 Cheng Huang†
Cheng Huang† Haiyan Luo†Baitao ZengChuanxin Feng
Haiyan Luo†Baitao ZengChuanxin Feng Jia ChenHuizhen YuanShuhui Huang
Jia ChenHuizhen YuanShuhui Huang Bicheng Yang*
Bicheng Yang* Yongyi Zou*Yanqiu Liu*
Yongyi Zou*Yanqiu Liu*- Department of Medical Genetics, Jiangxi Key Laboratory of Birth Defect Prevention and Control, Jiangxi Maternal and Child Health Hospital, Nanchang, Jiangxi, China
Background and purpose: Intellectual disability-7 (MRD7) is a subtype disorder of intellectual disability (MRD) involving feeding difficulties, hypoactivity, and febrile seizures at an age of early onset, then progressive intellectual and physical development deterioration. We purposed to identify the underlying causative genetic factors of three individuals in each Chinese family who presented with symptoms of intellectual disability and facial dysmorphic features. We provided prenatal diagnosis for the three families and genetic counseling for the prevention of this disease.
Methods: We collected retrospective clinical diagnostic evidence for the three probands in our study, which included magnetic resonance imaging (MRI), computerized tomography (CT), electroencephalogram (EEG), and intelligence tests for the three probands in our study. Genetic investigation of the probands and their next of kin was performed by Trio-whole exome sequencing (WES). Sanger sequencing or quantitative PCR technologies were then used as the next step to verify the variants confirmed with Trio-WES for the three families. Moreover, we performed amniocentesis to explore the state of the three pathogenic variants in the fetuses by prenatal molecular genetic diagnosis at an appropriate gestational period for the three families.
Results: The three probands and one fetus were clinically diagnosed with microcephaly and exhibited intellectual developmental disability, postnatal feeding difficulties, and facial dysmorphic features. Combining probands’ clinical manifestations, Trio-WES uncovered the three heterozygous variants in DYRK1A: a novel variant exon3_exon4del p.(Gly4_Asn109del), a novel variant c.1159C>T p.(Gln387*), and a previously presented but rare pathogenic variant c.1309C>T p.(Arg437*) (NM_001396.5) in three families, respectively. In light of the updated American College of Medical Genetic and Genomics (ACMG) criterion, the variant of exon3_exon4del and c.1159C>T were both classified as likely pathogenic (PSV1+PM6), while c1309C>T was identified as pathogenic (PVS1+PS2_Moderate+PM2). Considering clinical features and molecular testimony, the three probands were confirmed diagnosed with MRD7. These three discovered variants were considered as the three causal mutations for MRD7. Prenatal diagnosis detected the heterozygous dominant variant of c.1159C>T p.(Gln387*) in one of the fetuses, indicating a significant probability of MRD7, subsequently the gestation was intervened by the parents’ determination and professional obstetrical operation. On the other side, prenatal molecular genetic testing revealed wild-type alleles in the other two fetuses, and their parents both decided to sustain the gestation.
Conclusion: We identified two novel and one rare mutation in DYRK1A which has broadened the spectrum of DYRK1A and provided evidence for the diagnosis of MRD7 at the molecular level. Besides, this study has supported the three families with MRD7 to determine the causative genetic factors efficiently and provide concise genetic counseling for the three families by using Trio-WES technology.
Introduction
Intellectual disability-7 (MRD7 OMIM#614104) is caused by mutations in DYRK1A, which is an autosomal dominant hereditary disorder of autism and generally characterized by autism, intellectual disability (ID) features which cause primary microcephaly (−3 SD), nonverbal severe intellectual and physical disability, anxious behavior, and deformities. Facial abnormalities cover bilateral temporal stenosis, pointed nasal tips, large and underdeveloped ears, and deep-set eyes (van Bon et al., 2011). Early symptoms of MRD7 are feeding difficulties, febrile seizures, non-fluent motoric movements, and hypoactivity in infancy caused by central nervous system anomalies. The individual can develop other symptoms such as speech impairment, hypertonia, developmental delay, and autism behaviors, usually leading to the patient’s poor autonomy and adult social maladjustment. Less common features are known as optic nerve defects, cardiac anomalies, and contractures ataxia. Hitherto, there is no effective treatment strategy for MRD7, and receiving rehabilitation and anticonvulsant therapy can merely relieve the patients’ symptoms with MRD7, while poor compliance still remains a barrier.
The chromosome 21 at 21q22.13 positioned dual specificity tyrosine-phosphorylation-regulated kinase 1A (DYRK1A OMIM#600855), up to now, is the only known gene responsible for MRD7. DYRK1A, a gene function prominent in most of the characteristic features related to microcephaly, is highly conserved in the Down Syndrome critical region (DSCR). Loss-of-function mutations of DYRK1A can contribute to haploinsufficiency for the allelic gene leading to a decrease in surrounding ganglia about the soma and neurite complexity (Fotaki et al., 2002). Due to febrile seizures in fancy, presynaptic function in GABA releasing and re-uptaking and postsynaptic components’ function in GABA receptor trafficking and availability would transmit abnormally in GABAergic transmission (Kang et al., 2006; Qu and Leung, 2008). A primary diagnosis based on the clinical evidence included microcephaly, IUGR in the fetus period, intellectual disability, growth retardation, stereotype, facial gestalt, dysmorphic abnormalities, speech impairment, and clumsiness in fine motor patterns. As time goes on, with medical advances in various fields and the progress of DNA sequencing technologies, more and more DYRK1A variants have been identified in MRD7 patients, enabling further effective precise diagnosis for genetic confirmation. In 2008, DYRK1A was verified firstly as a factitive gene for MRD7 (Matsumoto et al., 1997; Møller et al., 2008). On the basis of the online data library Orphanet Reports Series (https://www.orpha.net), the MRD7 prevalence rate caused by a DYRK1A point mutation is less than 1/1000000. To date, 68 pathogenic variants in DYRK1A have been presented of this disease. There have been almost 200 mutations in DYRK1A presented, besides, a totality of 191 variants in DYRK1A have been presented and more than 80 variants resulting in MRD7 have been reported, based on the data library professional Human Gene Mutation Database (HGMD). Besides, further extensive explorations on the spectrum of DYRK1A resulting MRD7 are necessary, due to the diverse and large population condition of China. Hitherto, in China, only two reports on individuals were confirmed with MRD7 (Qiao et al., 2019; Ma et al., 2021).
In this study, using Trio-whole exome sequencing (WES), we confirmed the genetic incentive factors that two novel mutations and one rare mutation in three Chinese families of MRD7. Besides, for the prevention of MRD7 in these cases, we presented ethical issues concerning genetic counseling, such as prenatal diagnosis.
