al., 2021; Yang et al., 2021; Basith et al., 2022; Islam et al., 2022; Zhang et al., 2023a). Thus, we adopted RF algorithm to construct allergenic protein classifier. The process was as follows: 1) Random sampling: N new datasets are generated by random sampling with replacement, each of which has the same size as the original dataset. 2) Building decision trees: The CART decision tree algorithm is applied to each new dataset and builds a decision tree. Due to the characteristics of random sampling, each new dataset might only contain a part of samples and features of the original dataset, as well as the predictive ability of each tree might be different. 3) Integrating: N decision trees are combined into a strong classifier by voting or averaging. The RF strategy was involved in random sampling and random feature selection. Random sampling enables the differences among each new dataset and avoids model overfitting. Random feature selection enables the variability among decision trees and improves the generalization ability of the final model.
2.6 Classifier performance evaluation
For assessing machine learning models more accurately, the benchmark dataset was split into training and validation datasets with ratio of 4:1. Five-fold cross-validation was used in model training. We employed several common indexes to evaluate model performance (Hasan et al., 2022; Jeon et al., 2022; Shoombuatong et al., 2022; Thi Phan et al., 2022; Zhang et al., 2023b), including accuracy (Acc), sensitivity (Sn)/recall, specificity (Sp), precision, F1-score (F1), Matthews correlation coefficient (MCC), area under receiver operating characteristic (ROC) curve (AUC), area under precision-recall curve (AUPRC) (Su et al., 2023; Yang et al., 2023). The specific equations to calculate these measures were as follows:
where TP (true positive) and FP (false positive) denoted the numbers of sequences correctly and incorrectly classified as allergenic proteins, respectively. TN (true negative) and FN (false negative) were the numbers of samples correctly and incorrectly classified as non-allergenic proteins, respectively. The AUC and AUPRC values ranged from 0 to 1. Their higher values implied better predictive ability of models.
3 Results and discussion
3.1 Preliminary analysis results
In order to pick out the most appropriate feature extraction and model construction methods from the existing numerous algorithms, as well as to reduce experimental complexity and workload, we performed pre-analysis on the benchmark protein sequences by using iLearnPlus tool.
In this experiment part, we firstly chosen AAC, DPC, TPC, DDE, CKSAAP, GAAC, GDPC, GTPC, CKSAAGP features to build RF, LR, SVM, KNN classification models with default parameters, respectively. The prediction performance of these nine type features were assessed by combining with RF algorithm and shown in Figure 2. It was obvious that the best-performing feature extraction methods were CKSAAP, DPC and AAC and have achieved quite high AUC and AUPRC values of about 0.98. The performance of these features based on LR, SVM, KNN algorithms (see Supplementary Figures S1–S3) also indicated that CKSAAP, DPC and AAC were more preferable methods for encoding allergenic and non-allergenic protein sequences. Secondly, we tested each of the three feature extraction methods on each machine learning classifier with corresponding optimal parameters. The best number of decision tree for RF was set as 450, the best values of c and γ for SVM were
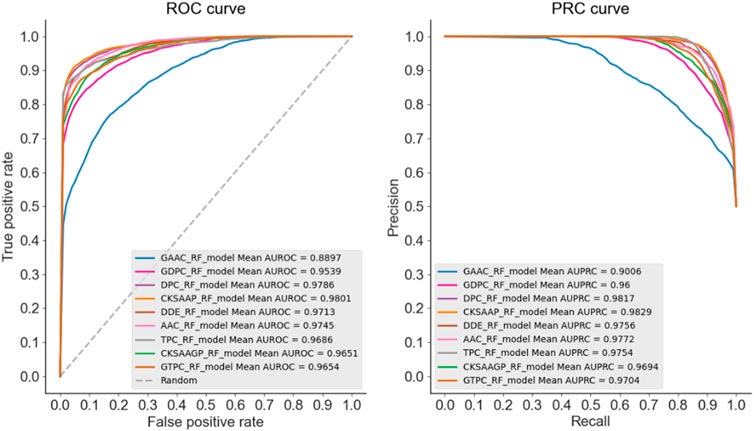
FIGURE 2. Performance of nine feature extraction methods based on RF algorithm.
