
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Genet. , 15 December 2023
Sec. Molecular Cytogenetics
Volume 14 - 2023 | https://doi.org/10.3389/fgene.2023.1293652
This article is part of the Research Topic Co-Occurrence of Numerical and Structural Aberration - Small Supernumerary Marker Chromosomes and B-Chromosomes View all 7 articles
 Thomas Liehr1*
Thomas Liehr1* Monika Ziegler1Luisa Person1Stefanie Kankel1Niklas Padutsch1
Monika Ziegler1Luisa Person1Stefanie Kankel1Niklas Padutsch1 Anja Weise1
Anja Weise1 Jörg Paul Weimer2Heather Williams3Susana Ferreira4
Jörg Paul Weimer2Heather Williams3Susana Ferreira4 Joana B. Melo4
Joana B. Melo4 Isabel M. Carreira4
Isabel M. Carreira4Introduction: With only 39 reported cases in the literature, carriers of a small supernumerary marker chromosome (sSMC) derived from chromosome 11 represent an extremely rare cytogenomic condition.
Methods: Herein, we present a review of reported sSMC(11), add 18 previously unpublished cases, and closely review eight cases classified as ‘centromere-near partial trisomy 11’ and a further four suited cases from DECIPHER.
Results and discussion: Based on these data, we deduced the borders of the pericentric regions associated with clinical symptoms into a range of 2.63 and 0.96 Mb for chromosome 11 short (p) and long (q) arms, respectively. In addition, the minimal pericentric region of chromosome 11 without triplo-sensitive genes was narrowed to positions 47.68 and 60.52 Mb (GRCh37). Furthermore, there are apparent differences in the presentation of signs and symptoms in carriers of larger sSMCs derived from chromosome 11 when the partial trisomy is derived from different chromosome arms. However, the number of informative sSMC(11) cases remains low, with overlapping presentation between p- and q-arm-imbalances. In addition, uniparental disomy (UPD) of ‘normal’ chromosome 11 needs to be considered in the evaluation of sSMC(11) carriers, as imprinting may be an influencing factor, although no such cases have been reported. Comprehensively, prenatal sSMC(11) cases remain a diagnostic and prognostic challenge.
A small supernumerary marker chromosome (sSMC) is a cytogenomic condition based on the presence of a numerical and structural aberration. In most cases, an sSMC, which can derive from any human chromosome, is present in addition to a normal karyotype of 46, XX or 46,XY. sSMCs are found in 0.044% of the human population, with ∼3.5 million sSMC carriers among the present population of ∼8 billion. Approximately 2.5 million (70%) sSMC carriers are clinically normal and located through chromosomal analyses for a different reason for referral. The remaining 30% have mild to severe clinical signs and symptoms due to the sSMC. Parental inheritance accounts for 30% and ∼50% of sSMC carriers are mosaic; mosaicism may also be ‘cryptic’, which means that molecular cytogenetic studies reveal submicroscopic differences in sSMC shape and content (Liehr, 2012; Liehr, 2023).
Using size, sSMCs are defined as generally smaller than a chromosome 20 within the same metaphase spread and can take different shapes, i.e., ring- (r), inverted duplication- (inv dup), or ‘centric-minute’- (min) shaped. Mitotic instability is elevated in min- and r-shaped sSMCs compared to inv-dup-shaped; thus, the former are more likely to lead to mosaicism (Hussein et al., 2014). In addition, sSMCs may be composed of material from a single chromosome or two or more chromosomes–the latter creates ‘complex sSMCs’ (Liehr et al., 2013). Suppose the sSMC material is derived from a single chromosome, but the order of the DNA stretches is disrupted or rearranged compared to a normal sister chromosome; in this case, it is termed ‘discontinuous sSMC’. These discontinuous sSMCs most likely form from a chromothripsis-related trisomic rescue event (Kurtas et al., 2019). In addition, an sSMC can carry no normal centromeric region but instead carry a neocentromere (Liehr et al., 2007). Finally, an sSMC can co-occur with uniparental disomy (UPD) of the normal sister chromosomes; co-occurrence accounts for ∼3% of de novo cases and ∼1.3% of all reported cases (Liehr et al., 2011). Patients may carry 2 to 7 sSMCs derived from different chromosomes or multiple sSMCs, which accounts for ∼1.2% of all cases (Robberecht et al., 2012). In sSMC cases with UPD, heterochromatic sSMCs not resulting in relevant copy number variations (CNVs) may still be causative for clinical presentation, either because (i) they carry genes involved with imprinting, and/or (ii) there is a (partial) isodisomy that leads to the activation of a parental recessive gene variant (Benn, 2021). Interestingly, in rare cases, mosaicism may also influence the phenotype (Liehr and Al-Rikabi, 2019).
Of the estimated 3.5 million sSMC carriers currently in the population, only ∼7,169 are documented in the literature (Liehr, 2023). The shape is not reported for 852 cases (∼12%), and 138 cases (∼2%) are neocentric. Of the remaining 6,179 reported cases, 3,795 (∼62%) have inv dup-, 1,563 (∼25%) min-, and 821 (∼13%) r-shaped (Liehr, 2023).
Few sSMCs are specifically attributed to a clinical syndrome, as outlined below (Liehr, 2023):
• +inv dup(5)(pter→q10::q10→pter) / tetrasomy 5p-syndrome (Blakey-Cheung et al., 2020, ORPHA:3309);
• +inv dup(8)(pter→q10::q10→pter) / tetrasomy 8p-syndrome (Napoleone et al., 1997, ORPHA:3307, OMIM #614290);
• +inv dup(9)(pter→q10∼12::q10∼12→pter) / tetrasomy 9p-syndrome (Süleyman et al., 2022, ORPHA:3310);
• +inv dup(12)(pter→q10∼12::q10∼12→pter) / tetrasomy 12p- or Pallister-Kilian-syndrome (ORPHA:884, OMIM #601803);
• +der(13 or 21)t(13 or 21;18)(q11.1;p11.1) / special form of trisomy 18p-syndrome (Liehr et al., 2013);
• +inv dup(15)(pter→q12∼13::q12∼13→pter) / proximal tetrasomy 15q-syndrome (Battaglia, 2008, ORPHA:3306);
• +inv dup(18)(pter→q10::q10→pter) / tetrasomy 18p-syndrome (ORPHA:3307, OMIM #614290);
• +inv dup(20)(pter→q10::q10→pter) / tetrasomy 20p-syndrome (Maziad and Seaver, 2015);
• +inv dup(22)(pter→q11.2::q11.2→pter) / proximal tetrasomy 22q- or cat eye-syndrome (ORPHA:195, OMIM #115470);
• +der(22)t(8;22)(q24;q11.1∼11.2) / supernumerary der(22)t(8;22)-syndrome (OMIM #613700);
• +der(22)t(11;22)(q23;q11.2) / derivative chromosome 22- or Emanuel-syndrome (ORPHA:96170, OMIM #609029).
These 11 syndromes constitute ∼34% of the reported cases. In addition, there are 786 sSMC cases reported in Turner syndrome mosaics (∼11%) (Li et al., 2021; Liehr, 2023).