Clinical presentations
In Jiangxi Maternal and Child Hospital (Jiangxi, China), the three probands from three respective nonconsanguineous healthy Chinese families were admitted to the medical genetics department and pediatric department. The three individuals’ clinical features included intellectual and physical disability and special facial features. Their families’ history had no evidence of skeletal, intellectual, neurological, metabolic, developmental, or any other heritable diseases. These three individuals were born with varying degrees of intrauterine growth restriction (IUGR) and microcephaly. Feeding difficulties, febrile seizures, and hypotonia were noticed during the infantile period, representing the typical facial gestalt in childhood, and all of them did not reach the normal intellectual development milestones. Besides, the brain CT, MRI, ECG examination, clinical investigations including intelligence testing, chromosomal karyotype analysis, serum amino acid determination, blood ammonia, and succinyl acetone level testing extracted in peripheral blood at 2.5 years old were conducted for the proband with exon3_exon4del variant in DYRK1A. Results of intelligence testing indicated intellectual disability (ID) in terms of social activity, language, fine motor skills, and sport. Analysis of serum amino acid determination indicated that the leucine and valine were moderately higher than normal levels. CT and MRI revealed bilaterally enlarged ventricles in the proband, while ECG showed no significant abnormalities. This proband also demonstrated the dysmorphic features of MRD7 including deep-set eyes, pointed nasal tip, and thin upper lip. Since the three probands’ mothers were in the process of gestation, they hoped to give birth to a healthy child. Thus, for professional prenatal genetic counseling, they sought guidance from the Medical Genetics Department. The three family trees are presented in Figure 1. The Ethics Committee of Jiangxi Maternal and Child Health Hospital authorized this study to utilize the clinical information and collection of samples, and informed consent forms were received from the patients.
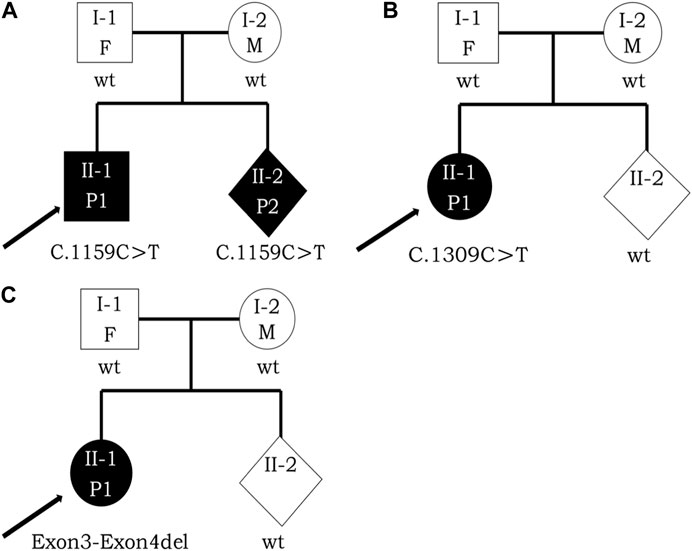
FIGURE 1. Pedigree analysis of these three families with DYRK1A variants. Opening symbols, unaffected; filled symbols, affected; squares, male; rounds, female; rhombus, fetus with unknown gender; arrowheads, the proband; F, father; M, mother; P1, patient 1; P2, patient 2; wt, wild type. Three black arrowheads represent the probands [(A), II-1, II-2; (B), II-1; (C), II-1) respectively. (A) I-1, proband’s father, 36 years old; I-2, proband’s mother, 30 years old; II-1, the proband, 5 years old; II-2, the fetus at the gestational age of 16 + 3 weeks. (B) I-1, proband’s father, 29 years old; I-2, proband’s mother, 29 years old; II-1, the proband, 4 years old; II-2, the fetus at the gestational age of 16 + 5 weeks. (C) I-1, proband’s father, 28 years old; I-2, proband’s mother, 26 years old; II-1, the proband, 4 years old; II-2, the fetus at the gestational age of 18 + 5 weeks.
Methods
Trio-WES
An amniotic fluid aspiration sample was adopted by amniocentesis and peripheral blood was collected from every participant after obtaining informed written consent or participation. Genomic DNA was collected from leukocytes, following manufacturers’ instructions. For analyzing and measuring participants’ DNA, we performed the NanoDrop spectrophotometer from Thermo Scientific in Wilmington, DE, United States. Above all, to formulate the WES libraries, 3 µg of participants’ genomic DNA clipped by performing ultrasonication (Covaris S220 Ultrasonicator). Next, by using the BGI V4 chip, we captured and concentrated the labeled regions containing exons and splicing sites of more than 20,000 genes in the human genome, after that, based on the manufacturer’s protocol, we implemented the sequencing by the MGISEQ-2000 platform (BGI, Shenzhen, China) with the pair-end sequencing. The quality control index of sequencing data met demand as follows: the average coverage depth for the labeled area was ≥180×, and the ratio of sites with an average depth >20× of the labeled regions was >95%. By applying the BWA to remove duplications, the sequenced parts were arrayed to the UCSC RefSeq database hg19 human reference genome. Then the GATK standard genotype tool was performed for variant searching of single nucleotide variations, insertions, or deletions (Indels) and copy number variations at the exons’ level using Exome Depth according to the algorithms based on probability and quality. Concisely, in the process, we filtered out the general pleomorphisms as dbSNP (minor allele frequency>0.01), 1,000 genomes (genotype frequency>0.005), gnomAD, EVC, and synonymous single nucleotide variants.
Variants annotations and interpretations
The gene nomenclature abides by the traditional patterns of the Human Genome Organization Gene Nomenclature Committee; the variant nomenclature abides by the traditional patterns of the Human Genome Variation Society and International Union of Pure and Applied Chemistry. Variant annotations and screenings were based on clinical phenotypes of the subject, population databanks (dbSNP, ExAC, 1,000 Genome), disease databanks (OMIM, HGMD, SMART, Clinvar), and biological resource prediction tools (SIFT, Mutation Taster, and Polyphen2). On the basis of the criteria of the American College of Medical Genetics and Genomics (ACMG) and the American Society for Molecular Pathology, interpretations of pathogenicity for variants were formulated. Detailed interpretations of the guidelines were consulted by the ClinGen Sequence Variation Interpretation Working Group and the British Society of Clinical Genomics (ACGS). When a novel/rare variant predicted to be pathogenic was identified in a sole acknowledged intellectual disability gene and possessed with the parallel pattern of inheritance, it should be deemed as a prospective candidate variant.
Confirmation of the three variants by Sanger sequencing and quantitative PCR
For the sake of validation about all the exons’ sequencing consequences and segregations’ analytical results, Sanger sequencing was operated by 3500DX Genetic Analyzer (Applied Biosystems) to verify the candidates for the two families with the point mutations, while quantitative PCR (qPCR) was performed on the family with exons deletion of cDNA using the QuantStudio 5 (Applied Biosystems). With normalized reference sequences and visualizations, the sequence analysis was performed by Seqman software (Technelysium, South Brisbane, QLD, Australia). Based on the online databank of the University of California, Santa Cruz (UCSC), we designed the reference sequences. Table 1 lists the four individual primer pairs concepted for verifying the three mutations.