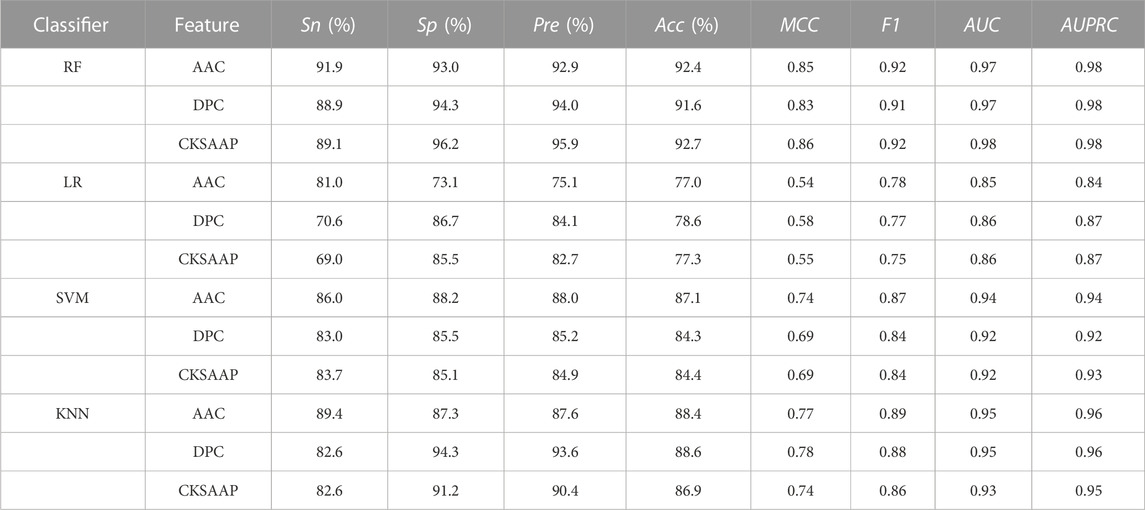
TABLE 1. Performance of each machine learning classifier based on AAC, DPC and CKSAAP features, respectively.
Through comprehensively comparing and analyzing the recognition performance of allergenic protein using different feature extraction methods and machine learning models, we selected AAC, DPC, CKSAAP features and RF algorithm with better performance for further detailed analysis.
3.2 Prediction results of multi-fusion features
To contain sequence composition information as comprehensive as possible, we fused AAC, DPC, CKSAAP features together. Each protein sequence was represented as a fused vector with 1,620 features. All the features were ranked in descending order by PCC and the top 200 features were selected out by using IFS strategy. To construct an effective and robust allergenic protein identification model, the PCA was used for further feature shrinkage. The top 100 principal features were ultimately screened out to build RF model. For determining the optimal decision tree parameter in RF algorithm, we tried to set it as 100, 200, 300, 400, 450, 500 with a cut-off value of 0.5. The prediction results (Table 2) demonstrated that the optimal value of decision tree should be set as 200, and the corresponding RF classifier could produce the highest accuracy of 91.4%.
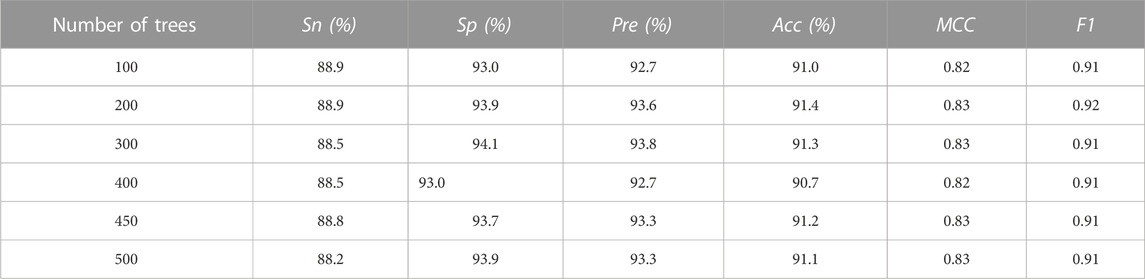
TABLE 2. Prediction ability comparison of random forest models with different number of decision trees.