For all other sSMCs, chromosome-specific genotype–phenotype correlations are based only on a paucity of cases. However, more and more evidence has emerged that proximal centromere-near regions of each human chromosome mainly carry triplo-insensitive genes (Liehr, 2023). Thus, in many cases, a proximal partial trisomy leads to no clinical symptoms; these sSMCs may be transmitted through generations but may adversely affect fertility, mainly in male carriers (Dalprà et al., 2005). Still, in sSMC cases comprised of centromere-near material distal to a specific region, the presence of triplo-sensitive genes leads to clinical signs in the carrier. Phenotypic effects can be chromosome-arm-specific differently, e.g., for cases with sSMC-induced proximal partial trisomy 11p or 11q.
The available data for triplo-sensitive regions is summarized herein, and a possible first genotype-phenotype correlation for sSMCs derived from proximal 11p and/or 11q is presented. Please note that sSMCs are grouped according to the origin of their centromere; thus, Emanuel syndrome cases with +der(22)t(11; 22) are not included (OMIM #609029).
All 57 sSMC cases were either studied in the laboratory of TL (Jena, Germany–for contributors see also in Acknowledgments) and/or previously published; they are also recorded in the sSMC database (Liehr, 2023). Herein, all 39 previously (by several authors) published sSMC(11) cases and 18 new cases studied in Jena are included (see Supplementary Table S1). The study’s own cases were acquired through routine diagnostic testing and patients were anonymized. Written informed consent was obtained from the individual(s) or minors’ legal guardian/next of kin for the publication of any potentially identifiable data included in this article. In addition, ethical approval was provided by the Ethical Commission of Jena University Hospital (code 4738-03/16) for this sSMC research. Lastly, eight cases from the literature with copy number gains near centromere 11 were also collected and summarized (see Supplementary Table S2). In total, 65 cases are listed in Tables 1, 2, with minimal clinical information and molecular(cyto)genetic study results–more data is provided in the corresponding Supplementary Tables S1, S2.
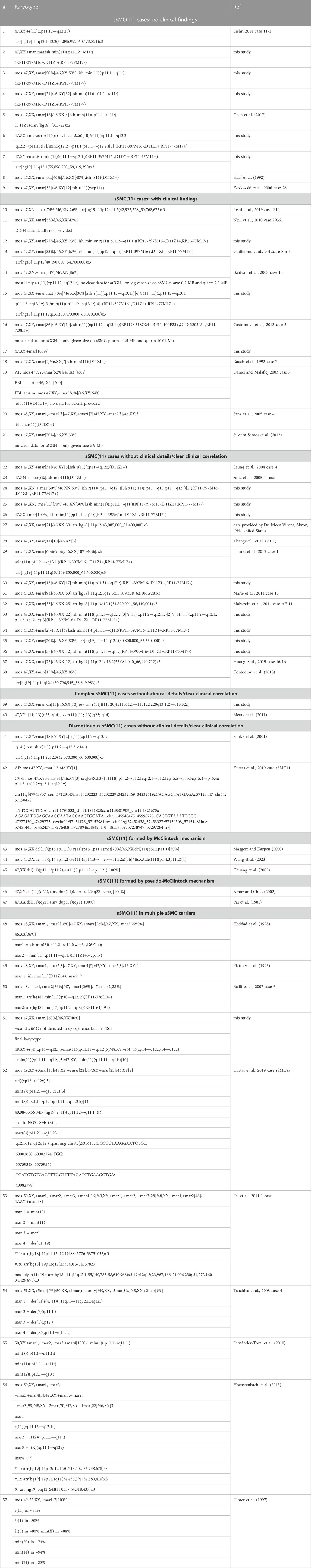
TABLE 1. Overview of the 57 sSMC(11) cases as characterized at present—for more details, see Supplementary Table S1; all molecular karyotyping data is given here in build GRCh37/hg19.
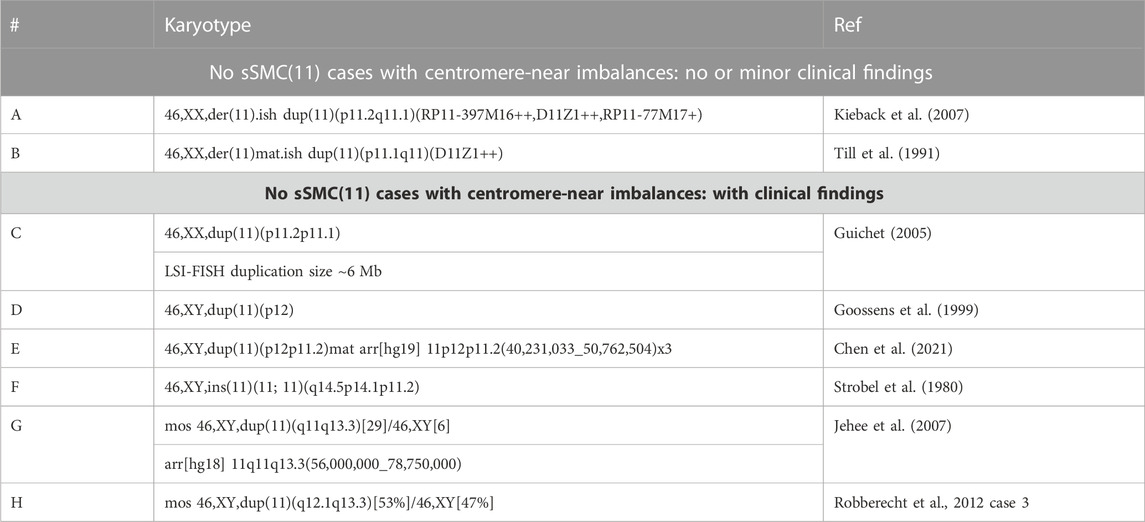
TABLE 2. Overview of the eight cases from the literature with centromere 11-near imbalances similar to those induced by an sSMC(11)—for more details, see Supplementary Table S1; all molecular karyotyping data is given here in build GRCh37/hg19.
The 18 cases presented here were identified by standard banding cytogenetics (Hliscs et al., 1997) and the corresponding sSMCs were characterized as derived from chromosome 11 by centromere-specific multicolor-FISH (cenM-FISH) (Nietzel et al., 2001). These results were confirmed by a commercial centromere-11-specific probe: CEP 11 (D11Z1) (Vysis/Abbott, Düsseldorf, Germany) and the euchromatic content was further determined by a subcenM-FISH-probe set as previously reported, i.e., a partial chromosome paint for 11p and 11q, probes for 11p11.2 (RP11-397M16–hg19: 48,303,671-48,479,496) and 11q12 (RP11-77M17–hg19: 57,352,936-57,521,103) that were applied, together with D11Z1 (Starke et al., 2003). The identification of sSMCs by chromosomal microdissection and subsequent reverse hybridization was carried out as published by Weimer et al. (1999).
Array-comparative genomic hybridization (aCGH) was completed with microdissected material from the corresponding sSMC as previously described (Melo et al., 2011). In some cases evaluated in this manner, the results of sizing the sSMC were not as exact as when using genomic DNA; thus, the values were rounded to the second digit after the decimal point as Mb.