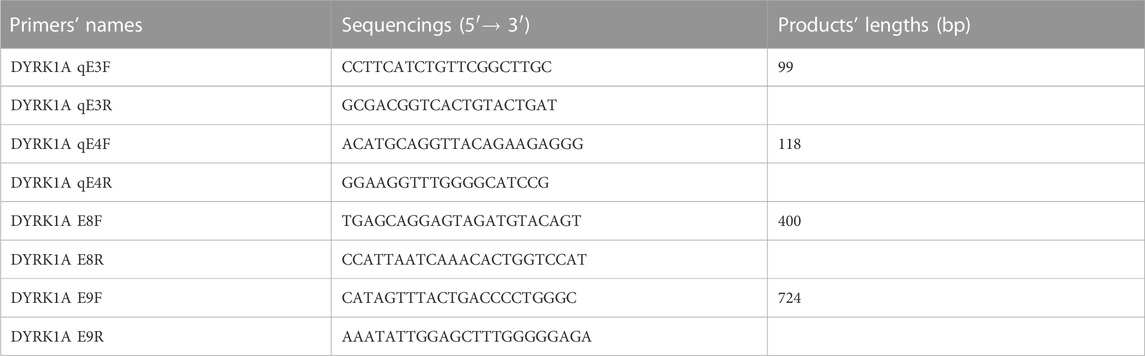
TABLE 1. Confirmation of the four primers purposed for the qPCR/Sanger sequencing of cDNA.
Prenatal diagnosis of the fetuses
For the three probands following the three factitive variants of DYRK1A were confirmed, those families expressed their anticipation to carry out the prenatal diagnosis for their fetuses at a fitted gestational period. Amniotic fluid samples in the sum of 20 mL were obtained at the gestational age of 16W + 3, 16W + 5, and 18W + 5 respectively from each pregnant mother’s uterine cavity from the three families following the B ultrasonography guidance. By using the QIAamp DNA Mini Kit (Qiagen, Germany), the extraction of fetal DNA from the amniotic fluid was implemented, according to the protocols of the manufacturer. In order to explore the mutations’ state of c.1159C>T and c.1309C>T by applying the forward and reverse sequencings, exon3_exon4del in DYRK1A was analyzed as presented before. Simultaneously, the matrilinear cell pollution has been removed by operating the quantitative fluorescent polymerase chain reaction, in light of the instruction presented before (Noveski et al., 2019).
Statistical analysis
The statistical analysis was performed using SPSS 20.0 software.
Results
Based on clinical symptoms and genetic discoveries, the three probands were diagnosed with MRD7. One of the fetuses was genetically diagnosed with MRD7 while the other two showed wild-type alleles in the detected locus by prenatal diagnosis.
Whole exome sequencing findings of patients’ phenotypes-related variants and Sanger sequencing validation
By using Trio-WES, the raw data sequencing production for each candidate approached at least 23,170.74 Mb. With the mean of 273.8.85×sequencing depth, to the human genome hg19, approximately 99.72% of sequencing reads were arranged. It is noteworthy that, in the three probands, the three heterozygous disorder-related variants in DYRK1A (NM_001396.5), 2 novel variants including c. 1159C>T and exon3_exon4del, and a rare variant c.1309C>T, passed the screening standards. About the three variants, in exon3 and exon4, variant exon3_exon4del gives rise to deletion of protein translation from residue 4 to residue 109 p.(Gly4_Asn109del), and the two variants c.1159C>T and c.1309C>T lead to the prophase termination in protein translation period at residue 387 p.(Gln387*) and residue 437 p.(Arg437*) by inducing a premature stop codon respectively. Furthermore, we collected and analyzed the incidental findings of other genes in insufficient pathogenic evidence for the three probands, which was placed in (Supplementary Table S1). Subsequently, Sanger sequencing and qPCR validated these three mutations in the three families.
Variant annotations and interpretations according to the ACMG guidelines
In the 1,000 Genomes databank the variant c. 1159C>T and exon3_exon4del were not searched, HGMD master databank, Genome Aggregation Database (gnomAD), nor in the Exome Variant Server. On the flip side, in our 200 healthy control cohorts, the two variants were not found. The variant exon3_exon4del is predicted to result in the 105-amino-acid-deletion from the 4th residue to the 109th residue in exon3 and exon4, whereas the variant c.1159C>T is speculated to produce a premature stop codon introduction by a substitution of glutamine that lead to a truncating protein at the 387th amino acid in DYRK1A. The DYRK1A protein family members’ alignments were revealed By the ClustalX online software that amino acids of DYRK1A from the 4th residue to the 387th amino acid are highly evolutionarily conserved, which is displayed in Figure 2. We predicted the the deleted protein structure resulted from two novel mutations and a premature truncation could be produced via selective degradation by the nonsense-mediated mRNA decay, respectively. By using algorithms of multiple bioinformatic methods which include SIFT (sift.jcvi.org), Mutation Taster (www.mutationtaster.org), and polyphen-2 (genetics.bwh.harvard.edu/pph2), these two novel variants were calculated to be deleterious. We build the homologous modeling three dimension structure by Swiss-model (SWISS-MODEL (expasy.org)) of wild type (Seq Identity: 100.00%, QMQE: 0.92, QMEAN: 0.87 ± 0.05), p.Gln387* (c.1159C>T) mutation (Seq Identity: 100.00%, QMQE: 0.92, QMEAN: 0.88 ± 0.05), p.Arg437* (c.1309C>T) mutation (Seq Identity: 100.00%, QMQE: 0.92, QMEAN: 0.87 ± 0.05) and p.Gly4_Asn109del (exon3_exon4del) mutation (Seq Identity: 99.60%, QMQE: 0.91, QMEAN: 0.87 ± 0.05) placed in Figure 3 (Camacho et al., 2009; Soundararajan et al., 2013; Bertoni et al., 2017; Bienert et al., 2017; Waterhouse et al., 2018; Steinegger et al., 2019; Studer et al., 2020; Studer et al., 2021).
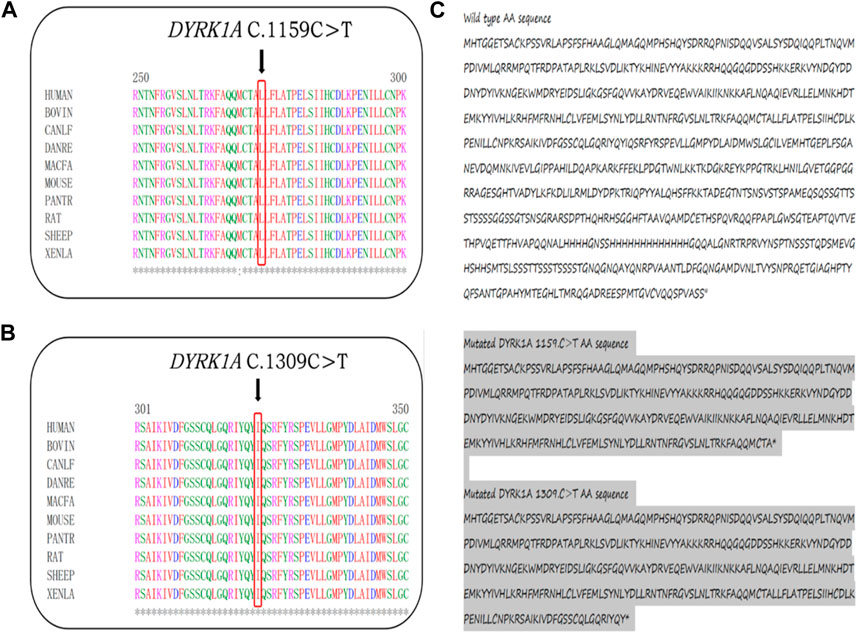
FIGURE 2. (A,B) The truncating amino acids’ location, pointed by black arrows, among species are highly evolutionarily conserved. (C) At the 387th and 437th of DYRK1A predicted premature termination codon.