To explore the generalization ability of these fused features, we constructed the optimal RF model based on the training dataset via 5-fold cross-validation (see Figure 3; Supplementary Table S1). Allergenic protein prediction was then conducted on both training and validation datasets. Performance evaluation results were enumerated in Table 3. The RF model could produce good performance on training protein sequences. The prediction accuracy, Sn, Sp, AUC and AUPRC values were 91.0%, 88.4%, 93.6%, 0.96 and 0.97, respectively (Figure 3). In addition, it could also produce pretty good performance on the validation dataset with the accuracy of 91.4%, Sn of 88.9%, Sp of 93.9%, AUC value of 0.97 and AUPRC value of 0.98 (Figure 4). All results implied that the presented RF classifier had good performance on generalization and robustness.
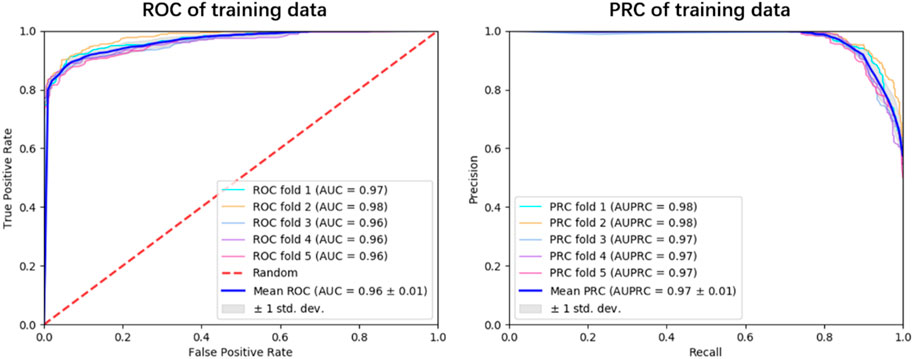
FIGURE 3. The ROC and PRC curves for the optimal RF classifier on training dataset. The blue solid line indicates the overall result of 5-fold cross-validation, the solid lines in other colors are results of each fold k = 1, 2, 3, 4, 5.
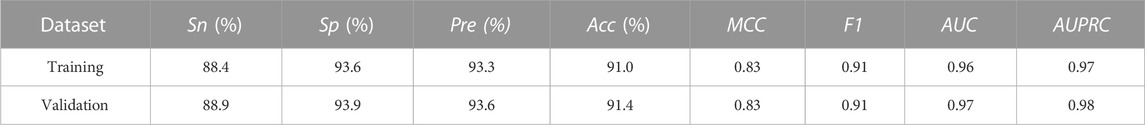
TABLE 3. Performance summary of the optimal RF model testing on training and validation datasets by 5-fold cross-validation.
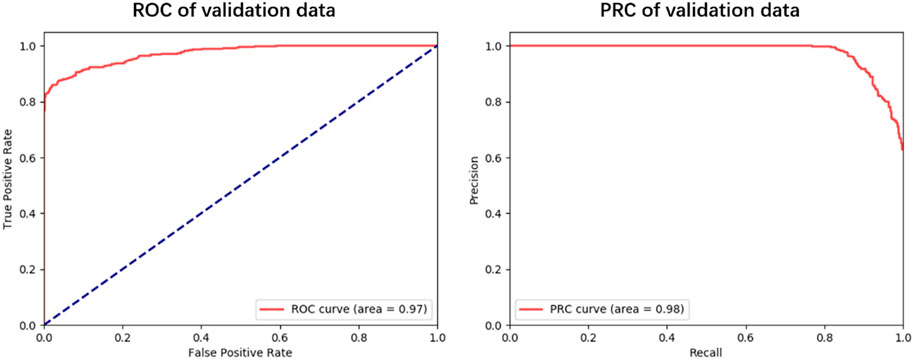
FIGURE 4. The ROC and PRC curves for the optimal RF classifier on validation dataset.