Molecular data from literature using other builds were translated into GRCh37/hg19 using the University of California Santa Cruz (UCSC) webpage using the function ‘View’ ‘In Other Genomes (Convert)’ and are included in Tables 3, 4. Data for Supplementary Table S4; Supplementary Figure S1 was acquired using the UCSC browser combined with OMIM or DECIPHER, Database of Genomic Variants, GnomAD, and localization of segmental duplications.
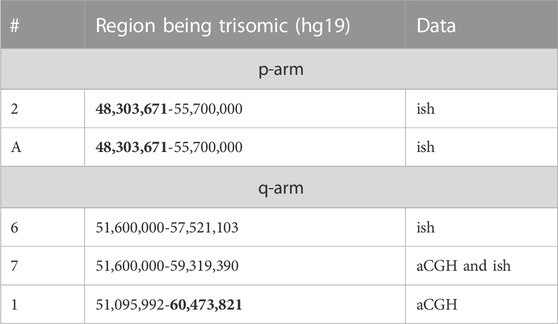
TABLE 3. Selected details on cases from Tables 1, 2 being suited to narrow down the triplo-insensitive regions of pericentric 11p and 11q. Data from cases providing the largest uncritical pericentric regions are highlighted in bold.
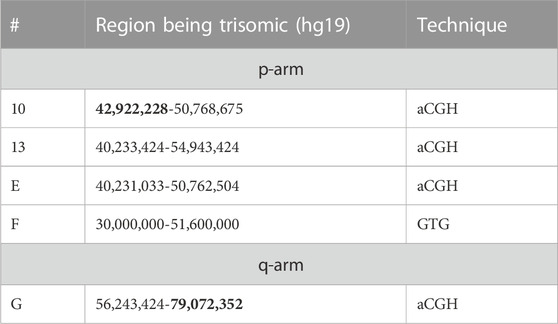
TABLE 4. Selected details on cases from Tables 1, 2 being suited to narrow down the borders between triplo-insensitive and sensitive regions of pericentric 11p and 11q. Data from cases providing the largest uncritical pericentric regions are highlighted in bold.
The 18 sSMC(11) cases presented herein were characterized for origin, shape, and content by cenM-FISH and subcenM-FISH; an example result is presented using case 2 (see Figure 1). The corresponding cases are in Table 1: cases 2-4, 6-7, 12, 15, 17, 24-26, 30, 33-36, 39, and 51. Together with the 39 cases from the literature, 57 sSMC(11) cases were available for analysis. In total, 12 cases were clinically normal (cases 1-9, 43-45) and 23 were clinically abnormal (cases 11-21, 46-57), and for the remaining 22, no clinical details were available and/or a clear clinical correlation to the presence of an sSMC was not possible. The male-to-female ratio for the 52/57 cases with known gender was 29:23.
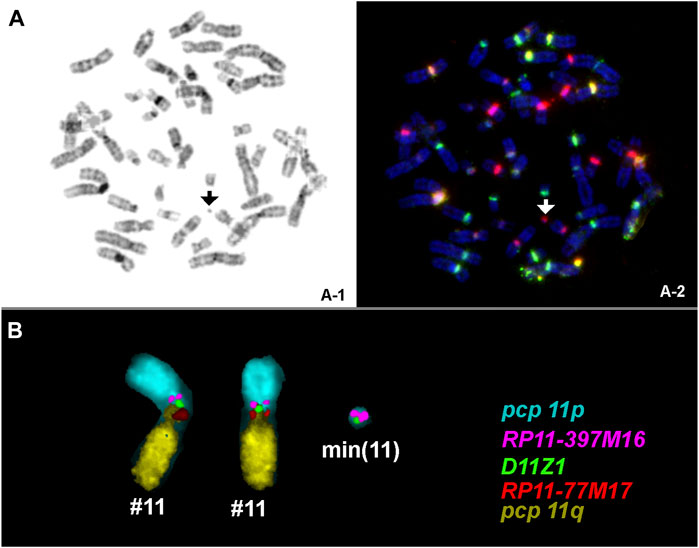
FIGURE 1. An sSMC was detected by GTG-banding (A) in all amnion cells studied; cenM-FISH identified chromosome-11-origin [arrowhead in (B)]; the presence of euchromatic content of chromosome 11p and the shape was determined by subcenM-FISH by applying the five probes as shown (C). A min(11) (:p11.12→q11:) was characterized. Abbreviations: pcp, partial chromosome paint.
In Table 1, all 57 cases are listed with genotype and size of the sSMC(11) and designated clinically normal (which may include infertility) or abnormal (for more details, see Supplementary Table S1).
For 43 of the 57 cases, the shape was reported: 50% had min-, 47% had r-, and 3% had inv dup-shaped. The sSMC(11) group distribution is shown in Figure 2 with observation of all shape subtypes excluding UPD(11)-associated sSMCs; several sSMCs could be attributed to more than one group and can be observed two or more times in Supplementary Table S3, which serves as the basis for the data presented in Figure 2.
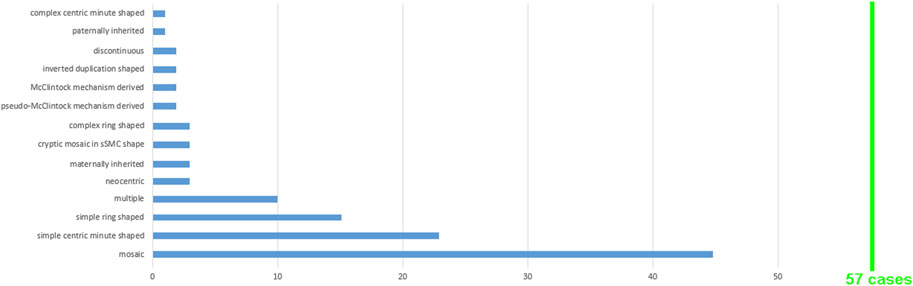
FIGURE 2. A total of 57 sSMC(11) cases were grouped according to the 14 features shown; for more details see Supplementary Table S3.
With the exception of case 41, all cases were constitutional sSMCs; case 41 was an acquired sSMC associated with atypical chronic myelogenous leukemia (aCML).
In Table 2, all eight cases from the literature with imbalances similar to those associated with sSMC(11) are listed with the genotype/size of the imbalance and clinical outcome (for more details, see Supplementary Table S2).
Only those cases from Tables 1, 2 with a good molecular(cyto)genetic characterization and/or a suitable clinical description were acceptable for inclusion in Tables 3–5. Similarly, cases with complex, discontinuous, and/or multiple sSMCs (11) could not be considered; sSMCs formed by (pseudo-)McClintock mechanism were also excluded.
Cases 1 to 9, A and B, demonstrated no relevant clinical signs associated with pericentric partial trisomy 11. Accordingly, Table 3 includes 5 cases (from 11 total clinically normal) that were suitable to determine the minimal pericentric triplo-insensitive region of chromosome 11; this region spans chromosome 11 positions 48,303,671 to 60,473,821 (hg19), at a minimum. This region comprises 305 genes, with only 11 OMIM morbid annotated genes, mostly activated by point mutations (Supplementary Table S4).
Cases 13-21 and C to H show relevant clinical signs most likely due to pericentric partial trisomy 11; Table 4 includes those five cases suitable to determine the region adjacent to the triplo-independent region (see Table 3). Thus, the triplo-sensitive region in 11p starts between 42,922,228-48,303,671, and the corresponding region in 11q starts between 60,473,821-79,072,352 Mb (hg19).
