 Yi Ren
Yi Ren Shuang Li
Shuang Li Jia-Jia Lei
Jia-Jia Lei Ru Li
Ru Li Bai-Xue Dong
Bai-Xue Dong Jing Yang
Jing Yang- 1Department of Endocrinology, The First Hospital of Shanxi Medical University, Taiyuan, China
- 2Department of First Clinical Medical School, Shanxi Medical University, Taiyuan, China
Background: Early detection and diagnosis are important crucial to prevent life-threatening acute attacks in patients with acute intermittent porphyria (AIP). We aim to provide comprehensive data on the clinical and hydroxymethylbilane synthase (HMBS) gene variant characteristics and genotype-phenotype association of Chinese patients with AIP in order to improve clinicians’ knowledge of AIP and reduce misdiagnosis and mistaken treatment.
Methods: We searched the literature on Chinese patients with AIP in PubMed, Web of Science, Wiley Online Library, ScienceDirect and Chinese literature databases up to August 2023 in our analysis to explore the clinical and HMBS gene variant characteristics of Chinese patients with AIP.
Results: A total of 41 original articles associated with Chinese AIP patients were included for analysis: 97 variants were detected in 160 unrelated families, including 35 missense, 29 frameshift, 24 splicing and 9 nonsense variants, with c.517C>T being the most common variant. Clinical data were reported in 77 of 160 patients: Most of them were female (67/77) and the age was 28.8 ± 9.9 years. The most common symptom was abdominal pain (73/77, 94.8%), followed by central nervous system symptoms (45/77, 58.4%). 13.0% (10/77) of patients experienced psychiatric symptoms. Hyponatremia was the most common electrolyte abnormality (42/77). 31 patients received carbohydrate loading therapy, and 30 of them were improved. 6 patients were treated with carbohydrate loading combined with hemin therapy and 5 eventually improved. All variants causing premature stop codons, frameshifts or enzyme activity center may experience more severe clinical phenotypes such as seizures, respiratory paralysis, intracranial hemorrhage disorder or respiratory failure.
Conclusion: The most common presenting symptom in Chinese AIP patients was abdominal pain, followed by central nervous system symptoms. The HMBS gene analysis in Chinese AIP patients revealed that the heterogeneity is strong and the most common variant was missense mutation, with c.517C>T being the most common variant. The genotype-phenotype association helps guide clinical diagnosis and treatment. However, the treatment for AIP in China is limited and monolithic, and more attention needs to be paid to the treatment.
1 Introduction
Acute intermittent porphyria (AIP) is an autosomal dominant disorder caused by partial deficiency of the third enzyme, hydroxymethylbilane synthase (HMBS), in heme synthesis (Stölzel et al., 2021). It has a low penetrance of only 1% based on all AIP heterozygotes (Chen et al., 2016). Most carriers remain disease-free for life and are known as latent AIP, while some patients experience life-threatening acute attacks, known as manifest AIP, due to common factors such as menstruation, smoking, drinking, infection, fasting and drug. The clinical manifestations of acute attacks of AIP are complex and varied involving multiple systems such as the gastrointestinal, neurological, and psychiatric systems. And there is substantial heterogeneity in severity, even in the same family.
The HMBS gene is considered to be the only gene responsible for the disease. Its housekeeping transcript consists of 14 exons and corresponding introns. Its pathogenic variant can lead to functional defects of HMBS. Genetic screening provides 95% sensitivity and about 100% specificity, which has been rapidly incorporated into good clinical practice (Kauppinen, 2004). It not only can diagnose the manifest AIP, but is also one of the most accurate methods to screen latent AIP. It has great significance for the early diagnosis of high-risk groups, effective prevention of acute attacks and improvement of the patients’ lives. With the development of gene sequencing test, numerous of mutations and their linked phenotypes have been identified, which has helped to establish genotype-phenotype correlations.
At present, there is a lack of epidemiological data on AIP worldwide, but Europe performed large scale prospective study to investigate the incidence of porphyrias (Elder et al., 2013). And multiple countries such as South Africa (Fortgens et al., 2017), the United States (Bonkovsky et al., 2014), Argentina (Cerbino et al., 2015), Colombia (Jaramillo-Calle and Aguirre Acevedo, 2019), and Russia (Goncharova et al., 2019) have reported cohort studies on AIP. And with the development of sequencing technology, more and more AIP patients have been reported in China. However, most of them are case reports, and there is still a lack of systematic analysis of the characteristics of Chinese AIP patients. A previous study identified 5 pathogenic and 20 likely pathogenic variants from the ChinaMAP database and preliminarily analyzed the epidemiological features of AIP in Hebei Province, China (Ma et al., 2022). However, there is insufficient knowledge and research on AIP in China, which can easily lead to misdiagnosis and mistaken treatment in clinical practice. How to provide timely and accurate diagnosis and treatment is a major challenge for healthcare worker. The aim of this study was to describe the clinical features and the characteristics of HMBS gene variants and genotype-phenotype association of Chinese AIP patients, in order to improve clinicians’ knowledge of AIP and to help clinicians in early identifying patients with this disease guide clinical management, and to provide genetic counselling and health education for asymptomatic heterozygotes.
2 Methods
2.1 Study design
This systematic review was conducted in accordance with the Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) statement checklist (Page et al., 2021) (Supplementary Material S1).
2.2 Strategy, criteria, and procedures for the literature search
We searched all literature about Chinese AIP patients for analysis in PubMed, Web of science, Wiley Online Library, ScienceDirect and Chinese databases CNKI, Wanfang and CQVIP up to August 2023. In the PubMed, Web of science, Wiley Online Library, ScienceDirect databases, we used the keywords “Acute Intermittent Porphyria” and “China or Chinese.” In the Chinese database, we used the keyword “Acute Intermittent Porphyria.” Inclusion criteria: (1) the cases were Chinese patients; (2) the diagnosis of AIP was confirmed by clinical and sequencing results (Zhang et al., 2020); (3) the variant was clearly reported. Exclusion criteria: (1) duplicated variant sites found in the same family or uncertain variant sites; (2) missing or uncomplete clinical data. The type of literature is not limited, all literature reporting correctly and complete information on AIP cases was accepted, such as original research, case reports, and briefs etc.
2.3 Data extraction
Data on the general information (age, sex, etc.), HMBS gene variants, clinical presentations, laboratory tests, treatment, and outcomes of the patients were retrieved. Microsoft Excel Spreadsheet software was used to organized and collated the extracted data. Two authors (SL, JL) identified the relevant original articles and extracted the data independently, while the third author (BD) checked the results. In case of disagreement, the relevant programs were repeated until a consensus was reached among the authors.
2.4 Data synthesis
The extracted data was then analyzed and interpreted by the SL and YR researchers. The primary outcomes assessed were the general information (hospital types, regions, age, sex, etc.), HMBS gene variants, clinical presentations, laboratory tests, treatment, and outcomes. A narrative (descriptive) method was conducted to synthesize this information.
2.5 The pathogenicity rating of HMBS variants and clinical phenotype
According to the standards and guidelines of American College of Medical Genetics and Genomics (ACMG) (Richards et al., 2015), each pathogenic criterion is weighted as very strong (PVS1), strong (PS1–4), moderate (PM1–6), or supporting (PP1–5). Each variant was classified for pathogenic (P), likely pathogenic (LP) and uncertain significance (VUS). AIP can present with a sudden life-threatening crisis characterized by severe abdominal pain and neuropsychiatric symptoms (Bissell et al., 2017). According to the main manifestation, the severity of the disease was classified into mild (abdominal pain is the main clinical manifestation), moderate (accompanied by neuropsychiatric symptoms) and severe (experiencing respiratory paralysis, intracranial hemorrhage, disseminated intravascular coagulation (DIC), acute heart failure (AHF), chronic renal failure (CRF) or respiratory failure, etc).