It is quite crucial to compare with existing methods for comprehensively evaluating a novel method. Therefore, we futher compared the prediction performance of our iAller with that of AlgPred 2.0 web server (https://webs.iiitd.edu.in/raghava/algpred2/) for testing on the same validation dataset (Zhang et al., 2007). The prediction accuracy of AlgPred 2.0 with setting Machine Learning Techique as “Hybrid” was 89.8%, which was inferior to that of iAller. Moreover, AllergenFP and ProAll-D servers were established on almost the same benchmark dataset as that of this work, and the accuracy of AllergenFP was 87.9%, the best AUC value of ProAll-D was 0.92 (Cui et al., 2007; Wang et al., 2013). It implied that iAller was superior to these existing tools and could provide reliable results for researches about allergenic protein predicting.
4 Conclusion
Identification of allergenic proteins from the perspective of bioinformatics can provide theoretical support for the relevant biological experimental research. Although many computational models for allergenic protein prediction have been developed, few of them have been widely validated and used in related researches. Improving the accuracy and effectiveness of allergenic protein prediction remains a challenging problem. This work attempted to explore more suitable feature extraction and selection methods as well as machine learning models for identifying allergenic proteins. After a series of trials and comparative analyses, we established an effective RF model based on 100 informative fusion features via 5-fold cross-validation. The accuracy and AUC value of the classifier on validation dataset reached 91.4% and 0.97. Evaluation results suggested that this computational model, iAller, was robust and its generalization ability was superior. It indicates that fusing different types of protein sequence features is a feasible strategy. However, there is still room for improvement. In future work, more types of information such as amino acid physicochemical properties, evolutionary information will be taken into consideration, and more feature selection methods will be attempted, as well as a web server shall be constructed for bringing more convenience to researchers.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding authors.
Author contributions
BL: Data curation, Investigation, Methodology, Validation, Visualization, Writing–original draft. ZY: Data curation, Formal Analysis, Investigation, Methodology, Writing–original draft. QL: Investigation, Writing–original draft. YZ: Data curation, Writing–original draft. HD: Conceptualization, Data curation, Supervision, Writing–review and editing. HL: Conceptualization, Visualization, Writing–review and editing. QL: Conceptualization, Funding acquisition, Project administration, Writing–review and editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. Southwest Medical University (2021ZKQN121), Chinese medicine research project of Sichuan Provincial Administration of Traditional Chinese Medicine (2021MS446), Office of Science and Technology and Talent Work of Luzhou (2022-JYJ-126), and National Natural Science Foundation of China (62371403).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2023.1294159/full#supplementary-material
References