To further refine the pericentric triplo-(in)sensitive regions of chromosome 11 ‘pathogenic’ and ‘likely pathogenic’ gains and duplication in the region were checked in OMIM for DECIPHER patients 411500 and 300792 (Supplementary Figure S1). Therefore, no pathogenic patient report is available for regions 45,048,321 to 61,479,322. In addition, population data for known benign copy number gains (Database of Genomic variants and gnomAD, records dgv1111n100 and nsv832175) also fit with data from Table 3 and DECIPHER data. Thus, the uncritical proximal triplo-insensitive region can be refined to 47,675,469 to 60,516,539 Mb, i.e., a region of 12.841,070 Mb (Supplementary Figure S1; Figure 3). However, the exact starting points of triplo-sensitive regions in 11p and 11q could only be narrowed to 45,048,321 to 47,675,469 Mb and 60,516,539 to 61,479,322, respectively; these are regions of 2.63 and 0.96 Mb.
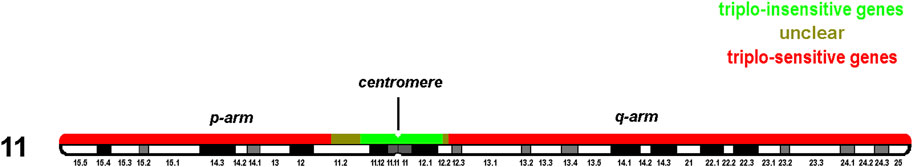
FIGURE 3. Regions containing dosage/duplication independent (green) and dependent genes (red); Olive stretches between those regions lack cases to narrow down the corresponding green and red regions.
Table 5 summarizes clinical signs and symptoms according to the nine cases (also based on data from Tables 3, 4) suitable for a preliminary genotype–phenotype correlation.
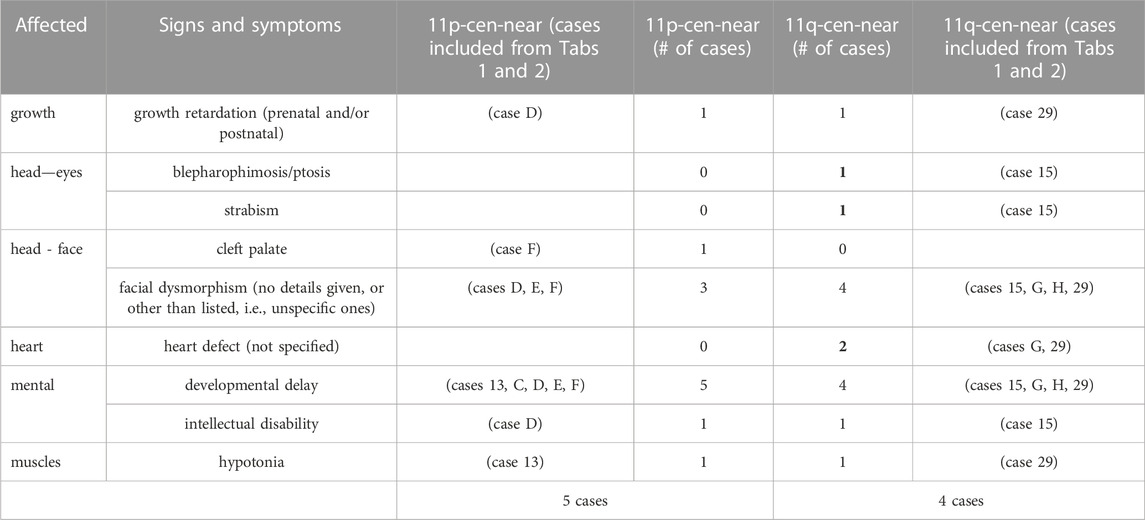
TABLE 5. Selected details on cases from Tables 1, 2 providing clinical data for a first genotype-phenotype correlation. Data from cases providing the largest uncritical pericentric regions are highlighted in bold.
This study adds 18 new sSMC(11) cases to the literature. Among the 57 cases evaluated herein, several types of sSMC shapes and subtypes are presented, as shown in Figure 2. Like non-acrocentric-derived sSMCs from other chromosomes, chromosome 11-derived sSMCs are predominantly r- and min-shaped, with very rare incidences of the inv dup-shaped (Liehr, 2012). The under- or over-representation of specific subgroups of sSMC(11) is likely attributable to the low case number availability. Accordingly, (i) no case with sSMC-associated UPD(11) is reported as of yet, (ii) 5/57 sSMC(11) cases (8.8%) were formed by (pseudo-)McClintock mechanism (Baldwin et al., 2008), which is > 8× more than observed among all sSMCs generally (Liehr, 2023), and (iii) 10/57 sSMC(11) (17.5%) were observed in multiple sSMC carriers in comparison to ∼1.2% observation among all sSMC(11)-carriers. The fact that ∼80% of sSMC(11) carriers show mosaic presentation aligns well with the observation that r- and min-shaped sSMCs are less mitotically stable (Hussein et al., 2014).
Clinical follow-up, particularly in prenatal cases is a problem (Detorri, 2011). As such, it is normal, that for 22/57 sSMC(11) cases (38.6%), no clinical data is available for analysis. Similarly, reliable data for parental origin was provided for only 4/55 cases, three of maternal origin and one of paternal origin; this aligns with the predominance of maternal sSMC inheritance (Dalprà et al., 2005). The sex of the sSMC(11) carriers was not reported in 5/57 cases (∼9%); a 1:1.26 male-to-female ratio does not seem remarkable for one or the other gender, as is established for all sSMCs (Liehr et al., 2011).
Unfortunately, limitations of sufficient clinical and molecular(cyto)genetic data negatively influenced the characterization and analysis of cases. For the 57 sSMC(11) cases summarized herein, these data were comprehensive for only six cases: 1, 2, 6, 7, 10, and 13 (see Tables 3, 4). Thus, to approach the goal of defining triplo-insensitive pericentric regions for chromosome 11 and provide the first preliminary genotype–phenotype correlation for 11p and 11q proximal trisomies, eight more cases from the literature were also included (Table 2), leading to four more cases (A, E, F, and G) in Tables 3, 4. These cases have similar centromere-near imbalances as induced by sSMC(11) from interstitial, intrachromosomal 11 duplications.
Euchromatic copy number gains, as induced by sSMC presence, are considered the main influencer of genotype–phenotype correlations (Liehr, 2012). However, there may be UPD of an sSMC sister-chromosome associated with disease causation even with a heterochromatic sSMC (Liehr, 2012). Finally, mosaicism has a major influence on genotype–phenotype correlation in these patients, which should be discussed, evaluated, and understood. For mosaicism, we must primarily consider that generally, only one to three tissues of the human body can be studied. In the prenatal evaluation, this is chorionic villus sampling, amniotic fluid, but may be fetal blood, too; postnatally, peripheral blood and buccal mucosa are easily accessible. The other ∼400 tissues of the human body remain a black box regarding the mosaic state of the sSMC. Studies have demonstrated that sSMCs seem to be present in all body tissues in different mosaic states (Fickelscher et al., 2007). Thus, it is not surprising that mosaicism levels generally have no major influence on the phenotype (Liehr, 2023). In cases where an sSMC is found in peripheral blood at 50% or more, if an sSMC is associated with a clinical impact, the patient will present with typical signs and symptoms. Considering this link, it seems justifiable to also include mosaic cases in a genotype–phenotype correlation study.