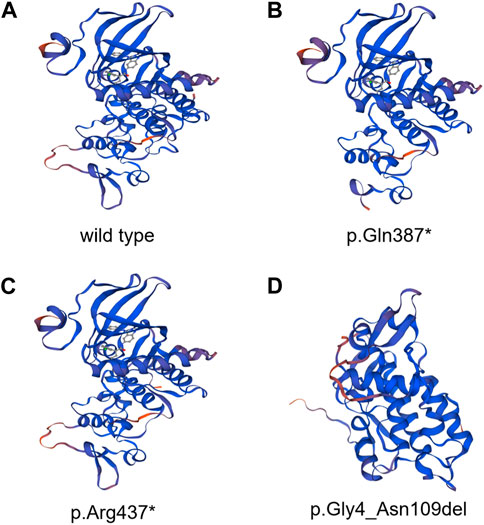
FIGURE 3. DYRK1A protein of homologous modeling three-dimensional structures by Swiss-model. (A) Whole structure of wild-type DYRK1A protein. (B) DYRK1A protein of p.Gln387* (c.1159C>T) mutation. (C) DYRK1A protein of p.Arg437* (c.1309C>T) mutation. (D) DYRK1A protein of p.Gly4_Asn109del (exon3_exon4del) mutation.
On the basis of the latest ACMG guideline, the two novel variants in our study were consequently classified as likely pathogenic (PSV1+PM6). Furthermore, we found the identified truncating variant c1309C>T inducing the alteration of the 437th Arg into a stop codon. This variant is considered pathogenic (PVS1+PS2_Moderate+PM2) in light of the ACMG guidelines. Overall, considering the clinical manifestations and examinations, the diagnosis of MRD7 was validated by the genetic analysis in the three families.
Genetic findings for the three fetuses by amniocentesis
Amniotic fluid analysis was produced after an additional 7 days, which identified one of the three fetuses was heterozygous autosomal dominant for c. 1159C>T possessing the equal genetic variant in their family as the proband. The other two fetuses were detected with wild-type alleles within the c.1309 and exon3_exon4 locus. In Figure 4, the chromatograms of Sanger sequence analysis and quantitative polymerase chain reaction testing for the members of the three families are displayed respectively. After prudent and adequate consideration, the first family resolved to intervene in the gestation at the earliest possible time and the other two families chose to continue the gestation. For these two families, subsequent interval follow-ups showed no anomaly in the two born children.
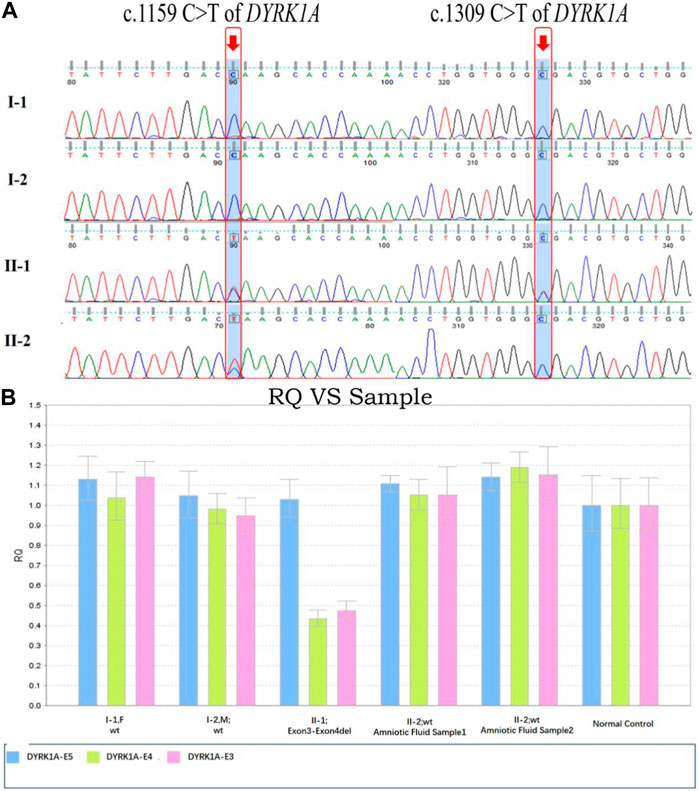
FIGURE 4. DYRK1A variants sequence consequences and variants analysis in distinct domains of DYRK1A. (A) The c.1159C>T and c.1309C>T variants of DYRK1A sequence chromatograms were identified for the two families’ members. (B) Fluorescence quantitative polymerase chain reaction (qPCR) bar chart for the exon3-exon4del variant of the DYRK1A detected in the family members, compared with normal control. Blue regions represent exon3, green regions represent exon4, and pink regions represent exon5.
Discussion
DYRK1A encodes a member of the dual specificity tyrosine-phosphorylation-regulated kinase (DYRK) family, a 58 kDa tyrosine-phosphorylation-regulated kinase possessing with serine/threonine and tyrosine kinase activities. Besides, on chromosome 21 DYRK1A resides in the Down syndrome critical region (DSCR) and is reckoned responsible for intellectual disabilities as a mighty housekeeping gene related to Down syndrome (Guimerá et al., 1996; Matsumoto et al., 1997; Eisenberg and Levanon, 2013). Of the five mammalian DYRKs, which is the highly conserved DYRK family, the DYRK1A regulates neuronal morphogenesis by phosphorylating cytoskeletal elements and also participates in the development of olfactory, visual, and central nerve systems. DYRK1A includes a conserved catalytic kinase domain prior to a distinctive DYRK homology (DH) box, two nuclear localization signals (NLS), one preceding and the other one with the kinase domain, a PEST domain, a speckle targeting signal (STS), a histidine repeat and a serine/threonine repeat (Evers et al., 2017). The guardian-like functional DYRK-homology box (DH box) stabilizes the kinase domain in a middling structure during the process of protein maturation with tyrosine autophosphorylation activity (Kinstrie et al., 2010), as shown in Figure 5. The NLS is discerned by importin protein in the cytosol, which could conduct the transfer of the protein to the nuclear pores, while the PEST indicates the signal sequence of degradation, a characteristic of short-lived proteins. Within the nucleus, the histidine repeat region labels the protein to bonding factor assembly (Alvarez et al., 2003). DYRK1A is expressed in neuroepithelia precursor cells during the prophase of embryonic development in Drosophila, which sets the conversion step from proliferations to neurogenic divisions, and mutations of DYRK1A could induce a microcephaly phenotype and abnormal retinal forming progression (Tejedor et al., 1995; Fotaki et al., 2002; Hämmerle et al., 2002; Laguna et al., 2008; Brault et al., 2021). In our case, the three mutations were confirmed to the family co-segregation by Sanger sequencing and fluorescence quantitative polymerase chain reaction. We analyzed the homologous alignment of protein sequences, which reveals that the two variants and one sequence are highly conserved between humans and other species. DYRK1A protein constructed using Swiss-model homologous modeling software, the three-dimensional structure of wild type differs from variant forms placed in Figure 3. c.1159C>T and c.1309C>T lead to two premature stop codons at residue 387 and residue 437 causing the termination of the primary structures of DYRK1A proteins. The nonsense variant c.1159C>T and c.1309C>T are located in exon8 and exon9 respectively, which is the interior of the kinase domain and lead to the elimination of the helix-turn-helix functional component, disrupting the critical activation segment. The variant exon3_exon4del is located in the NLS domain, which could interfere with the transport of proteins into the nucleus. The nonsense variant c.1159C>T and c.1309C>T variant lead to the elimination of and deletion of inframe shift exon3_exon4del interfere with the nuclear transport, and then the abnormal assembly of helix-turn-helix functional component and NLS domain, which damages the stability and flexibility of DYRK1A protein structure ultimately affect the function of the tyrosine autophosphorylation activity. Based on the DYRK1A protein prediction models of wild type, the variant forms cannot complete the protein structure due to the nonsense mutations or inframe shift, leading to an early end and deletion of the translation process, which has a significant impact on protein function. It has been reported that haploinsufficiency of DYRK1A is related to intellectual disability (ID), autism spectrum disorder (ASD), intrauterine growth restriction (IUGR), and development delay (DD) (van Bon et al., 2016; Earl et al., 2017; Arbones et al., 2019; Morison et al., 2022).