Ahmad, K., Waris, M., and Hayat, M. (2016). Prediction of protein submitochondrial locations by incorporating dipeptide composition into chou's general pseudo amino acid composition. J. Membr. Biol. 249 (3), 293–304. doi:10.1007/s00232-015-9868-8
Ao, C., Jiao, S., Wang, Y., Yu, L., and Zou, Q. (2022). Biological sequence classification: a review on data and general methods. Research 2022. doi:10.34133/research.0011
Ao, C., Ye, X., Sakurai, T., Zou, Q., and Yu, L. (2023). m5U-SVM: identification of RNA 5-methyluridine modification sites based on multi-view features of physicochemical features and distributed representation. Bmc Biol. 21 (1), 93. doi:10.1186/s12915-023-01596-0
Basith, S., Lee, G., and Manavalan, B. (2022). STALLION: a stacking-based ensemble learning framework for prokaryotic lysine acetylation site prediction. Brief. Bioinform 23 (1), bbab412. doi:10.1093/bib/bbab412
Bhasin, M., and Raghava, G. P. S. (2004). Classification of nuclear receptors based on amino acid composition and dipeptide composition. J. Biol. Chem. 279 (22), 23262–23266. doi:10.1074/jbc.M401932200
Chen, Z., Zhao, P., Li, C., Li, F., Xiang, D., Chen, Y.-Z., et al. (2021). iLearnPlus: a comprehensive and automated machine-learning platform for nucleic acid and protein sequence analysis, prediction and visualization. Nucleic Acids Res. 49 (10), e60. doi:10.1093/nar/gkab122
Cui, J., Han, L. Y., Li, H., Ung, C. Y., Tang, Z. Q., Zheng, C. J., et al. (2007). Computer prediction of allergen proteins from sequence-derived protein structural and physicochemical properties. Mol. Immunol. 44 (4), 514–520. doi:10.1016/j.molimm.2006.02.010
Dao, F.-Y., Lv, H., Wang, F., Feng, C.-Q., Ding, H., Chen, W., et al. (2018). Identify origin of replication in Saccharomyces cerevisiae using two-step feature selection technique. Bioinformatics 35 (12), 2075–2083. doi:10.1093/bioinformatics/bty943
Dao, F. Y., Liu, M. L., Su, W., Lv, H., Zhang, Z. Y., Lin, H., et al. (2023). AcrPred: a hybrid optimization with enumerated machine learning algorithm to predict Anti-CRISPR proteins. Int. J. Biol. Macromol. 228, 706–714. doi:10.1016/j.ijbiomac.2022.12.250
Dimitrov, I., Flower, D. R., and Doytchinova, I. (2013). AllerTOP--a server for in silico prediction of allergens. BMC Bioinforma. 14 (6), S4. doi:10.1186/1471-2105-14-S6-S4
Dimitrov, I., Naneva, L., Doytchinova, I., and Bangov, I. (2014). AllergenFP: allergenicity prediction by descriptor fingerprints. Bioinformatics 30 (6), 846–851. doi:10.1093/bioinformatics/btt619
Fiers, MWEJ, Kleter, G. A., Nijland, H., Peijnenburg, AACM, Nap, J. P., and van Ham, RCHJ (2004). Allermatch, a webtool for the prediction of potential allergenicity according to current FAO/WHO Codex alimentarius guidelines. BMC Bioinforma. 5, 133. doi:10.1186/1471-2105-5-133
Galli, S. J., Tsai, M., and Piliponsky, A. M. (2008). The development of allergic inflammation. Nature 454 (7203), 445–454. doi:10.1038/nature07204
Guo, C., Tang, Y., Li, Q., Yang, Z., Guo, Y., Chen, C., et al. (2023). Deciphering the immune heterogeneity dominated by natural killer cells with prognostic and therapeutic implications in hepatocellular carcinoma. Comput. Biol. Med. 158, 106872. doi:10.1016/j.compbiomed.2023.106872
Gupta, S., Ansari, H. R., Gautam, A., Open Source Drug Discovery, C., and Raghava, G. P. (2013). A rare case of benign multicystic peritoneal mesothelioma: a clinical dilemma. Biol. direct 8, 27–29. doi:10.1007/s12262-011-0314-6