However, there are also rare exceptions, e.g., Pallister-Kilian syndrome patients, who are not found to have sSMC(12) in peripheral blood, unlike all other tissues (Liehr and Al-Rikabi, 2019). Furthermore, there can also be normal carriers of otherwise deleterious sSMCs, e.g., inv dup(9p) with the marker chromosome in nearly 100% of the peripheral blood cells (Liehr and Al-Rikabi, 2019).
For copy number variations (CNVs), it is well known that they may span up to several euchromatic, gene-containing megabases in size and can be placed into two groups based on current knowledge: 1) not associated with disease causation, and 2) associated with microdeletion/microduplication syndromes (Itsara et al., 2009). They are neither clearly distinguished by size nor by number of genes involved. The only difference established is that CNVs leading to the aforementioned syndromes contain at least one dosage-sensitive gene (Rice and McLysaght, 2017). The best example may be hereditary neuropathy with liability to pressure palsies (HNPP) and Charcot-Marie Tooth disease type 1A (CMT1A) associated with a 1.4 Mb microdeletion or microduplication in 17p11.2, respectively. The 1.4 Mb of DNA includes dozens of genes; however, only the PMP22 gene is dosage-sensitive and results in the respective syndrome when present in one or three copies instead of two (van Paassen et al., 2014). Similarly, previous research provided evidence that pericentric CNVs/copy number gains induced by sSMC presence are comparable with either a harmless CNV or a microduplication syndrome (Qi et al., 2013; Liehr, 2023). Simply stated, the proximal centromere-near regions of all chromosomes seem to be no issue for human cells if they are present in more than two copies, as long as they only contain DNA/genes that are dosage insensitive. The main goal of a genotype–phenotype correlation of an sSMC must be to determine the regions, which are unproblematic if present in more than two copies. In the suggested region, 305 genes are included, which are obviously insensitive to copy numbers gains (Supplementary Table S4). Finally, the regions that contain the first centromere-near dosage-dependent or better triplo-sensitive gene(s) should be determined. The more informative sSMC cases that are available, the better such a link can be.
Even though only a few cases of chromosome 11 were available for such an undertaking, herein, a first refinement of clinical and molecular(cyto)genomic data was performed. As outlined in Tables 3, 4 (see also Supplementary Figure S1), the minimal pericentric region of chromosome 11 that does not contain dosage-sensitive, or more precisely, duplication-sensitive genes, is between positions 47.68 and 60.52 Mb (GRCh37/hg19). Furthermore, clinical symptoms are expected if an sSMC(11) contains greater than 2.63 Mb or 0.96 Mb of DNA for the short or long arm, respectively. Increasing the publication of more informative sSMC(11) cases will further delineate these regions.
Positing a preliminary first genotype–phenotype correlation of deleterious partial trisomy 11p compared to 11q, the data from only nine cases are presented in Table 5. As to be expected, clinically abnormal sSMC(11) cases demonstrate rather unspecific common symptoms like dysmorphism, developmental delay, intellectual disability, and hypotonia. Of interest, eye and heart problems were isolated to partial trisomy 11q cases only.
Overall, genotype–phenotype correlations for microduplication syndromes are hampered by clinical variance in the presentation of symptoms (Watson et al., 2014). This is also the case for sSMCs, which may also be more difficult to interpret due to mosaicism (Liehr and Al-Rikabi, 2019), UPD (Liehr et al., 2011), an insufficiently characterized sSMC (see Table 1), and/or the presence of a harmless sSMC with an uncharacterized variant in any human gene associated with disease causation (Nelle et al., 2010). Still, herein, the first steps needed to distinguish a harmful sSMC from a harmless sSMC(11) are presented.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
The studies involving humans were approved by the Ethical Commission of Jena University Hospital (code 4738-03/16). The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants’ legal guardians/next of kin.
TL: Conceptualization, Data curation, Formal Analysis, Project administration, Resources, Writing–original draft, Writing–review and editing. MZ: Formal Analysis, Methodology, Writing–review and editing. LP: Formal Analysis, Methodology, Writing–review and editing. SK: Formal Analysis, Methodology, Writing–review and editing. NP: Formal Analysis, Methodology, Writing–review and editing. AW: Formal Analysis, Investigation, Software, Writing–review and editing. JW: Formal Analysis, Methodology, Resources, Writing–review and editing. HW: Writing–original draft, Writing–review and editing. SF: Formal Analysis, Methodology, Writing–review and editing. JM: Formal Analysis, Methodology, Resources, Writing–review and editing. IC: Formal Analysis, Methodology, Resources, Writing–review and editing.
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
sSMC(11)-cases were provided by C. Altus (Magdeburg, Germany), C. Alves (Porto, Portugal), B. Belitz (Berlin, Germany), H.-C. Duba (Linz, Austria) P. Kozlowski (Düsseldorf, Germany), A. Junge (Dresden, Germany), M. Lemmens (Aachen, Germany), E. Manolakis (Athens, Greece), T. Martin (Homburg, Germany), M. Sagi (Jerusalem, Israel).
Author HW was employed by Cache DNA, Inc.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2023.1293652/full#supplementary-material
Supplementary Figure S1 | The pericentric region of chromosome 11 is shown in the UCSC genome browser. The orange rectangle indicates the triplo-insensitive region as determined in Table 3. The depicted Decipher track was filtered for “pathogenic” and “likely pathogenic” patients. The OMIM track only shows OMIM morbid annotated genes. The database of genomic variants and gnomAD were filtered for duplications/gains of >0.1 Mb in the population, which fits well with the concentration of segmental duplication (bottom track).