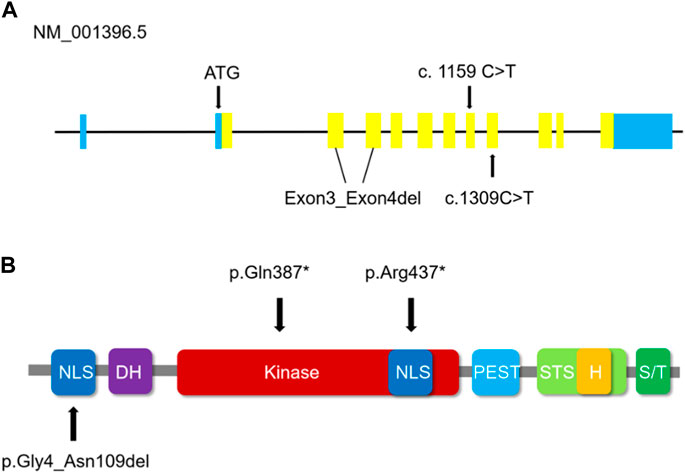
FIGURE 5. (A) Schematic representation of DYRK1A (NM_001396.5) coding regions illustrates the positions of the three variants in this study. Blue regions represent non-coding regions, yellow regions represent the coding sequence. (B) The domains in which the validated DYRK1A variants are positioned. The variant exon3_exon4del has an impact on the first NLS domain of the DYRK1A protein. The truncating amino acids alteration p.(Gln387*) of the c.1159 C>T variant and p.(Arg437*) of the c.1309 C>T variant are positioned in the Kinase domain and the second NLS domain respectively of the DYRK1A protein. NLS indicates the nuclear localization signals in the dark blue domain. DH indicates the DYRK homology box in the purple domain. STS indicates the speckle targeting signal in the light green domain. H indicates the histidine repeat in the yellow domain, and S/T indicates the serine/threonine repeat in the dark green domain. Kinase indicates protein kinase in the red domain, and the PEST is concentrated in proline, glutamic acids, serine, and threonine in the light blue domain.
Intellectual disability-7 was first reported in 1997, and MRD7 patients often show microcephaly, severe intellectual disability, dysmorphic features and feeding difficulties, and febrile seizures in childhood (Matsumoto et al., 1997; Møller et al., 2008). Other clinical manifestations may include intrauterine growth restriction, speech, anxious autistic behavior, and hand stereotypies. These symptoms mentioned above often lead to neurodevelopmental dysplasia (Widowati et al., 2018; Arbones et al., 2019; Mewasingh et al., 2020). The majority of patients with MRD7 present the imaging feature of diffuse cortical atrophy on MRI at a relatively early age (Courcet et al., 2012). In a large series of 31 MRD7 patients as listed in Table 2, although microcephaly (96.7%), DD (96.7%), and ID (96.7%) are the most general features of this disease, there is still a phenotype variability, with other general clinical discoveries like seizures (90%), feeding problems (83.4%), facial gestalt (80%), short stature (50%), IUGR (46.7%), ASD (40%), gait disturbance (26.7%), sleep disturbance (13.4%), hypertonia (13.4%) and dental anomalies (3.3%). Besides, there are uncommon clinical features but severe in significant organs of this disease including eye abnormalities (33.3%), musculoskeletal features (16.7%), cardiac defects (6.7%), and gastrointestinal problems (6.7%). However, it is notable that the probands in our cases did not show evidence of eye anomalies, cardiac defects, or gastrointestinal problems. As placed in (Supplementary Table S1), in the incidental findings of insufficient pathogenic evidence genes for the three probands by trio-WES, we found the c.-115C>G and c.*286A>T variants of TUBB3 in AD heterozygote inherited pattern estimated as VUS for the proband of c.1159C>T, c.194C>T and c.874A>G variants of NALCN in AD heterozygote inherited pattern estimated as VUS for the proband of c.1309C>T, 16p12.2del variants of C16orf52, CDR2, EF2K, POLR3E, VWA3A, and UQCRC2 in heterozygote inherited pattern estimated as pathogenic for the proband of exon3_exon4del, which have the related symptoms about neurological and psychiatric developmental abnormalities to MRD7. The c.-115C>G and c.*286A>T variants of TUBB3 could give rise to the dysfunction of neuronal-specific β-tubulin isotype III, which may lead to compound cortical dysplasia with other brain developmental abnormalities type 1 associated with the syndrome of MRD7, however, the location of TUBB3 variant is in the non-coding region and comprehensively estimated as LP. The c.194C>T and c.874A>G variants of NALCN were inherited from proband’s father and were estimated as VUS, her father did not show the symptoms of the same variants of NALCN and the certain variants’ pathogenic proof were not retrieved in the database presented as before (Radwitz et al., 2022; Puri et al., 2023). The 16p12.2del variants were inherited from the proband’s mother, which was estimated as pathogenic for her mother’s corresponding symptoms. Furthermore, the variant in UQCRC2 is related to Mitochondrial complex III deficiency, nuclear type 5, which is a mitochondrial respiratory chain disorder characterized by highly variable clinical phenotypes including mitochondrial encephalopathy, psychomotor delay, ataxia, severe dysplasia, liver dysfunction, metabolic acidosis, renal tubular disease, muscle weakness and exercise intolerance (Hernando et al., 2002; Ballif et al., 2007; Bansept et al., 2023). For the proband who possesses the mutations of DYRK1A and UQCRC2 prescribed as before, the symptoms caused by these two gene mutations have a certain degree of similarity, while the missing fragments caused by 16p12.2del are relatively functional, therefore we estimated this mutation as pathogenic. However, the common symptoms of her mother include ID and DD, but did not show any evidence of the microcephaly, seizures, feeding difficulties, and facial gestalt, which were diagnosed in the proband of exon3_exon4del. The experimental validation of DYRK1A for the three probands’ mutations requires we conduct further research.