Han, Y. M., Yang, H., Huang, Q. L., Sun, Z. J., Li, M. L., Zhang, J. B., et al. (2022). Risk prediction of diabetes and pre-diabetes based on physical examination data. Math. Biosci. Eng. 19 (4), 3597–3608. doi:10.3934/mbe.2022166
Hasan, M. M., Tsukiyama, S., Cho, J. Y., Kurata, H., Alam, M. A., Liu, X., et al. (2022). Deepm5C: a deep-learning-based hybrid framework for identifying human RNA N5-methylcytosine sites using a stacking strategy. Mol. Ther. 30, 2856–2867. doi:10.1016/j.ymthe.2022.05.001
Islam, M. S., Awal, M. A., Laboni, J. N., Pinki, F. T., Karmokar, S., Mumenin, K. M., et al. (2022). HGSORF: henry gas solubility optimization-based random forest for C-section prediction and XAI-based cause analysis. Comput. Biol. Med. 147, 105671. doi:10.1016/j.compbiomed.2022.105671
Ivanciuc, O., Schein, C. H., and Braun, W. (2003). SDAP: database and computational tools for allergenic proteins. Nucleic Acids Res. 31 (1), 359–362. doi:10.1093/nar/gkg010
Jeon, Y. J., Hasan, M. M., Park, H. W., Lee, K. W., and Manavalan, B. (2022). TACOS: a novel approach for accurate prediction of cell-specific long noncoding RNAs subcellular localization. Brief. Bioinform 23 (4), bbac243. doi:10.1093/bib/bbac243
Ju, Z., and Wang, S.-Y. (2020). Prediction of lysine formylation sites using the composition of k-spaced amino acid pairs via Chou's 5-steps rule and general pseudo components. Genomics 112 (1), 859–866. doi:10.1016/j.ygeno.2019.05.027
Karl Pearson, F. R. S. (1901). LIII on lines and planes of closest fit to systems of points in space. Philos. Mag. Ser. 2, 559–572. doi:10.1080/14786440109462720
Kimber, I., Dearman, R. J., Penninks, A. H., Knippels, L. M. J., Buchanan, R. B., Hammerberg, B., et al. (2003). Assessment of protein allergenicity on the basis of immune reactivity: animal models. Environ. Health Perspect. 111 (8), 1125–1130. doi:10.1289/ehp.5813
Ladics, G. S., and Selgrade, M. K. (2009). Identifying food proteins with allergenic potential: evolution of approaches to safety assessment and research to provide additional tools. Regul. Toxicol. Pharmacol. 54 (3), S2–S6. doi:10.1016/j.yrtph.2008.10.010
Lathwal, A., Kumar, R., and Raghava, G. P. S. (2021). In-silico identification of subunit vaccine candidates against lung cancer-associated oncogenic viruses. Comput. Biol. Med. 130, 104215. doi:10.1016/j.compbiomed.2021.104215
Le, N. Q. K. (2023). Explainable artificial intelligence for protein function prediction: a perspective view. Curr. Bioinforma. 18 (3), 205–207. doi:10.2174/1574893618666230220120449
Lu, H., Shang, C., Zou, S., Cheng, L., Yang, S., and Wang, L. (2022). A novel method for predicting essential proteins by integrating multidimensional biological attribute information and topological properties. Curr. Bioinforma. 17 (4), 369–379. doi:10.2174/1574893617666220304201507
Miescher, S. M., and Vogel, M. (2002). Molecular aspects of allergy. Mol. Asp. Med. 23 (6), 413–462. doi:10.1016/s0098-2997(02)00009-2
Muh, H. C., Tong, J. C., and Tammi, M. T. (2009). AllerHunter: a SVM-pairwise system for assessment of allergenicity and allergic cross-reactivity in proteins. PLoS One 4 (6), e5861. doi:10.1371/journal.pone.0005861
Nguyen, M. N., Krutz, N. L., Limviphuvadh, V., Lopata, A. L., Gerberick, G. F., and Maurer-Stroh, S. (2022). AllerCatPro 2.0: a web server for predicting protein allergenicity potential. Nucleic Acids Res. 50 (W1), W36–W43. doi:10.1093/nar/gkac446