Aalfs, C. M., Fantes, J. A., Wenniger-Prick, L. J., Sluijter, S., Hennekam, R. C., van Heyningen, V., et al. (1997). Tandem duplication of 11p12-p13 in a child with borderline development delay and eye abnormalities: dose effect of the PAX6 gene product? Am. J. Med. Genet. 73, 267–271. doi:10.1002/(sici)1096-8628(19971219)73:3<267::aid-ajmg7>3.0.co;2-p
Amor, D. J., and Choo, K. H. (2002). Neocentromeres: role in human disease, evolution, and centromere study. Am. J. Hum. Genet. 71, 695–714. doi:10.1086/342730
Baldwin, E. L., May, L. F., Justice, A. N., Martin, C. L., and Ledbetter, D. H. (2008). Mechanisms and consequences of small supernumerary marker chromosomes: from Barbara McClintock to modern genetic-counseling issues. Am. J. Hum. Genet. 82, 398–410. doi:10.1016/j.ajhg.2007.10.013
Ballif, B. C., Hornor, S. A., Sulpizio, S. G., Lloyd, R. M., Minier, S. L., Rorem, E. A., et al. (2007). Development of a high-density pericentromeric region BAC clone set for the detection and characterization of small supernumerary marker chromosomes by array CGH. Genet. Med. 9, 150–162. doi:10.1097/gim.0b013e3180312087
Battaglia, A. (2008). The inv dup (15) or idic (15) syndrome (Tetrasomy 15q). Orphanet J. Rare Dis. 3, 30. doi:10.1186/1750-1172-3-30
Benn, P. (2021). Uniparental disomy: origin, frequency, and clinical significance. Prenat. Diagn. 41, 564–572. doi:10.1002/pd.5837
Blakey-Cheung, S., Parker, P., Schlaff, W., Monseur, B., Keppler-Noreuil, K., and Al-Kouatly, H. B. (2020). Diagnosis and clinical delineation of mosaic tetrasomy 5p. Eur. J. Med. Genet. 63, 103634. doi:10.1016/j.ejmg.2019.02.006
Castronovo, C., Valtorta, E., Crippa, M., Tedoldi, S., Romitti, L., Amione, M. C., et al. (2013). Design and validation of a pericentromeric BAC clone set aimed at improving diagnosis and phenotype prediction of supernumerary marker chromosomes. Mol. Cytogenet. 6, 45. doi:10.1186/1755-8166-6-45
Chen, C. P., Chen, M., Wang, P. T., Chern, S. R., Chen, S. W., Lai, S. T., et al. (2017). Prenatal diagnosis and molecular cytogenetic characterization of mosaicism for a small supernumerary marker chromosome derived from chromosome 11. Taiwan J. Obstet. Gynecol. 56, 394–397. doi:10.1016/j.tjog.2017.04.025
Chen, X., Xu, H., Shi, W., Wang, F., Xu, F., Zhang, Y., et al. (2021). 11p11.12p12 duplication in a family with intellectual disability and craniofacial anomalies. BMC Med. Genomics 14, 99. doi:10.1186/s12920-021-00945-8
Chuang, L., Wakui, K., Sue, W. C., Su, M. H., Shaffer, L. G., and Kuo, P. L. (2005). Interstitial deletion 11(p11.12p11.2) and analphoid marker formation results in inherited Potocki-Shaffer syndrome. Am. J. Med. Genet. 133A, 180–183. doi:10.1002/ajmg.a.30362
Dalprà, L., Giardino, D., Finelli, P., Corti, C., Valtorta, C., Guerneri, S., et al. (2005). Cytogenetic and molecular evaluation of 241 small supernumerary marker chromosomes: cooperative study of 19 Italian laboratories. Genet. Med. 7, 620–625. doi:10.1097/01.gim.0000182876.57766.2d
Daniel, A., and Malafiej, P. (2003). A series of supernumerary small ring marker autosomes identified by FISH with chromosome probe arrays and literature review excluding chromosome 15. Am. J. Med. Genet. 117A, 212–222. doi:10.1002/ajmg.a.10100
Dettori, J. R. (2011). Loss to follow-up. Evid. Based. Spine Care J. 2, 7–10. doi:10.1055/s-0030-1267080
Fei, X., Qi, M., Zhao, Y., and Li-Ling, J. (2011). Identification and characterization of a complex pure mosaic of small supernumerary marker chromosomes involving 11p11.12 → q12.1 and 19p12 → q12 regions in a child featuring multiple congenital anomalies. Am. J. Med. Genet. 155A, 3116–3121. doi:10.1002/ajmg.a.34346
Fernández-Toral, J., Rodríguez, L., Plasencia, A., Martínez-Frías, M. L., Ewers, E., Hamid, A. B., et al. (2010). Four small supernumerary marker chromosomes derived from chromosomes 6, 8, 11 and 12 in a patient with minimal clinical abnormalities: a case report. J. Med. Case Rep. 4, 239. doi:10.1186/1752-1947-4-239
Fickelscher, I., Starke, H., Schulze, E., Ernst, G., Kosyakova, N., Mkrtchyan, H., et al. (2007). A further case with a small supernumerary marker chromosome (sSMC) derived from chromosome 1--evidence for high variability in mosaicism in different tissues of sSMC carriers. Prenat. Diagn. 27, 783–785. doi:10.1002/pd.1776
Goossens, E., Cayenberghs, R., and Fryns, J. P. (1999). Moderate mental retardation without dysmorphic symptoms in intrachromosomal 11p12 duplication. Genet. Couns. 10, 137–140. n.a.
Guichet, A. (2005). Pericentromeric 11p11 duplication in a girl with isolated learning disability, particularly dyscalculia. Chromosome Res. 13 (1), 32. Abstractno 1.39-P.
Guilherme, R. S., Klein, E., Venner, C., Hamid, A. B., Bhatt, S., Melaragno, M. I., et al. (2012). Human ring chromosomes and small supernumerary marker chromosomes-do they have telomeres? Chromosome Res. 20, 825–835. doi:10.1007/s10577-012-9316-x
Haaf, T., Sumner, A. T., Köhler, J., Willard, H. F., Schmid, M., and Summer, A. T. (1992). A microchromosome derived from chromosome 11 in a patient with the CREST syndrome of scleroderma. Cytogenet. Cell. Genet. 60, 12–17. doi:10.1159/000133284
Haddad, B. R., Schröck, E., Meck, J., Cowan, J., Young, H., Ferguson-Smith, M. A., et al. (1998). Identification of de novo chromosomal markers and derivatives by spectral karyotyping. Hum. Genet. 103, 619–625. doi:10.1007/s004390050878
Hamid, A. B., Kreskowski, K., Weise, A., Kosayakova, N., Mrasek, K., Voigt, M., et al. (2012). How to narrow down chromosomal breakpoints in small and large derivative chromosomes--a new probe set. J. Appl. Genet. 53, 259–269. doi:10.1007/s13353-012-0098-9
Hliscs, R., Mühlig, P., and Claussen, U. (1997). The spreading of metaphases is a slow process which leads to a stretching of chromosomes. Cytogenet. Cell. Genet. 76, 167–171. doi:10.1159/000134537
Hochstenbach, R., van Gijn, M. E., Krijtenburg, P. J., Raemakers, R., van 't Slot, R., Renkens, I., et al. (2013). Gain of FAM123B and ARHGEF9 in an obese man with intellectual disability, congenital heart defects and multiple supernumerary ring chromosomes. Mol. Syndromol. 3, 274–283. doi:10.1159/000345241
Huang, M. H., Lee, C., Chang, J. S., Wang, H. C., Lai, H. L., Chang, C. C., et al. (2019). Retrospectively investigating the 12-year experience of prenatal diagnosis of small supernumerary marker chromosomes through array comparative genomic hybridization. Taiwan J. Obstet. Gynecol. 58, 139–144. doi:10.1016/j.tjog.2018.11.026
Hussein, S. S., Kreskowski, K., Ziegler, M., Klein, E., Hamid, A. B., Kosyakova, N., et al. (2014). Mitotic stability of small supernumerary marker chromosomes depends on their shape and telomeres - a long term in vitro study. Gene 552, 246–248. doi:10.1016/j.gene.2014.09.041
Itsara, A., Cooper, G. M., Baker, C., Girirajan, S., Li, J., Absher, D., et al. (2009). Population analysis of large copy number variants and hotspots of human genetic disease. Am. J. Hum. Genet. 84, 148–161. doi:10.1016/j.ajhg.2008.12.014
Jehee, F. S., Bertola, D. R., Yelavarthi, K. K., Krepischi-Santos, A. C., Kim, C., Vianna-Morgante, A. M., et al. (2007). An 11q11-q13.3 duplication, including FGF3 and FGF4 genes, in a patient with syndromic multiple craniosynostoses. Am. J. Med. Genet. 143A, 1912–1918. doi:10.1002/ajmg.a.31863
Joshi, A., Lall, M., Agarwal, S., Paliwal, P., Saviour, P., Mahajan, S., et al. (2019). Molecular characterization of supernumerary marker chromosomes found as unexpected chromosome abnormalities in nine prenatal and nine postnatal samples. Obstet. Gynecol. Int. J. 10, 211–221. doi:10.15406/ogij.2019.10.00446
Kieback, P., Hennig, C., Jauch, A., and Liehr, T. (2007). Prenatal diagnosis of a direct intrachromosomal duplication 11p12->11q11.2∼12.1, a one year follow up. Med. Gen. 19, 83. Abstractno P080.