3 Results
3.1 Search results
The screening process is shown in Figure 1. A total of 602 publications were identified after searching PubMed (n = 52), Web of Science (n = 36), Wiley Online Library (n = 12), ScienceDirect (n = 144) and Chinese databases CNKI (n = 175), Wanfang (n = 115) and CQVIP (n = 68). After removing duplicates, titles and abstracts of 434 publications were screened. The full text of the remaining 209 studies was downloaded and evaluated, and another 168 studies were further excluded due to no variants reported or variants of the same family reported. Finally, 41 studies (Lam et al., 2001; Yang et al., 2008; Lam et al., 2011; Xie, 2012; Kong et al., 2013; Zhou, 2014; Li et al., 2015; Li et al., 2015; Chen et al., 2015; Jiao et al., 2015; Yang et al., 2015; You et al., 2015; Yuan et al., 2015; Li et al., 2016; Yang et al., 2016; Lei et al., 2017; Li et al., 2017; Yang et al., 2017; Hu et al., 2018a; Zheng et al., 2018; Wang et al., 2019; Wang et al., 2019; Hu, 2019; Zhang and Gao, 2019; Yang et al., 2020; Yang et al., 2020; Zhang et al., 2020; Sun et al., 2020; Teng et al., 2020; Fu et al., 2021; Gao et al., 2021; Huang et al., 2021; Zhang et al., 2021; Haiqing, 2022; Hu et al., 2022; Li, 2022; Li et al., 2022; Yang et al., 2022; Zhou et al., 2022; Guo and Luo, 2023; Liang and Li, 2023) were obtained for analysis (21 English-language and 20 Chinese-language articles).
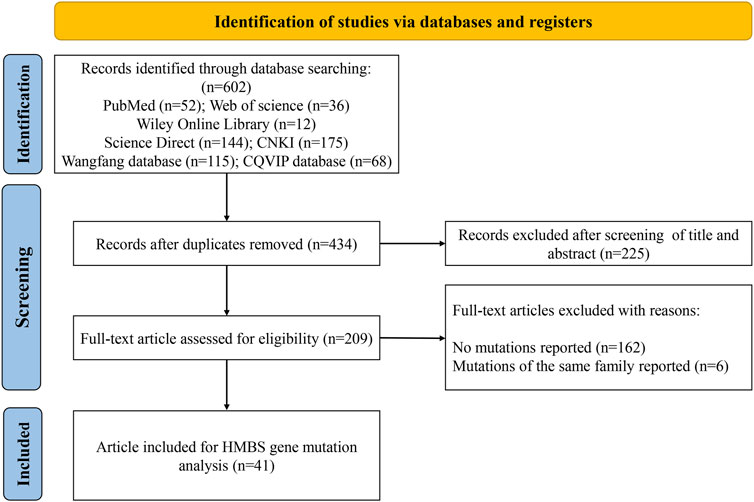
FIGURE 1. Flow chart of the study selection process.
3.2 Genetic analysis of the HMBS gene in Chinese with AIP
A total of 41 articles involving 160 patients were enrolled in the study, most of which were case reports. The majority of reports were sourced from grade III hospitals, predominantly originating from Hebei, Taiwan, and Beijing, with sporadic cases identified in other provinces. Totally, 97 variants were detected in 160 patients (Table 1), including 35 missense mutations (36.1%), 29 frameshift mutations (29.9%), 24 splicing mutations (24.7%) and 9 nonsense mutations (9.3%). Exon variants were mainly concentrated in exons 11 and 14 (34.0%). No variants were found in exon 1 (Figure 2). The majority of the mutations were family specificity, but 21.6% (21/97) variants occurred in several families, among these, c.517C>T was the most common variant, which was found in 16 unrelated families (Figure 2). According to the ACMG, among 97 variants, 45 variants are pathogenic, 14 variants are likely pathogenic and 38 variants are uncertain significance (Table 1).
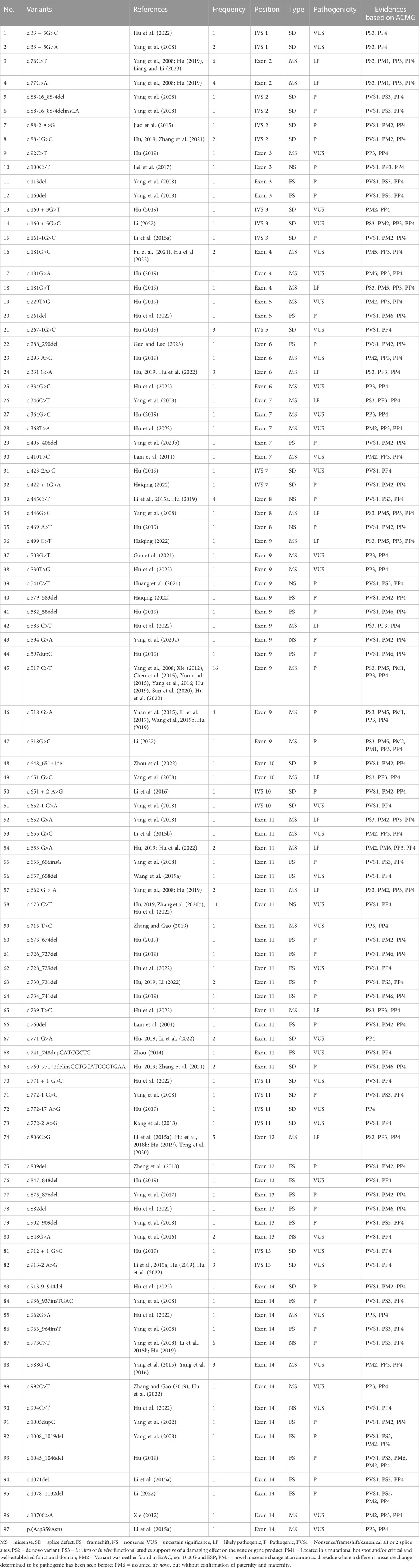
TABLE 1. 97 HMBS gene mutations identified in 160 Chinese patients with AIP.