Oseroff, C., Sidney, J., Tripple, V., Grey, H., Wood, R., Broide, D. H., et al. (2012). Analysis of T cell responses to the major allergens from German cockroach: epitope specificity and relationship to IgE production. J. Immunol. 189 (2), 679–688. doi:10.4049/jimmunol.1200694
Platts-Mills, T. A. (2001). The role of immunoglobulin E in allergy and asthma. Am. J. Respir. Crit. Care Med. 164 (2), S1–S5. doi:10.1164/ajrccm.164.supplement_1.2103024
Saha, S., and Raghava, G. P. (2006). AlgPred: prediction of allergenic proteins and mapping of IgE epitopes. Nucleic acids Res. 34, W202–W209. doi:10.1093/nar/gkl343
Sahoo, S., Mahapatra, S. R., Parida, B. K., Rath, S., Dehury, B., Raina, V., et al. (2021). DBCOVP: a database of coronavirus virulent glycoproteins. Comput. Biol. Med. 129, 104131. doi:10.1016/j.compbiomed.2020.104131
Shanthappa, P. M., and Kumar, R. (2022). ProAll-D: protein allergen detection using long short term memory - a deep learning approach. ADMET DMPK 10 (3), 231–240. doi:10.5599/admet.1335
Sharma, N., Patiyal, S., Dhall, A., Devi, N. L., and Raghava, G. P. S. (2021a). ChAlPred: a web server for prediction of allergenicity of chemical compounds. Comput. Biol. Med. 136, 104746. doi:10.1016/j.compbiomed.2021.104746
Sharma, N., Patiyal, S., Dhall, A., Pande, A., Arora, C., and Raghava, G. P. S. (2021b). AlgPred 2.0: an improved method for predicting allergenic proteins and mapping of IgE epitopes. Brief. Bioinform 22 (4), bbaa294. doi:10.1093/bib/bbaa294
Shoombuatong, W., Basith, S., Pitti, T., Lee, G., and Manavalan, B. (2022). THRONE: a new approach for accurate prediction of human rna N7-methylguanosine sites. J. Mol. Biol. 434 (11), 167549. doi:10.1016/j.jmb.2022.167549
Soeria-Atmadja, D., Lundell, T., Gustafsson, M. G., and Hammerling, U. (2006). Computational detection of allergenic proteins attains a new level of accuracy with in silico variable-length peptide extraction and machine learning. Nucleic Acids Res. 34 (13), 3779–3793. doi:10.1093/nar/gkl467
Stigler, S. M. (1989). Francis galton's account of the invention of correlation. Stat. Sci. 4 (2), 73–79. doi:10.1214/ss/1177012580
Su, W., Xie, X. Q., Liu, X. W., Gao, D., Ma, C. Y., Zulfiqar, H., et al. (2023). iRNA-ac4C: a novel computational method for effectively detecting N4-acetylcytidine sites in human mRNA. Int. J. Biol. Macromol. 227, 1174–1181. doi:10.1016/j.ijbiomac.2022.11.299
Thi Phan, L., Woo Park, H., Pitti, T., Madhavan, T., Jeon, Y. J., and Manavalan, B. (2022). MLACP 2.0: an updated machine learning tool for anticancer peptide prediction. Comput. Struct. Biotechnol. J. 20, 4473–4480. doi:10.1016/j.csbj.2022.07.043
Wang, J., Yu, Y., Zhao, Y., Zhang, D., and Li, J. (2013). Evaluation and integration of existing methods for computational prediction of allergens. BMC Bioinforma. 14 (4), S1. doi:10.1186/1471-2105-14-S4-S1
Wang, J., Zhou, Y., Zhang, H., Hu, L., Liu, J., Wang, L., et al. (2023a). Pathogenesis of allergic diseases and implications for therapeutic interventions. Signal Transduct. Target. Ther. 8 (1), 138. doi:10.1038/s41392-023-01344-4
Wang, X., Wang, S., Fu, H., Ruan, X., and TangDeepFusion-Rbp, X. (2021). DeepFusion-RBP: using deep learning to fuse multiple features to identify RNA-binding protein sequences. Curr. Bioinforma. 16 (8), 1089–1100. doi:10.2174/1574893616666210618145121
Wang, Y., Zhai, Y., Ding, Y., and Zou, Q. (2023b). SBSM-pro: support bio-sequence machine for proteins. Available at: https://arXiv:2308.10275.