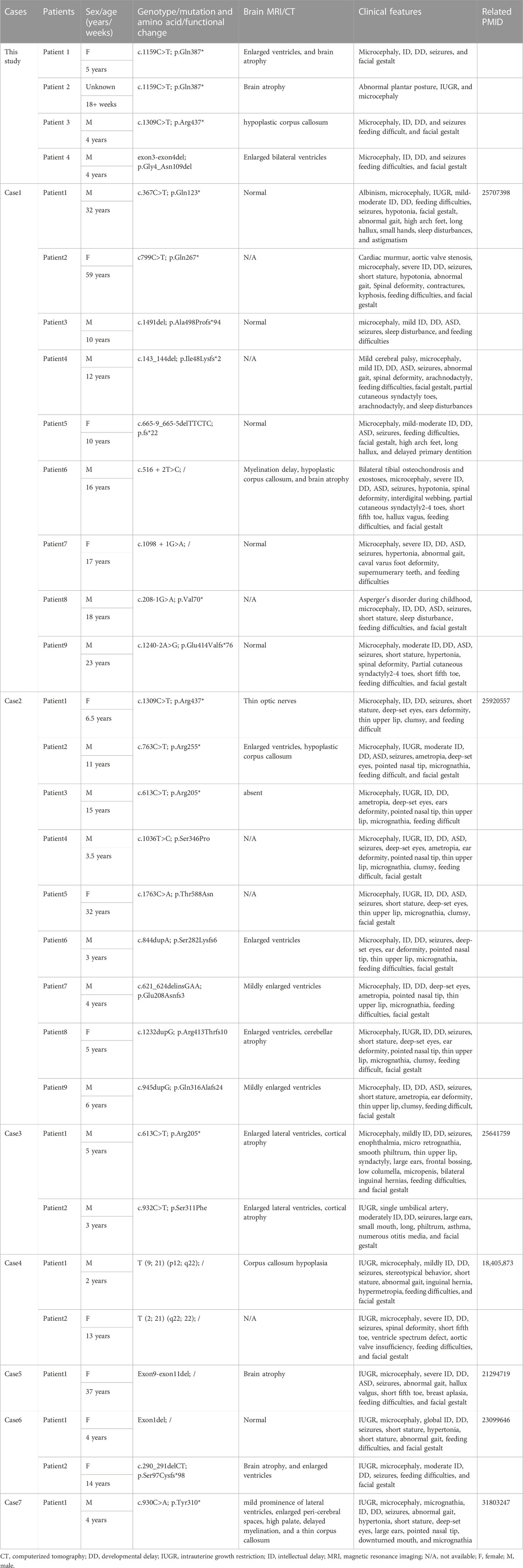
TABLE 2. DYRK1A mutations screening and clinical manifestations of patients.
To date, the majority of reported MRD7 patients have been sporadic cases resulting from a de novo pathogenic variant of the DYRK1A gene (Hamdan et al., 2014). With accordingly negligible risks of recurrence, apparent de novo mutations have been reckoned as the primary occurrences for germline or zygotic incidents (Xu et al., 2023). Nevertheless, for the pathogenic variant, those with a high ratio are truly inherited from a parent mosaic of which there is little evidence to prove. In our study, the fetus was detected with a heterozygous pathogenic variant of c.1159C>T p.(Gln387*) for DYRK1A, the same as the proband, which indicates a high probability of parental mosaicism by the occurrences of a same de novo mutation twice in a family. For a family with a childbearing history induced by an obvious de novo pathogenic variant affected by a genetic disease, the indetermination about the existence and the proportion of parental mosaicism gives cause counseling on the risk of recurrence to be imprecise and challenging. Furthermore, the risk of recurrence relies on whether the parental mosaic mutation exists in the paternal or maternal germ cell system and on the mutated reproductive cells’ harboring ratio (Campbell et al., 2014). It has been known that Sanger sequencing and the other conventional molecular technologies are limited in identifying the levels of somatic cell mosaicism for variant allele fraction or variant allele frequency (VAF) higher than 10%–20%. The normative exome sequencing variant-calling pipelining cannot routinely detect the existence of VAF at less than 10% level (Gambin et al., 2020). Nevertheless, in recent years progress in genomic technologies has improved the detection capability to be able to identify and analyze mosaicism at a low level (Pareja et al., 2022). Quantitative polymerase chain reaction (qPCR), high-depth sequencing, droplet digital PCR (ddPCR), single-base extension assays, and Multiple Independent Primer PCR Sequencing (MIPP-seq) all already proved efficient in improving the identification and quantification for mosaicism of low-level (Uchiyama et al., 2016; Doan et al., 2021; Zemet et al., 2022). In this study, due to the limitation of local technology and the burden of economic spending for the family of parent mosaicism detection, this family refused to detect the parent mosaicism. We respected their choices and informed them of the genetic cause of this situation.
Nowadays, diagnosis of MRD7 is generally established on the integration of typical clinical features and genetic analysis. Since DYRK1A was first defined as the inducing gene for MRD7 by Moller in 2008, clinical testing such as the ultrasonic evaluation of intrauterine fetal growth, and neurological, psychiatric, and auxiliary examinations was promoted and has helped a lot in the clinical diagnosis of MRD7 (Wong, 2009; Edwards and Hui, 2018; Davidson et al., 2021; Masselli et al., 2021). In recent years, WES has speedily developed as the most popular targeted enrichment method, which is peculiarly used to operate for monogenic (“Mendelian”) disorders in genetic testing at the molecular level. The exome includes about 85% of mutations with vast effects on disease-related traits, and shares only nearly 2% human genome. In our described cases, Trio-WES was performed for the unbiased analysis of the whole genes, time-efficient to sift the candidate genes preceding to detecting and reduced the demand for an economic-consuming (Singleton, 2011; Petrovski et al., 2019). According to the integration of Trio-WES and clinical background, the diagnosis pattern of MRD7 has been promoted enhancing the symptomatic diagnosis, prenatal diagnosis, and genetic counseling.
Conclusion
We presented two novel likely pathogenic variants and one rare likely pathogenic variant inducing heterozygous presentations of DYRK1A-associated MRD7 and described the clinical heterogeneity for those affected populations. We described the process of providing precise genetic counseling according to the fetus’ pathogenic evidence, leading to a decision whether to terminate the abnormal gestation, and informed those three families of professional fertility counseling and guidance for the next gestation.
Data availability statement
The original contributions presented in the study are publicly available. This data can be found here: https://doi.org/10.6084/m9.figshare.24598266.v1.
Ethics statement
The studies involving humans were approved by the No. 20200086 Ethics Committee of Jiangxi Maternal and Child Health Hospital. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants’ legal guardians/next of kin. Written informed consent was obtained from the minor(s)’ legal guardian/next of kin for the publication of any potentially identifiable images or data included in this article.