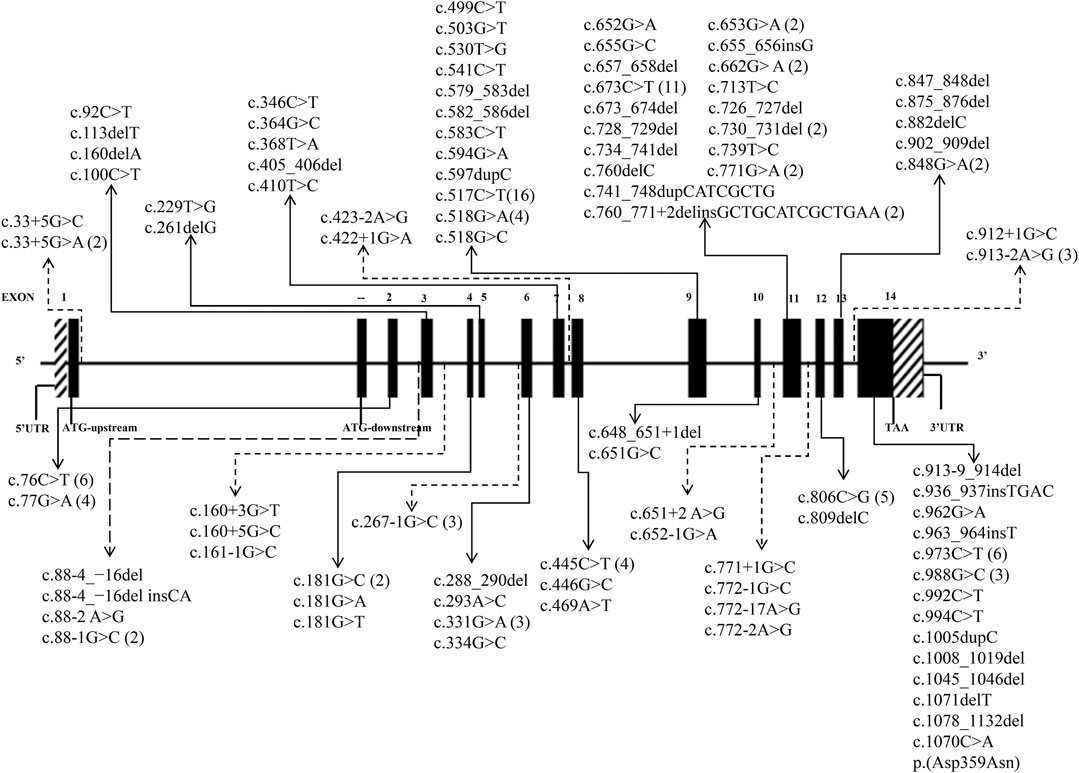
FIGURE 2. 97 HMBS gene mutations spectrum in transcript (NM_000190.4). (The solid lines indicate exon mutation sites. Dashed lines indicate intron mutation sites. The parentheses indicate the frequency of mutations).
3.3 Clinical characteristic of Chinese people with AIP
Clinical data were clearly reported for 77 out of 160 patients. Among the 77 AIP patients, there were 65 females (female/male ratio: 6.7:1), with an average age of 28.8 ± 9.9 years (range: 1–59 years), and the majority (83.1%) were aged between 18-39 years old (Table 2). The most common symptom was abdominal pain (73/77, 94.8%), often accompanied with nausea, anorexia, vomiting, constipation, and ileus. 58.4% (45/77) of AIP patients suffered from central nervous system symptoms, such as consciousness disturbances, seizures, respiratory paralysis, dizziness, headache, and dysphagia. One patient presented with dizziness and was diagnosed with “acute cerebral infarction,” while another patient presented with consciousness disturbances being diagnosed with “intracranial hemorrhage.” 32.2% (24/77) of patients experienced peripheral nervous system symptoms, such as weakness, numbness or stiffness in limbs, somatic pain, and limb paralysis. 13.0% (10/77) of patients experienced psychiatric symptoms, such as anxiety, depression, irritability, hallucination, and delirium. 48.1% (37/77) of patients suffered from autonomic nervous system symptoms, such as tachycardia, hypertension, urinary retention, and hypotension. Hyponatremia was the most common laboratory abnormality (42/77). Other abnormalities observed were liver dysfunction (18/77), kidney dysfunction (8/77), anemia (13/77), hypokalemia (3/77), sex hormonal imbalance (2/77), cortisol rhythm disturbance (1/77). Urinary PBG testing was performed on 38 patients, all results were positive (Table 2). Treatment strategy and outcome were reported in 42 patients (Table 2). 31 patients received the administration of high glucose, and 30 of them were improved. One severe patient with respiratory paralysis died. 6 patients were treated with high glucose combined with hemin therapy, 5 eventually improved and one died due to multi-organ failure. One case was complicated by depression, and after effective antidepressant treatment, her AIP symptoms were also controlled. After receiving prophylactic hemin infusion treatment, two patients exhibited a reduction in the frequency of acute attacks. In addition, three patients who experienced menstrual-associated acute attacks were administered gonadotropin-releasing hormone analogue (GnRH-a) for menstrual suppression to mitigate these episodes. Symptomatic treatments including fluid restriction, sedation, analgesia, hepatoprotection, and antiepileptic therapy were provided based on individual patient conditions.
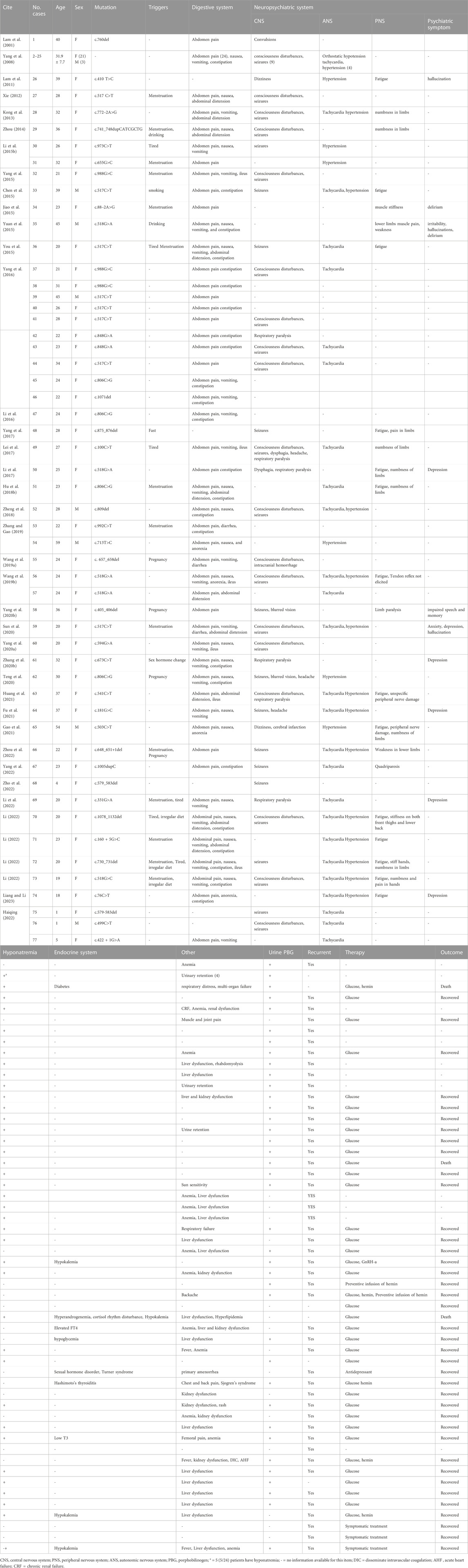
TABLE 2. Clinical data of 77 Chinese patients with AIP.
3.4 Genotype-phenotype association of Chinese people with AIP
A total of 53 Chinese AIP patients with 36 variants were included in the genotype-phenotype association analysis (reported cases 2–25 were excluded because the variants did not match with the patient’s phenotype). As shown in Tables 1, 3, patients with nonsense variants (c.100C>T, c.541C>T, c.594G>A, c.673C>T, c.848G>A, and c.973C>T) had only moderate and severe phenotypes. Patients with frameshift variant (c.405_406del, c.579_583del,c.648_651+1del,c.730_731del,c.760del,c.741_748dupCATCGCTG, c.809del, and c.1005dupC) mainly experienced moderate phenotype, while c. 657_658del and c.875_876del were found in severely affected patients and 1071del was found in mildly affected patient. And patients with missense variant and splice defect were associated with mild, moderate and severe phenotype, while affecting enzyme activity center (c.76C>T, c.517C>T, c.518G>A, and c.518G>C) mainly experienced moderate and severe phenotype.