Kontodiou, M., Paspaliaris, V., Dagklis, T., Siomou, E., Al-Rikabi, A. H., Tsita, K., et al. (2018). Identification of a small supernumerary marker chromosome involving 11p14.1q12.1 in a prenatal case: clinical and molecular characterization. OBM Transpl. 2, 1. doi:10.21926/obm.genet.1803035
Kozlowski, P., Grund, I., Hickmann, G., Stressig, R., and Knippel, A. J. (2006). Quantitative fluorescent polymerase chain reaction versus cytogenetics: risk-related indication and clinical implication of nondetected chromosomal disorders. Fetal diagn. Ther. 21, 217–223. doi:10.1159/000089306
Kurtas, N. E., Xumerle, L., Leonardelli, L., Delledonne, M., Brusco, A., Chrzanowska, K., et al. (2019). Small supernumerary marker chromosomes: a legacy of trisomy rescue? Hum. Mutat. 40, 193–200. doi:10.1002/humu.23683
Leung, W. C., Waters, J. J., and Chitty, L. (2004). Prenatal diagnosis by rapid aneuploidy detection and karyotyping: a prospective study of the role of ultrasound in 1589 second-trimester amniocenteses. Prenat. Diagn. 24, 790–795. doi:10.1002/pd.985
Li, C., Luo, W., Xiao, T., Yang, X., Ou, M., Zhang, L., et al. (2021). Case report: genetic analysis of a small supernumerary marker chromosome in a unique case of mosaic Turner syndrome. Front. Pediatr. 10, 799284. doi:10.3389/fped.2022.799284
Liehr, T. (2012). Small supernumerary marker chromosomes (sSMC) - a guide for human geneticists and clinicians; with contributions by UNIQUE (The Rare Chromosome Disorder Support Group). Berlin: Springer.
Liehr, T. (2014). Small supernumerary marker chromosomes detected in connection with infertility. Zhonghua Nan Ke Xue 20, 771–780. n.a.
Liehr, T. (2023). Small supernumerary marker chromosomes. https://cs-tl.de/DB/CA/sSMC/0-Start.html (accessed on August 20, 2023).
Liehr, T., and Al-Rikabi, A. (2019). Mosaicism: reason for normal phenotypes in carriers of small supernumerary marker chromosomes with known adverse outcome. A systematic review. Front. Genet. 10, 1131. doi:10.3389/fgene.2019.01131
Liehr, T., Cirkovic, S., Lalic, T., Guc-Scekic, M., de Almeida, C., Weimer, J., et al. (2013). Complex small supernumerary marker chromosomes - an update. Mol. Cytogenet. 6, 46. doi:10.1186/1755-8166-6-46
Liehr, T., Ewers, E., Hamid, A. B., Kosyakova, N., Voigt, M., Weise, A., et al. (2011). Small supernumerary marker chromosomes and uniparental disomy have a story to tell. J. Histochem. Cytochem. 59, 842–848. doi:10.1369/0022155411412780
Liehr, T., Utine, G. E., Trautmann, U., Rauch, A., Kuechler, A., Pietrzak, J., et al. (2007). Neocentric small supernumerary marker chromosomes (sSMC)--three more cases and review of the literature. Cytogenet. Genome Res. 118, 31–37. doi:10.1159/000106438
Maggert, K. A., and Karpen, G. H. (2000). Acquisition and metastability of centromere identity and function: sequence analysis of a human neocentromere. Genome Res. 10, 725–728. doi:10.1101/gr.10.6.725
Malvestiti, F., De Toffol, S., Grimi, B., Chinetti, S., Marcato, L., Agrati, C., et al. (2014). De novo small supernumerary marker chromosomes detected on 143,000 consecutive prenatal diagnoses: chromosomal distribution, frequencies, and characterization combining molecular cytogenetics approaches. Prenat. Diagn. 34, 460–468. doi:10.1002/pd.4330
Marle, N., Martinet, D., Aboura, A., Joly-Helas, G., Andrieux, J., Flori, E., et al. (2014). Molecular characterization of 39 de novo sSMC: contribution to prognosis and genetic counselling, a prospective study. Clin. Genet. 85, 233–244. doi:10.1111/cge.12138
Maziad, A. S., and Seaver, L. H. (2015). Mosaic tetrasomy 20p associated with osteoporosis and recurrent fractures. Am. J. Med. Genet. 167A, 1582–1586. doi:10.1002/ajmg.a.37074
Melo, J. B., Backx, L., Vermeesch, J. R., Santos, H. G., Sousa, A. C., Kosyakova, N., et al. (2011). Chromosome 5 derived small supernumerary marker: towards a genotype/phenotype correlation of proximal chromosome 5 imbalances. J. Appl. Genet. 52, 193–200. doi:10.1007/s13353-011-0035-3
Metay, C., Lecerf, L., Tosca, L., Briand-Suleau, A., Ortonne, V., Serero, S., et al. (2011). Rare modes of paternal malsegregation of a translocation t(11;13)(q25;q14) in two patients with intellectual disability. Chromosome Res. 19 (S1), S100–S101. (Abstractno 1.P114.
Napoleone, R. M., Varela, M., and Andersson, H. C. (1997). Complex congenital heart malformations in mosaic tetrasomy 8p: case report and review of the literature. Am. J. Med. Genet. 73, 330–333. doi:10.1002/(sici)1096-8628(19971219)73:3<330::aid-ajmg19>3.0.co;2-k
Neill, N. J., Torchia, B. S., Bejjani, B. A., Shaffer, L. G., and Ballif, B. C. (2010). Comparative analysis of copy number detection by whole-genome BAC and oligonucleotide array CGH. Mol. Cytogenet. 3, 11. doi:10.1186/1755-8166-3-11
Nelle, H., Schreyer, I., Ewers, E., Mrasek, K., Kosyakova, N., Merkas, M., et al. (2010). Presence of harmless small supernumerary marker chromosomes hampers molecular genetic diagnosis: a case report. Mol. Med. Rep. 3, 571–574. doi:10.3892/mmr_00000299
Nietzel, A., Rocchi, M., Starke, H., Heller, A., Fiedler, W., Wlodarska, I., et al. (2001). A new multicolor-FISH approach for the characterization of marker chromosomes: centromere-specific multicolor-FISH (cenM-FISH). Hum. Genet. 108, 199–204. doi:10.1007/s004390100459
OMIM (2023). An online catalog of human genes and genetic disorders. Available at: https://www.omim.org/ (accessed on August 20, 2023).