Author contributions
CH: Data curation, Investigation, Writing–original draft. HL: Data curation, Investigation, Writing–original draft. BZ: Data curation, Investigation, Writing–original draft. CF: Data curation, Writing–original draft, Validation. JC: Data curation, Writing–original draft. HY: Data curation, Writing–original draft. SH: Writing–original draft. BY: Writing–original draft, Funding acquisition, Resources. YZ: Funding acquisition, Resources, Writing–review and editing. YL: Formal Analysis, Funding acquisition, Resources, Visualization, Writing–review and editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This study is supported by the Jiangxi Province Graduate Innovation Special Fund Project (YC2022-s237 to CH), Nature Science Research Project of Jiangxi province (Grant No. 20232BAB216025 to HL) and Jiangxi Provincial Key Laboratory of Birth Defect for Prevention and Control (No. 20202BCD42017). National Natural Science Foundation of China (Grant No. 82160318), Jiangxi Provincial Clinical Research Center for Birth Defects (20223BCG74002), Science Foundation of Jiangxi Province (No. 20224BAB206037). Jiangxi Province Key Research and Development Project (Grant No. 20232BBG70023).
Acknowledgments
We appreciate those patients’ families for sharing the medical information of their own free will.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2023.1290949/full#supplementary-material
References
Alvarez, M., Estivill, X., and de la Luna, S. (2003). DYRK1A accumulates in splicing speckles through a novel targeting signal and induces speckle disassembly. J. Cell Sci. 116 (15), 3099–3107. doi:10.1242/jcs.00618
Arbones, M. L., Thomazeau, A., Nakano-Kobayashi, A., Hagiwara, M., and Delabar, J. M. (2019). DYRK1A and cognition: a lifelong relationship. Pharmacol. Ther. 194, 199–221. doi:10.1016/j.pharmthera.2018.09.010
Ballif, B. C., Hornor, S. A., Jenkins, E., Madan-Khetarpal, S., Surti, U., Jackson, K. E., et al. (2007). Discovery of a previously unrecognized microdeletion syndrome of 16p11.2-p12.2. Nat. Genet. 39 (9), 1071–1073. doi:10.1038/ng2107
Bansept, C., Gaignard, P., Lebigot, E., Eyer, D., Delplancq, G., Hoebeke, C., et al. (2023). UQCRC2-related mitochondrial complex III deficiency, about 7 patients. Mitochondrion 68, 138–144. doi:10.1016/j.mito.2022.12.001
Bertoni, M., Kiefer, F., Biasini, M., Bordoli, L., and Schwede, T. (2017). Modeling protein quaternary structure of homo- and hetero-oligomers beyond binary interactions by homology. Sci. Rep. 7 (1), 10480. doi:10.1038/s41598-017-09654-8
Bienert, S., Waterhouse, A., de Beer, T. A. P., Tauriello, G., Studer, G., Bordoli, L., et al. (2017). The SWISS-MODEL Repository-new features and functionality. Nucleic Acids Res. 45 (D1), D313–d319. doi:10.1093/nar/gkw1132
Brault, V., Nguyen, T. L., Flores-Gutiérrez, J., Iacono, G., Birling, M. C., Lalanne, V., et al. (2021). Dyrk1a gene dosage in glutamatergic neurons has key effects in cognitive deficits observed in mouse models of MRD7 and Down syndrome. PLoS Genet. 17 (9), e1009777. doi:10.1371/journal.pgen.1009777
Camacho, C., Coulouris, G., Avagyan, V., Papadopoulos, J., and Bealer, K. (2009). BLAST+: architecture and applications. BMC Bioinforma. 10, 421. doi:10.1186/1471-2105-10-421
Campbell, I. M., Stewart, J. R., James, R. A., Lupski, J. R., Stankiewicz, P., Olofsson, P., et al. (2014). Parent of origin, mosaicism, and recurrence risk: probabilistic modeling explains the broken symmetry of transmission genetics. Am. J. Hum. Genet. 95 (4), 345–359. doi:10.1016/j.ajhg.2014.08.010
Courcet, J. B., Faivre, L., Malzac, P., Masurel-Paulet, A., Lopez, E., Callier, P., et al. (2012). The DYRK1A gene is a cause of syndromic intellectual disability with severe microcephaly and epilepsy. J. Med. Genet. 49 (12), 731–736. doi:10.1136/jmedgenet-2012-101251
Davidson, J. R., Uus, A., Matthew, J., Egloff, A. M., Deprez, M., Yardley, I., et al. (2021). Fetal body MRI and its application to fetal and neonatal treatment: an illustrative review. Lancet Child. Adolesc. Health 5 (6), 447–458. doi:10.1016/S2352-4642(20)30313-8
Doan, R. N., Miller, M. B., Kim, S. N., Rodin, R. E., Ganz, J., Bizzotto, S., et al. (2021). MIPP-Seq: ultra-sensitive rapid detection and validation of low-frequency mosaic mutations. BMC Med. Genomics 14 (1), 47. doi:10.1186/s12920-021-00893-3
Earl, R. K., Turner, T. N., Mefford, H. C., Hudac, C. M., Gerdts, J., Eichler, E. E., et al. (2017). Clinical phenotype of ASD-associated DYRK1A haploinsufficiency. Mol. Autism 8, 54. doi:10.1186/s13229-017-0173-5
Edwards, L., and Hui, L. (2018). First and second trimester screening for fetal structural anomalies. Semin. Fetal Neonatal Med. 23 (2), 102–111. doi:10.1016/j.siny.2017.11.005
Eisenberg, E., and Levanon, E. Y. (2013). Human housekeeping genes, revisited. Trends Genet. 29 (10), 569–574. doi:10.1016/j.tig.2013.05.010
Evers, J. M., Laskowski, R. A., Bertolli, M., Clayton-Smith, J., Deshpande, C., Eason, J., et al. (2017). Structural analysis of pathogenic mutations in the DYRK1A gene in patients with developmental disorders. Hum. Mol. Genet. 26 (3), 519–526. doi:10.1093/hmg/ddw409
Fotaki, V., Dierssen, M., Alcántara, S., Martínez, S., Martí, E., Casas, C., et al. (2002). Dyrk1A haploinsufficiency affects viability and causes developmental delay and abnormal brain morphology in mice. Mol. Cell Biol. 22 (18), 6636–6647. doi:10.1128/mcb.22.18.6636-6647.2002
Gambin, T., Liu, Q., Karolak, J. A., Grochowski, C. M., Xie, N. G., Wu, L. R., et al. (2020). Low-level parental somatic mosaic SNVs in exomes from a large cohort of trios with diverse suspected Mendelian conditions. Genet. Med. 22 (11), 1768–1776. doi:10.1038/s41436-020-0897-z
Guimerá, J., Casas, C., Pucharcòs, C., Solans, A., Domènech, A., Planas, A. M., et al. (1996). A human homologue of Drosophila minibrain (MNB) is expressed in the neuronal regions affected in Down syndrome and maps to the critical region. Hum. Mol. Genet. 5 (9), 1305–1310. doi:10.1093/hmg/5.9.1305