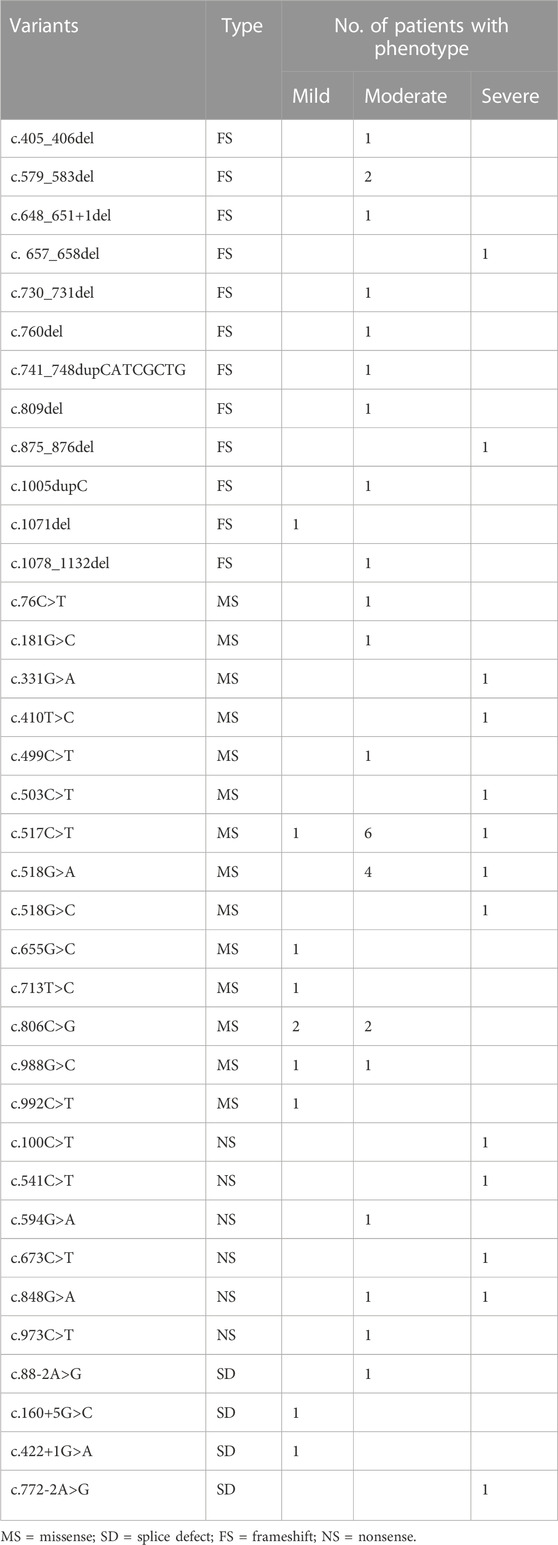
TABLE 3. Genotype-phenotype association of Chinese AIP patient.
4 Discussion
This is the first study to provide a comprehensive description of the clinical and genetic features of Chinese patients with AIP. The findings of this study may have significant implications for the management of this disease. Identifying the most common clinical features can help clinicians to recognize AIP patients across different departments. Since the symptoms of AIP are not specific, diagnosis is usually delayed (Anderson et al., 2005), and molecular analysis of the HMBS gene has become the most useful diagnostic test for identifying asymptomatic AIP family members and those in the intermission of attacks. Improved understanding of the molecular heterogeneity of the HMBS gene can help clinicians provide patients with clinical counseling and health education to prevent life-threatening acute attacks. Additionally, genotype-phenotype association analysis may predict severe clinical phenotypes in future patients.
The clinical manifestations of AIP are diverse and are characterized by life-threatening acute intermittent attacks, caused by porphyrin accumulation in the visceral, central, peripheral, and autonomic nervous systems (Stölzel et al., 2021). Abdominal pain is the most common clinical manifestation during AIP attacks (Ramanujam and Anderson, 2015), and it is also the first symptom to appear (Kothadia et al., 2023). A large population survey in the United States in 2020 showed that approximately 1/4 of patients with a history of abdominal pain had symptoms similar to acute hepatic porphyria (Lakhoo et al., 2021). These gastrointestinal symptoms can often cause decreased appetite and impaired energy intake and absorption, resulting in a negative energy balance in patients, which in turn further exacerbates the onset of AIP. 94.8% (73/77) of the AIP patients included in this study experienced abdominal pain, often accompanied by nausea, vomiting, abdominal distension, and constipation. A prospective, multinational, natural History study showed abdominal pain is the most common symptom during acute attacks (92%) (Gouya et al., 2020), which is consistent with our results. However, the incidence of abdominal pain in the Chinese patients is significantly higher than that in Brazilian patients (77/172,52%) (Souza et al., 2023).
The second major clinical manifestation was neuropsychiatric symptoms, such as conscious disorder, convulsions, weakness, stiffness and numbness of the extremities, anxiety, depression, irritability, hallucinations, and delirium, and the convulsions in some patients may be related to hyponatremia. AIP should be remembered as an important differential diagnosis for neuromuscular disorders (de Souza et al., 2021). In our study, hyponatremia is the most common biochemical abnormality, 55.5% of patients had hyponatremia which may be associated with syndrome of inappropriate antidiuretic hormone secretion (SIADH) (Li et al., 2015; Yang et al., 2016). It has been shown that large accumulations of porphyrin precursors cause damage to the hypothalamus, resulting in increased secretion of vasopressin and retention of large amounts of body fluid, causing dilutional hyponatremia (Aksoy Ö et al., 2020). Therefore, the key to correcting hyponatremia in AIP patients is to limit their fluid intake.
Disorders of the endocrine metabolic system may arise due to abnormal porphyrin metabolism. Our study findings suggest that some patients with AIP might exhibit endocrine disorders, including hyperprolactinemia, hyperandrogenism, disruption of cortisol rhythms, thyroid dysfunction, and abnormal glucose metabolism. Limited research has been conducted on the impact of porphyrin metabolism on the endocrine system; however, it is hypothesized that these alterations could be associated with damage to the hypothalamus, pituitary gland, and other endocrine glands caused by porphyrin accumulation. The precise mechanism underlying these changes is still being investigated.