ORPHA (2023). The portal for rare diseases and orphan drugs. Available at: https://www.orpha.net/consor/cgi-bin/Disease.php?lng=EN (accessed on August 20, 2023).
Pai, G. S., Thomas, G. H., and Benke, P. J. (1981). Absence of constitutive heterochromatin in a partially identified supernumerary marker chromosome. J. Med. Genet. 18, 392–394. doi:10.1136/jmg.18.5.392
Plattner, R., Heerema, N. A., Yurov, Y. B., and Palmer, C. G. (1993). Efficient identification of marker chromosomes in 27 patients by stepwise hybridization with alpha-satellite DNA probes. Hum. Genet. 91, 131–140. doi:10.1007/BF00222713
Qi, M., Zhao, Y., Wang, Y., and Li, T. (2013). A new small supernumerary marker chromosome involving 14pter → q12 in a child with severe neurodevelopmental retardation: case report and literature review. Gene 531, 457–461. doi:10.1016/j.gene.2013.08.084
Rauch, A., Pfeiffer, R. A., Trautmann, U., Liehr, T., Rott, H. D., and Ulmer, R. (1992). A study of ten small supernumerary (marker) chromosomes identified by fluorescence in situ hybridization (FISH). Clin. Genet. 42, 84–90. doi:10.1111/j.1399-0004.1992.tb03145.x
Rice, A. M., and McLysaght, A. (2017). Dosage sensitivity is a major determinant of human copy number variant pathogenicity. Nat. Commun. 8, 14366. doi:10.1038/ncomms14366
Robberecht, C., Voet, T., Utine, G. E., Schinzel, A., de Leeuw, N., Fryns, J. P., et al. (2012). Meiotic errors followed by two parallel postzygotic trisomy rescue events are a frequent cause of constitutional segmental mosaicism. Mol. Cytogenet. 5, 19. doi:10.1186/1755-8166-5-19
Sanz, R., Sousa, A., and Gonzáles, A. (2005). Identification of the chromosomal origin of small supernumerary marker chromosomes and its phenotypic effect. Chromosome Res. 13 (1), 69. Abstractno 1.126-P.
Silveira-Santos, R., Sousa, A. C., Avila, M., Serafim, S., Custódio, S., Sousa, A. B., et al. (2012). Back to the karyotype: a case of mosaic marker chromosome 11 detected by aCGH. Eur. J. Hum. Genet. 20 (1). Abstractno P03.013.
Starke, H., Nietzel, A., Weise, A., Heller, A., Mrasek, K., Belitz, B., et al. (2003). Small supernumerary marker chromosomes (SMCs): genotype-phenotype correlation and classification. Hum. Genet. 114, 51–67. doi:10.1007/s00439-003-1016-3
Starke, H., Raida, M., Trifonov, V., Clement, J. H., Loncarevic, I. F., Heller, A., et al. (2001). Molecular cytogenetic characterization of an acquired minute supernumerary marker chromosome as the sole abnormality in a case clinically diagnosed as atypical Philadelphia-negative chronic myelogenous leukaemia. Br. J. Haematol. 113, 435–438. doi:10.1046/j.1365-2141.2001.02787.x
Strobel, R. J., Riccardi, V. M., Ledbetter, D. H., and Hittner, H. M. (1980). Duplication 11p11.3 leads to 14.1 to meiotic crossing--over. Am. J. Med. Genet. 7, 15–20. doi:10.1002/ajmg.1320070105
Süleyman, M., Oğuz, S., Kaykı, G., Çelik, H. T., Şimsek-Kiper, P. Ö., Utine, G. E., et al. (2022). A very rare case of a newborn with tetrasomy 9p and literature review. Turk. J. Pediatr. 64, 171–178. doi:10.24953/turkjped.2021.685
Thangavelu, M., Tepperberg, J. H., Hume, J., and Huang, B. (2011). “A 3.44 MB interstitial duplication of chromosome 3 with no apparent phenotype detected by SNP array,” in Abstracts of the 12th international congress of human genetics 2011 (Montreal: Canada). Abstractno 1257T.
Till, M., Rafat, A., Charrin, C., Plauchu, H., and Germain, D. (1991). Duplication of chromosome 11 centromere in fetal and maternal karyotypes: a new variant? Prenat. Diagn. 11, 481–482. doi:10.1002/pd.1970110713
Tsuchiya, K. D., Opheim, K. E., Hannibal, M. C., Hing, A. V., Glass, I. A., Raff, M. L., et al. (2008). Unexpected structural complexity of supernumerary marker chromosomes characterized by microarray comparative genomic hybridization. Mol. Cytogenet. 1, 7. doi:10.1186/1755-8166-1-7
Ulmer, R., Pfeiffer, R. A., Wiest, E., Goelz, R., and Trautmann, U. (1997). Multiple (up to seven) different accessory small marker chromosomes: prenatal diagnosis and follow-up. Ann. Genet. 40, 109–114. n.a.
van Paassen, B. W., van der Kooi, A. J., van Spaendonck-Zwarts, K. Y., Verhamme, C., Baas, F., and de Visser, M. (2014). PMP22 related neuropathies: Charcot-Marie-Tooth disease type 1A and hereditary neuropathy with liability to pressure palsies. Orphanet J. Rare Dis. 9, 38. doi:10.1186/1750-1172-9-38
Wang, Y., Lazier, J., Myles-Reid, D., Noor, A., Chitayat, D., and Greenfeld, E. (2023). Role of comprehensive cytogenomic investigation in successful reproductive outcome of parental small neocentromeric supernumerary ring chromosome: a case report. Clin. Case Rep. 11, e6632. doi:10.1002/ccr3.6632
Watson, C. T., Marques-Bonet, T., Sharp, A. J., and Mefford, H. C. (2014). The genetics of microdeletion and microduplication syndromes: an update. Annu. Rev. Genomics Hum. Genet. 15, 215–244. doi:10.1146/annurev-genom-091212-153408
Keywords: small supernumerary marker chromosomes (sSMCs), chromosome 11, triplosensitive genes, pericentric region, copy number variation (CNV), uniparental disomy (UPD), imprinting
Citation: Liehr T, Ziegler M, Person L, Kankel S, Padutsch N, Weise A, Weimer JP, Williams H, Ferreira S, Melo JB and Carreira IM (2023) Small supernumerary marker chromosomes derived from human chromosome 11. Front. Genet. 14:1293652. doi: 10.3389/fgene.2023.1293652
Received: 13 September 2023; Accepted: 28 November 2023;
Published: 15 December 2023.
Edited by:
Paulo Ricardo Gazzola Zen, Federal University of Health Sciences of Porto Alegre, BrazilReviewed by:
Samarth S. Bhatt, Neuberg Supratech Reference Laboratory, IndiaCopyright © 2023 Liehr, Ziegler, Person, Kankel, Padutsch, Weise, Weimer, Williams, Ferreira, Melo and Carreira. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Thomas Liehr, dGhvbWFzLmxpZWhyQG1lZC51bmktamVuYS5kZQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.