Wei, L., He, W., Malik, A., Su, R., Cui, L., and Manavalan, B. (2021). Computational prediction and interpretation of cell-specific replication origin sites from multiple eukaryotes by exploiting stacking framework. Brief. Bioinform 22 (4), bbaa275. doi:10.1093/bib/bbaa275
Wu, D., Fang, X., Luan, K., Xu, Q., Lin, S., Sun, S., et al. (2023). Identification of SH2 domain-containing proteins and motifs prediction by a deep learning method. Comput. Biol. Med. 162, 107065. doi:10.1016/j.compbiomed.2023.107065
Yan, C., Li, M., Ma, J., Liao, Y., Luo, H., Wang, J., et al. (2022). A novel feature selection method based on MRMR and enhanced flower pollination algorithm for high dimensional biomedical data. Curr. Bioinforma. 17 (2), 133–149. doi:10.2174/1574893616666210624130124
Yang, H., Luo, Y., Ren, X., Wu, M., He, X., Peng, B., et al. (2021). Risk Prediction of Diabetes: big data mining with fusion of multifarious physical examination indicators. Inf. Fusion 75, 140–149. doi:10.1016/j.inffus.2021.02.015
Yang, H., Luo, Y. M., Ma, C. Y., Zhang, T. Y., Zhou, T., Ren, X. L., et al. (2023). A gender specific risk assessment of coronary heart disease based on physical examination data. NPJ Digit. Med. 6 (1), 136. doi:10.1038/s41746-023-00887-8
Zhang, H., Chi, M., Su, D., Xiong, Y., Wei, H., Yu, Y., et al. (2023b). A random forest-based metabolic risk model to assess the prognosis and metabolism-related drug targets in ovarian cancer. Comput. Biol. Med. 153, 106432. doi:10.1016/j.compbiomed.2022.106432
Zhang, Y.-F., Wang, Y.-H., Gu, Z.-F., Pan, X.-R., Li, J., Ding, H., et al. (2023a). Bitter-RF: a random forest machine model for recognizing bitter peptides. Front. Med. (Lausanne). 10, 1052923. doi:10.3389/fmed.2023.1052923
Zhang, Z. H., Koh, J. L. Y., Zhang, G. L., Choo, K. H., Tammi, M. T., and Tong, J. C. (2007). AllerTool: a web server for predicting allergenicity and allergic cross-reactivity in proteins. Bioinformatics 23 (4), 504–506. doi:10.1093/bioinformatics/btl621
Zhao-Yue Zhang, Z.-J. S., Yu-He, YANG, and Hao, L. I. N. (2022). Towards a better prediction of subcellular location of long non-coding RNA. Front. Comput. Sci. 16 (5), 165903. doi:10.1007/s11704-021-1015-3
Zhou, Q., Li, S., Yan, X., Zhu, H., Liu, W., Guo, Y., et al. (2023). Characterization, potential prognostic value, and immune heterogeneity of cathepsin C in diffuse glioma. Curr. Bioinforma. 18 (1), 76–91. doi:10.2174/1574893618666221101144857
Keywords: allergenic protein, multi-feature fusion, feature selection, random forest, prediction
Citation: Liu B, Yang Z, Liu Q, Zhang Y, Ding H, Lai H and Li Q (2023) Computational prediction of allergenic proteins based on multi-feature fusion. Front. Genet. 14:1294159. doi: 10.3389/fgene.2023.1294159
Received: 15 September 2023; Accepted: 11 October 2023;
Published: 19 October 2023.
Edited by:
Gajendra PS. Raghava, Indraprastha Institute of Information Technology Delhi, IndiaReviewed by:
Balachandran Manavalan, Sungkyunkwan University, Republic of KoreaSumeet Patiyal, National Cancer Institute (NIH), United States
Copyright © 2023 Liu, Yang, Liu, Zhang, Ding, Lai and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hongyan Lai, bGFpaHlAY3F1cHQuZWR1LmNu; Qun Li, cXVuZXIxOTg0QDE2My5jb20=
†These authors have contributed equally to this work