Glucose loading therapy is effective for most acute attacks. In China, the majority of patients recovered with intravenous glucose infusions. However, glucose can only control acute attacks, and there is a need to develop new specific medicine to treat severely patients and prevent acute attacks of AIP. Hemin is the first line therapy for AIP, and at present, Givosiran also has been emerged as first line therapy for AHP in the last years, however, in China, they are much expensive and difficult to obtain, therefore, it has not yet been put into clinical treatment for AIP patients, only a very small number of Chinese AIP patients receive intravenous hemin infusions to control and prevent acute attacks of AIP. In terms of treatment, avoiding triggers is the key to controlling acute attacks of AIP. Some patients in this study experienced acute attacks related to menstruation and depression, which were reduced with GnRH-a and antidepressant treatment. The majority of patients are able to recover after treatment, but a few severe patients may die due to prolonged delay or worsening of the disease. Therefore, early diagnosis and effective treatment are crucial for patient prognosis. Glucose-loading therapy has demonstrated efficacy in managing the majority of acute attacks. In China, intravenous glucose infusions have led to successful recovery in most patients. However, glucose alone can only control acute attacks, highlighting the urgent need for the development or introduction of new specific medications that can effectively treat severe cases and prevent acute attacks of AIP. Currently, hemin is considered the first-line therapy for AIP, while Givosiran has emerged as the first-line therapy for AHP in recent years. Nevertheless, these medications are often prohibitively expensive and difficult to access in China, which hinders their clinical use among AIP patients. Consequently, only a small fraction of Chinese AIP patients receive intravenous hemin infusions to manage and prevent acute attacks. To effectively manage such attacks, it is crucial to identify triggers and implement appropriate interventions. Notably, some patients in this study experienced acute attacks associated with menstruation and depression; however, these symptoms were alleviated through GnRH-a administration and antidepressant treatment respectively. While most patients achieve recovery after treatment initiation, a few severe cases may succumb due to delayed diagnosis or disease progression. Therefore, early diagnosis coupled with effective treatment strategies play a pivotal role in determining patient prognosis. According to the Human Gene Mutation Database (HGMD), more than 500 variants have been reported, the majority of which were missense mutations (31.9%), followed by small deletions, insertions and duplications (Bustad et al., 2021). A number of foreign studies also showed that missense mutations accounted for the largest proportion, which was consistent with our findings. In this study, 97 variants were detected in 160 unrelated families, including 35 missense, 29 frameshift, 24 splicing and 9 nonsense variants. We investigated the pathogenicity of 97 variants including in this study, among them, 45 variants are pathogenic, 14 variants are likely pathogenic and 38 variants are uncertain significance (Table 1). In China, most variants were reported in case reports, and there is insufficient function study on HMBS variants. Only 1/3 of the variants have undergone preliminary function study (Table 1). Further research is needed to elucidate the pathogenicity of these variants.
The distribution of variants in the HMBS gene exhibits some degree of variation among different countries. The number of variants in exons 9 and 11 exceeded others, mainly due to their size (Kauppinen and von und zu Fraunberg, 2002; Kauppinen, 2004). A study of 121 unrelated French Caucasian AIP families identified 78 different variants, 60% of which were in exons 9, 11, and 13 (Puy et al., 1997). In China, the variants in exons 11 and 14 were more widely distributed, demonstrating the heterogeneity (Figure 2). Most of the variants were family specific, that was, the occurrence of the same variant in several families was very low. In our study, c.517C>T occurred in 16 unrelated families, and 22 other variants also occurred in more than one family, and most of them are missense mutations (Figure 2). c.517C>T was a known pathogenic variant in Nova Scotia, Canada, with a high frequency due to the founder effect (Greene-Davis et al., 1997). In 2009, Sharon D identified 123 different variants on 283 patients in the UK, most variants were present in fewer than 3 families, but c.517C>T was present in 35 families (12%) (Whatley et al., 2009). It has also been reported that a variant was often shared by several families because of the founder effect, such as p. (Trp198*) from Sweden, p. (Gly111Arg) from Argentina, p. (Trp283*) from Switzerland, c. 669_698del from Spain (Guillén-Navarro et al., 2004), p.Arg116Trp from the Netherlands (de Rooij et al., 2009).
The correlation between genotype and phenotype should be cautiously interpreted, considering the clinical phenotype observed in HMBS gene variants (Table 2), which highlights the impact of genotype on phenotype. Variations in clinical presentation among patients carrying the same variant may suggest the involvement of modifier genes or environmental factors. Previous studies on glutathione synthetase deficiency have demonstrated that mutations leading to frameshifts, premature stop codons, or aberrant splicing are associated with moderate to severe clinical phenotypes (Njålsson et al., 2005). Consistent with these findings, our study also reveals that variants causing premature stop codons, frameshifts, or disruption of enzyme activity center are more likely to result in severe clinical manifestations such as respiratory paralysis, intracranial hemorrhage, disseminated intravascular coagulation (DIC), acute hepatic failure (AHF), or respiratory failure.
Among all the AIP patients we collected, there were obvious regional differences. They were mainly reported from Hebei, Taiwan and Beijing, and almost all the reports came from third-class hospitals. The regional medical level was a significant factor. Many patients chose to go to first-class hospitals because of unclear diagnosis and recurrent attacks. This suggests that we still lack awareness of this disease, especially in primary hospitals. In addition, our research was based only on HMBS variants from literature reports, so it was highly influenced by reporting bias. Further epidemiological data may provide more accurate information. Although there were some disadvantages in our study, this was the first investigation of HMBS gene variant in China, which fully revealed the characteristics of HMBS variant.
In the process of literature retrieval, we found that many patients with a clinical diagnosis of AIP did not receive related genetic testing; all the patients had clinically proven elevated urinary porphyrin precursors. Most patients recovered with carbohydrate intake. Therefore, variant screening was not necessary for patients with clinical onset and was not recommended in the normal population. However, in addition to AIP, other hepatic porphyria such as hereditary coproporphyrin (HCP) and variegate porphyria (VP) also had elevated urinary porphyrin precursors, so it had limitations in identifying of acute porphyria (Stölzel et al., 2019). More importantly, it might be the only way for early diagnosis of other asymptomatic patients in AIP family. Thus, large-scale mutation screening was recommended among the AIP family members of a proband case. This might be especially true in populations where no founder effects has been identified in AIP patients (Kauppinen, 2004).
This study provides a preliminary analysis of the genetic and phenotypic characteristics of Chinese AIP patients. Our results increase clinicians’ understanding of AIP, which could provide clinical guidance for AIP patients and reduce misdiagnosis and mistaken treatment in China. However, whether these variants are pathogenic, further verification at the molecular level is required to provide a reliable basis for the clinical diagnosis, and might provide new ideas and methods for the treatment of acute attacks and long-term complications of AIP. At present, the research on rare diseases in China was still in the preliminary stage, so we should enhance the knowledge of this disease in both clinical and scientific research, and further develop a nationwide epidemiological survey of AIP, which will be a difficult task for us in the future.
There are certain limitations in this study. Firstly, due to the inherent nature of this systematic review, it is inevitable to encounter publication bias. Secondly, the analysis of clinical features did not include many Chinese AIP patients diagnosed solely by biochemistry, as a clear HMBS variant was lacking. Therefore, the data for analyzing clinical features may not be sufficiently comprehensive.
5 Conclusion
The analysis of the HMBS gene in Chinese AIP patients revealed that missense mutations were the most common variant, with c.517C>T being the predominant alteration. However, due to the strong heterogeneity of AIP, screening for a single variant is not recommended in suspected patients; instead, whole gene sequencing should be performed. Abdominal pain was identified as the most frequent presenting symptom, followed by central nervous system manifestations. Women aged 18–40 years with recurrent abdominal pain and/or neuropsychiatric symptoms associated with menstruation or dietary factors should be alerted to the possibility of AIP. Variants causing premature stop codons, frameshifts or enzyme activity center disorder may lead to more severe clinical phenotypes such as respiratory paralysis, intracranial hemorrhage, DIC, AHF, CRF or respiratory failure.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding authors.
Author contributions
YR: Conceptualization, Funding acquisition, Writing–original draft. SL: Data curation, Formal Analysis, Writing–original draft. J-JL: Data curation, Formal Analysis, Writing–original draft. RL: Data curation, Writing–original draft. B-XD: Formal Analysis, Writing–original draft. JY: Writing–review and editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by Key research and development project of Shanxi Province, China (201903D321127), Special Project on the Transformation and Guidance of Scientific and Technological Achievements in Shanxi Province, China (201804D131044).
Acknowledgments
The authors thank the Key research and development project of Shanxi Province, China (201903D321127), Special Project on the Transformation and Guidance of Scientific and Technological Achievements in Shanxi Province, China (201804D131044) for the support.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2023.1291719/full#supplementary-material
References
Aksoy Ö, Y., Gündüz, M., Ünal, Ö., Bostancı, F., Çaycı, F., and Bayrakcı, U. S. (2020). A mysterious case with abdominal pain and syndrome of inappropriate anti-diuretic hormone secretion. Turk J. Pediatr. 62 (3), 487–490. doi:10.24953/turkjped.2020.03.018
Anderson, K. E., Bloomer, J. R., Bonkovsky, H. L., Kushner, J. P., Pierach, C. A., Pimstone, N. R., et al. (2005). Recommendations for the diagnosis and treatment of the acute porphyrias. Ann. Intern Med. 142 (6), 439–450. doi:10.7326/0003-4819-142-6-200503150-00010
Bissell, D. M., Anderson, K. E., and Bonkovsky, H. L. (2017). Porphyria. N. Engl. J. Med. 377 (9), 862–872. doi:10.1056/NEJMra1608634
Bonkovsky, H. L., Maddukuri, V. C., Yazici, C., Anderson, K. E., Bissell, D. M., Bloomer, J. R., et al. (2014). Acute porphyrias in the USA: features of 108 subjects from porphyrias consortium. Am. J. Med. 127 (12), 1233–1241. doi:10.1016/j.amjmed.2014.06.036
Bustad, H. J., Kallio, J. P., Vorland, M., Fiorentino, V., Sandberg, S., Schmitt, C., et al. (2021). Acute intermittent porphyria: an overview of therapy developments and future perspectives focusing on stabilisation of HMBS and proteostasis regulators. Int. J. Mol. Sci. 22 (2), 675. doi:10.3390/ijms22020675
Cerbino, G. N., Gerez, E. N., Varela, L. S., Melito, V. A., Parera, V. E., Batlle, A., et al. (2015). Acute intermittent porphyria in Argentina: an update. Biomed. Res. Int. 2015, 946387. doi:10.1155/2015/946387
Chen, B., Solis-Villa, C., Hakenberg, J., Qiao, W., Srinivasan, R. R., Yasuda, M., et al. (2016). Acute intermittent porphyria: predicted pathogenicity of HMBS variants indicates extremely low penetrance of the autosomal dominant disease. Hum. Mutat. 37 (11), 1215–1222. doi:10.1002/humu.23067
Chen, M. C., Chang, C. J., Lu, Y. H., Niu, D. M., Lou, H. Y., and Chang, C. C. (2015). R173W mutation of hydroxymethylbilane synthetase is associated with acute intermittent porphyria complicated with rhabdomyolysis: the first report. J. Clin. Gastroenterol. 49 (3), 256–257. doi:10.1097/mcg.0000000000000264
de Rooij, F. W., Kavelaars, F. G., Koole-Lesuis, H., and Wilson, J. H. (2009). Evidence for an ancestral founder of the common R116W mutation in the hydroxymethylbilane synthase gene in acute intermittent porphyria in The Netherlands. Cell Mol. Biol. (Noisy-le-grand) 55 (2), 64–69.
de Souza, P. V. S., Badia, B. M. L., Farias, I. B., Pinto, W., and Oliveira, A. S. B. (2021). Acute hepatic porphyria: pathophysiological basis of neuromuscular manifestations. Front. Neurosci. 15, 715523. doi:10.3389/fnins.2021.715523
Elder, G., Harper, P., Badminton, M., Sandberg, S., and Deybach, J. C. (2013). The incidence of inherited porphyrias in Europe. J. Inherit. Metab. Dis. 36 (5), 849–857. doi:10.1007/s10545-012-9544-4
Fortgens, P., Pienaar, E., Corrigall, A., Sonderup, M., Spearman, C. W., and Meissner, P. (2017). Molecular characterisation of acute intermittent porphyria in a cohort of South African patients and kinetic analysis of two expressed mutants. J. Clin. Pathol. 70 (6), 515–520. doi:10.1136/jclinpath-2016-203907
Fu, L., Wang, Z., Wang, M., Guo, J., and Yu, X. (2021). Acute intermittent porphyria with skin damage as the initial manifestation: a case report. Chin. J. Neuroimmunol. Neurology 28 (1), 85–86. doi:10.3969/j.issn.1006-2963.2021.01.017
Gao, P., Chen, X., Li, F., Wang, L., and Zhang, H. (2021). Acute intermittent porphyria with cerebral infarction as the initial manifestation: a case report. Chin. J. Stroke 16 (1), 69–72. doi:10.3969/j.issn.1673-5765.2021.01.012
Goncharova, M., Pshenichnikova, O., Luchinina, Y., Pustovoit, Y., Karpova, I., and Surin, V. (2019). Molecular genetic study of acute intermittent porphyria in Russia: HMBS gene mutation spectrum and problem of penetrance. Clin. Genet. 96 (1), 91–97. doi:10.1111/cge.13558
Gouya, L., Ventura, P., Balwani, M., Bissell, D. M., Rees, D. C., Stölzel, U., et al. (2020). EXPLORE: a prospective, multinational, natural history study of patients with acute hepatic porphyria with recurrent attacks. Hepatology 71 (5), 1546–1558. doi:10.1002/hep.30936
Greene-Davis, S. T., Neumann, P. E., Mann, O. E., Moss, M. A., Schreiber, W. E., Welch, J. P., et al. (1997). Detection of a R173W mutation in the porphobilinogen deaminase gene in the Nova Scotian "foreign Protestant" population with acute intermittent porphyria: a founder effect. Clin. Biochem. 30 (8), 607–612. doi:10.1016/s0009-9120(97)00114-8
Guillén-Navarro, E., Carbonell, P., Glover, G., Sánchez-Solís, M., and Fernández-Barreiro, A. (2004). Novel HMBS founder mutation and significant intronic polymorphism in Spanish patients with acute intermittent porphyria. Ann. Hum. Genet. 68, 509–514. doi:10.1046/j.1529-8817.2003.00114.x
Guo, Y., and Luo, M. (2023). Exploration of GnRH-a treatment for acute intermittent porphyria related to menstrual cycle. J. Reproductive Med. 32 (03), 438–442. doi:10.3969/j.issn.1004-3845.2023.03.022
Haiqing, Z. (2022). Clinical characteristics of childhood onset acute intermittent porphyria and correlation analysis of HMBS genotypes and phenotypes. Shandong: Shandong university. Master.
Hu, Y. (2019). Molecular genetics study on 56 families with acute intermittent porphyrin disease in China. Hebei: Hebei Medical University. Master.
Hu, Y., Kang, N., Wu, J., Liu, X., Zhang, X., and Zhang, S. (2018a). Acute intermittent porphyria: a case report and literature review. Clin. Focus 33 (6), 536–539. doi:10.3969/j.issn.1004-583X.2018.06.018
Hu, Y., Kang, N., Wu, J., Liu, X., Zhang, X., and Zhang, S. (2018b). Acute intermittent porphyria: a case report and literature review. Clin. Focus 33 (06), 536–539. doi:10.3969/j.issn.1004-583X.2018.06.018
Hu, Y., Li, W., Kang, N., Ma, L., Teng, Q., Mo, G., et al. (2022). Identification and molecular analysis of 17 novel variants of hydroxymethylbilane synthase in Chinese patients with acute intermittent porphyria. Clin. Genet. 101 (1), 116–121. doi:10.1111/cge.14063
Huang, S., Li, R., and Yuan, Y. (2021). Severe neuropathic attack in a woman with acute intermittent porphyria: a case report. J. Int. Med. Res. 49 (1), 300060520983143. doi:10.1177/0300060520983143
Jaramillo-Calle, D. A., and Aguirre Acevedo, D. C. (2019). Acute hepatic porphyrias in Colombia: an analysis of 101 patients. JIMD Rep. 44, 65–72. doi:10.1007/8904_2018_125
Jiao, H., Xianfeng, Z., Hui, H., Yuhong, Z., and Chu, Z. (2015). A novel mutation, IVS2-2AgG, associated with acute intermittent porphyria in a Chinese family. J. Pak Med. Assoc. 65 (8), 898–900.
Kauppinen, R. (2004). Molecular diagnostics of acute intermittent porphyria. Expert Rev. Mol. Diagn 4 (2), 243–249. doi:10.1586/14737159.4.2.243
Kauppinen, R., and von und zu Fraunberg, M. (2002). Molecular and biochemical studies of acute intermittent porphyria in 196 patients and their families. Clin. Chem. 48 (11), 1891–1900. doi:10.1093/clinchem/48.11.1891
Kong, X. F., Han, Y., Li, X. H., Gao, D. Y., Zhang, X. X., and Gong, Q. M. (2013). Recurrent porphyria attacks in a Chinese patient with a heterozygous PBGD mutation. Gene 524 (2), 401–402. doi:10.1016/j.gene.2013.03.130
Kothadia, J. P., LaFreniere, K., and Shah, J. M. (2023). “Acute hepatic porphyria,” in StatPearls. (Treasure island (FL) ineligible companies: StatPearls publishing copyright © 2023) (Florida, United States: StatPearls Publishing LLC).
Lakhoo, K., Almario, C. V., Khalil, C., and Spiegel, B. M. R. (2021). Prevalence and characteristics of abdominal pain in the United States. Clin. Gastroenterol. Hepatol. 19 (9), 1864–1872.e5. doi:10.1016/j.cgh.2020.06.065
Lam, C. W., Lau, K. C., Mak, C. M., Tsang, M. W., and Chan, Y. W. (2011). Circulating fluorocytes at the first attack of acute intermittent porphyria: a missing link in the pathogenesis. Clin. Chim. Acta 412 (1-2), 208–212. doi:10.1016/j.cca.2010.09.005
Lam, C. W., Poon, P. M., Tong, S. F., Lo, A. W., Lai, C. K., Choi, K. L., et al. (2001). Novel mutation and polymorphisms of the HMBS gene detected by denaturing HPLC. Clin. Chem. 47 (2), 343–346. doi:10.1093/clinchem/47.2.343
Lei, L., Guo, Y., and Yang, Y. (2017). Recurrent abdominal pain as principal clinical manifestation of acute intermittent porphyria: a case report and literature review. World Chin. J. Dig. 25 (12), 1128–1134. doi:10.11569/wcjd.v25.i12.1128
Li, F., Zhu, D., Chen, D., and Lin, X. (2017). Clinical characteristics of acute intermittent porphyria (report of 1 case). J. Clin. Neurology 30 (06), 468–470.
Li, J., Wu, Y., Hu, D., He, C., and Wu, L. (2016). A case report and literature review of acute intermittent porphyria. Prog. Mod. Biomed. 16 (17), 3286–3290. doi:10.13241/j.cnki.pmb.2016.17.020
Li, P., Zhang, S., Kang, N., Jiao, L., Li, J., Li, G., et al. (2022). A servere acute intermittent porphyria patient: successful treatment and management. Chin. J. Endocrinol. Metabolism 38 (4), 335–338. doi:10.3760/cma.j.cn311282-20210913-00594
Li, Q. (2022). Analysis of the clinical characteristics of four cases of acute intermittent porphyria and literature review. Taiyuan, China: Shanix Medical University. Master.
Li, X., Liu, G., Shu, H., Wu, D., Lie, G., and Qian, J. (2015a). Clinical and genetic characteristics of 36 patients with acute intermittent porphyria. Med. J. Peking Union Med. Coll. Hosp. 6 (02), 110–114. doi:10.3969/j.issn.1674-9008.2015.02.007
Li, Y., Qu, H., Wang, H., Deng, H., and Liu, Z. (2015b). Novel A219P mutation of hydroxymethylbilane synthase identified in a Chinese woman with acute intermittent porphyria and syndrome of inappropriate antidiuretic hormone. Ann. Hum. Genet. 79 (4), 310–312. doi:10.1111/ahg.12107
Liang, Y., and Li, D. (2023). Depressive acute intermittent porphyria: a case report. Jiangsu Med. J. 49 (4), 428–430. doi:10.19460/j.cnki.0253-3685.2023.04.026
Ma, L., Tian, Y., Qi, X., Li, P., Li, J., Teng, Q., et al. (2022). Acute intermittent porphyria: prevalence of pathogenic HMBS variants in China, and epidemiological survey in Hebei Province, China. Ann. Transl. Med. 10 (10), 560. doi:10.21037/atm-22-1600
Njålsson, R., Ristoff, E., Carlsson, K., Winkler, A., Larsson, A., and Norgren, S. (2005). Genotype, enzyme activity, glutathione level, and clinical phenotype in patients with glutathione synthetase deficiency. Hum. Genet. 116 (5), 384–389. doi:10.1007/s00439-005-1255-6
Page, M. J., McKenzie, J. E., Bossuyt, P. M., Boutron, I., Hoffmann, T. C., Mulrow, C. D., et al. (2021). The PRISMA 2020 statement: an updated guideline for reporting systematic reviews. Bmj 372, n71. doi:10.1136/bmj.n71
Puy, H., Deybach, J. C., Lamoril, J., Robreau, A. M., Da Silva, V., Gouya, L., et al. (1997). Molecular epidemiology and diagnosis of PBG deaminase gene defects in acute intermittent porphyria. Am. J. Hum. Genet. 60 (6), 1373–1383. doi:10.1086/515455
Ramanujam, V. S., and Anderson, K. E. (2015). Porphyria diagnostics-Part 1: a brief overview of the porphyrias. Curr. Protoc. Hum. Genet. 86, 1–17. doi:10.1002/0471142905.hg1720s86
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of medical genetics and Genomics and the association for molecular pathology. Genet. Med. 17 (5), 405–424. doi:10.1038/gim.2015.30
Souza, P. V. S., Afonso, G., de Rezende Pinto, W. B. V., de Lima Serrano, P., de Mattos Lombardi Badia, B., Farias, I. B., et al. (2023). Brazilian registry of patients with porphyria: REBRAPPO study. Orphanet J. Rare Dis. 18 (1), 49. doi:10.1186/s13023-023-02653-1
Stölzel, U., Doss, M. O., and Schuppan, D. (2019). Clinical guide and update on porphyrias. Gastroenterology 157 (2), 365–381. doi:10.1053/j.gastro.2019.04.050
Stölzel, U., Stauch, T., and Kubisch, I. (2021). Porphyria. Internist (Berl) 62 (9), 937–951. doi:10.1007/s00108-021-01066-1
Sun, S., Fang, L., Zhao, C., Zhong, Q., Gao, X., and Li, L. (2020). Acute intermittent porphyria with Posterior reversible encephalopathy syndrome: a case report and literature review. J. Shandong Univ. Sci. 58 (02), 118–121. doi:10.6040/j.issn.1671-7554.0.2019.1042
Teng, Q., Ma, L., Ma, Y., Zhang, Y., Kang, N., Hu, Y., et al. (2020). The challenge of managing comorbidities: a case report of primary Sjogren's syndrome in a patient with acute intermittent porphyria. Intractable Rare Dis. Res. 9 (3), 137–140. doi:10.5582/irdr.2020.03064
Wang, F., Jin, D., Ding, Y., and Zhou, J. (2019a). The 474th case of early pregnancy - wandering abdominal pain - cerebral hemorrhage - genetic testing - porphyria. Natl. Med. J. China 99 (33), 2624–2626. doi:10.3760/cma.j.issn.0376-2491.2019.33.015
Wang, X., Zhao, X., Jiao, S., Wang, T., Sun, Q., and Liu, X. (2019b). The different clinical manifestations of porphyria between the same genotype twin sisters. J. Apoplexy Nerv. Dis. 36 (03), 217–222. doi:10.19845/j.cnki.zfysjjbzz.2019.03.006
Whatley, S. D., Mason, N. G., Woolf, J. R., Newcombe, R. G., Elder, G. H., and Badminton, M. N. (2009). Diagnostic strategies for autosomal dominant acute porphyrias: retrospective analysis of 467 unrelated patients referred for mutational analysis of the HMBS, CPOX, or PPOX gene. Clin. Chem. 55 (7), 1406–1414. doi:10.1373/clinchem.2008.122564
Xie, X. (2012). Pedigree investigation and literature review of an actue intermittent porphyria syndrome patient. Dalian, China: Dalian Medical University. Master.
Yang, A. L., Ma, L. M., Zhang, H. J., and Zhang, J. W. (2022). Novel mutation of hydroxymethylbilane synthase in a case of acute intermittent porphyria presenting with posterior reversible encephalopathy syndrome. J. Coll. Physicians Surg. Pak 32 (12), Ss102–ss104. doi:10.29271/jcpsp.2022.Supp0.SS102
Yang, C. C., Kuo, H. C., You, H. L., Wang, J., Huang, C. C., Liu, C. Y., et al. (2008). HMBS mutations in Chinese patients with acute intermittent porphyria. Ann. Hum. Genet. 72, 683–686. Pt 5. doi:10.1111/j.1469-1809.2008.00463.x
Yang, J., Chen, Q., Yang, H., Hua, B., Zhu, T., Zhao, Y., et al. (2016). Clinical and laboratory features of acute porphyria: a study of 36 subjects in a Chinese tertiary referral center. Biomed. Res. Int. 2016, 3927635. doi:10.1155/2016/3927635
Yang, J., Han, F., Chen, Q., Zhu, T., Zhao, Y., Yu, X., et al. (2020a). Reversible splenial lesion syndrome (RESLES) due to acute intermittent porphyria with a novel mutation in the hydroxymethylbilane synthase gene. Orphanet J. Rare Dis. 15 (1), 98. doi:10.1186/s13023-020-01375-y
Yang, J., Wang, H., Yin, K., Hua, B., Zhu, T., Zhao, Y., et al. (2015). A novel mutation in the porphobilinogen deaminase gene in an extended Chinese family with acute intermittent porphyria. Gene 565 (2), 288–290. doi:10.1016/j.gene.2015.04.027
Yang, J., Yang, H., Chen, Q., Hua, B., Zhu, T., Zhao, Y., et al. (2017). Reversible MRI findings in a case of acute intermittent porphyria with a novel mutation in the porphobilinogen deaminase gene. Blood Cells Mol. Dis. 63, 21–24. doi:10.1016/j.bcmd.2016.12.005
Yang, Y., Chen, X., Wu, H., Peng, H., Sun, W., He, B., et al. (2020b). A novel heterozygous mutation in the HMBS gene in a patient with acute intermittent porphyria and posterior reversible encephalopathy syndrome. Mol. Med. Rep. 22 (1), 516–524. doi:10.3892/mmr.2020.11117
You, Y., Liang, M., Gong, C., and Shu, S. (2015). Acute intermittent porphyria complicated with Acute kidney injury: a case report. Chin. J. Nephrol. 31 (5), 395–396. doi:10.3760/cma.j.issn.1001-7097.2015.05.014
Yuan, T., Li, Y. H., Wang, X., Gong, F. Y., Wu, X. Y., Fu, Y., et al. (2015). Acute intermittent porphyria: a diagnostic challenge for endocrinologist. Chin. Med. J. Engl. 128 (14), 1980–1981. doi:10.4103/0366-6999.160621
Zhang, J., and Gao, Q. (2019). Efficacy of prophylactic haemoglobin infusion in the treatment of severe acute intermittent porphyria. Health Care Today (8), 95–96. doi:10.3969/j.issn.1671-0223(x).2019.08.067
Zhang, L., Han, B., and Chen, M. (2020a). Exports consensus on diagnosis and treatment of porphyria in China. Natl. Med. J. China 100 (14), 1051–1056. doi:10.3760/cma.j.cn112137-20200219-00349
Zhang, S., Wu, J., Teng, Q., Zhang, Y., Hu, Y., and Kang, N. (2020b). An extremely rare combination of acute intermittent porphyria and Turner syndrome. Intractable Rare Dis. Res. 9 (3), 141–144. doi:10.5582/irdr.2020.03065
Zhang, Y., Xiao, H., Xiong, Q., Wu, C., and Li, P. (2021). Two novel hydroxymethylbilane synthase splicing mutations predispose to acute intermittent porphyria. Int. J. Mol. Sci. 22 (20), 11008. doi:10.3390/ijms222011008
Zheng, Y., Xu, J., Liang, S., Lin, D., and Banerjee, S. (2018). Whole exome sequencing identified a novel heterozygous mutation in HMBS gene in a Chinese patient with acute intermittent porphyria with rare type of mild anemia. Front. Genet. 9, 129. doi:10.3389/fgene.2018.00129
Zho, H., Li, B., Yang, X., Zhang, T., Li, J., Zhao, Y., et al. (2022). Acute intermittent porphyria with infantile spasms: a case report and literature review. J. Shandong Univ. Heal. Sci. 60 (6), 82–89. doi:10.6040/j.issn.1671-7554.0.2021.1317
Zhou, X. (2014). Misdiagnosis of hematoporphyria as functional abdominal pain: a case report. Chin. J. Clin. Ration. Drug Use 7 (13), 10. doi:10.15887/j.cnki.13-1389/r.2014.13.092
Keywords: acute intermittent porphyria, hydroxymethylbilane synthase, China, mutation analysis, abdominal pain
Citation: Ren Y, Li S, Lei J-J, Li R, Dong B-X and Yang J (2023) Clinical feature and genetic analysis of HMBS gene in Chinese patients with acute intermittent porphyria: a systematic review. Front. Genet. 14:1291719. doi: 10.3389/fgene.2023.1291719
Received: 10 September 2023; Accepted: 22 November 2023;
Published: 11 December 2023.
Edited by:
Pilar Giraldo, University of Zaragoza, SpainReviewed by:
Kitiwan Rojnueangnit, Thammasat University Hospital, ThailandPaulo Victor Sgobbi Souza, Federal University of São Paulo, Brazil
Copyright © 2023 Ren, Li, Lei, Li, Dong and Yang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jing Yang, eWFuZ2psbUAxMjYuY29t; Yi Ren, cmVueWlfMF8wQDE2My5jb20=