Hamdan, F. F., Srour, M., Capo-Chichi, J. M., Daoud, H., Nassif, C., Patry, L., et al. (2014). De novo mutations in moderate or severe intellectual disability. PLoS Genet. 10 (10), e1004772. doi:10.1371/journal.pgen.1004772
Hämmerle, B., Vera-Samper, E., Speicher, S., Arencibia, R., Martínez, S., and Tejedor, F. J. (2002). Mnb/Dyrk1A is transiently expressed and asymmetrically segregated in neural progenitor cells at the transition to neurogenic divisions. Dev. Biol. 246 (2), 259–273. doi:10.1006/dbio.2002.0675
Hernando, C., Plaja, A., Rigola, M. A., Pérez, M. M., Vendrell, T., Egocue, J., et al. (2002). Comparative genomic hybridisation shows a partial de novo deletion 16p11.2 in a neonate with multiple congenital malformations. J. Med. Genet. 39 (5), E24. doi:10.1136/jmg.39.5.e24
Kang, J. Q., Shen, W., and Macdonald, R. L. (2006). Why does fever trigger febrile seizures? GABAA receptor gamma2 subunit mutations associated with idiopathic generalized epilepsies have temperature-dependent trafficking deficiencies. J. Neurosci. 26 (9), 2590–2597. doi:10.1523/JNEUROSCI.4243-05.2006
Kinstrie, R., Luebbering, N., Miranda-Saavedra, D., Sibbet, G., Han, J., Lochhead, P. A., et al. (2010). Characterization of a domain that transiently converts class 2 DYRKs into intramolecular tyrosine kinases. Sci. Signal 3 (111), ra16. doi:10.1126/scisignal.2000579
Laguna, A., Aranda, S., Barallobre, M. J., Barhoum, R., Fernández, E., Fotaki, V., et al. (2008). The protein kinase DYRK1A regulates caspase-9-mediated apoptosis during retina development. Dev. Cell 15 (6), 841–853. doi:10.1016/j.devcel.2008.10.014
Ma, L., Wu, Z., Tang, Q., Ji, X., Mei, Y., Peng, T., et al. (2021). Generation of an induced pluripotent stem cell line from a patient with global development delay carrying DYRK1A mutation (c.1730T>A) and a gene correction isogenic iPSC line. Stem Cell Res. 53, 102305. doi:10.1016/j.scr.2021.102305
Masselli, G., Cozzi, D., Ceccanti, S., Laghi, F., Giancotti, A., and Brunelli, R. (2021). Fetal body MRI for fetal and perinatal management. Clin. Radiol. 76 (9), 708.e1–708.e8. doi:10.1016/j.crad.2021.05.006
Matsumoto, N., Ohashi, H., Tsukahara, M., Kim, K. C., Soeda, E., and Niikawa, N. (1997). Possible narrowed assignment of the loci of monosomy 21-associated microcephaly and intrauterine growth retardation to a 1.2-Mb segment at 21q22.2. Am. J. Hum. Genet. 60 (4), 997–999.
Mewasingh, L. D., Chin, R. F. M., and Scott, R. C. (2020). Current understanding of febrile seizures and their long-term outcomes. Dev. Med. Child. Neurol. 62 (11), 1245–1249. doi:10.1111/dmcn.14642
Møller, R. S., Kübart, S., Hoeltzenbein, M., Heye, B., Vogel, I., Hansen, C. P., et al. (2008). Truncation of the Down syndrome candidate gene DYRK1A in two unrelated patients with microcephaly. Am. J. Hum. Genet. 82 (5), 1165–1170. doi:10.1016/j.ajhg.2008.03.001
Morison, L. D., Braden, R. O., Amor, D. J., Brignell, A., van Bon, B. W. M., and Morgan, A. T. (2022). Social motivation a relative strength in DYRK1A syndrome on a background of significant speech and language impairments. Eur. J. Hum. Genet. 30 (7), 800–811. doi:10.1038/s41431-022-01079-w
Noveski, P., Terzic, M., Vujovic, M., Kuzmanovska, M., Sukarova Stefanovska, E., and Plaseska-Karanfilska, D. (2019). Multilevel regression modeling for aneuploidy classification and physical separation of maternal cell contamination facilitates the QF-PCR based analysis of common fetal aneuploidies. PLoS One 14 (8), e0221227. doi:10.1371/journal.pone.0221227
Pareja, F., Ptashkin, R. N., Brown, D. N., Derakhshan, F., Selenica, P., da Silva, E. M., et al. (2022). Cancer-causative mutations occurring in early embryogenesis. Cancer Discov. 12 (4), 949–957. doi:10.1158/2159-8290.CD-21-1110
Petrovski, S., Aggarwal, V., Giordano, J. L., Stosic, M., Wou, K., Bier, L., et al. (2019). Whole-exome sequencing in the evaluation of fetal structural anomalies: a prospective cohort study. Lancet 393 (10173), 758–767. doi:10.1016/S0140-6736(18)32042-7
Puri, D., Barry, B. J., and Engle, E. C. (2023). TUBB3 and KIF21A in neurodevelopment and disease. Front. Neurosci. 17, 1226181. doi:10.3389/fnins.2023.1226181
Qiao, F., Shao, B., Wang, C., Wang, Y., Zhou, R., Liu, G., et al. (2019). A de novo mutation in DYRK1A causes syndromic intellectual disability: a Chinese case report. Front. Genet. 10, 1194. doi:10.3389/fgene.2019.01194
Qu, L., and Leung, L. S. (2008). Mechanisms of hyperthermia-induced depression of GABAergic synaptic transmission in the immature rat hippocampus. J. Neurochem. 106 (5), 2158–2169. doi:10.1111/j.1471-4159.2008.05576.x
Radwitz, J., Hausrat, T. J., Heisler, F. F., Janiesch, P. C., Pechmann, Y., Rübhausen, M., et al. (2022). Tubb3 expression levels are sensitive to neuronal activity changes and determine microtubule growth and kinesin-mediated transport. Cell Mol. Life Sci. 79 (11), 575. doi:10.1007/s00018-022-04607-5
Singleton, A. B. (2011). Exome sequencing: a transformative technology. Lancet Neurol. 10 (10), 942–946. doi:10.1016/S1474-4422(11)70196-X
Soundararajan, M., Roos, A. K., Savitsky, P., Filippakopoulos, P., Kettenbach, A. N., Olsen, J. V., et al. (2013). Structures of Down syndrome kinases, DYRKs, reveal mechanisms of kinase activation and substrate recognition. Structure 21 (6), 986–996. doi:10.1016/j.str.2013.03.012
Steinegger, M., Meier, M., Mirdita, M., Vöhringer, H., Haunsberger, S. J., and Söding, J. (2019). HH-suite3 for fast remote homology detection and deep protein annotation. BMC Bioinforma. 20 (1), 473. doi:10.1186/s12859-019-3019-7
Studer, G., Rempfer, C., Waterhouse, A. M., Gumienny, R., Haas, J., and Schwede, T. (2020). QMEANDisCo-distance constraints applied on model quality estimation. Bioinformatics 36 (8), 1765–1771. doi:10.1093/bioinformatics/btz828
Studer, G., Tauriello, G., Bienert, S., Biasini, M., Johner, N., and Schwede, T. (2021). ProMod3-A versatile homology modelling toolbox. PLoS Comput. Biol. 17 (1), e1008667. doi:10.1371/journal.pcbi.1008667
Tejedor, F., Zhu, X. R., Kaltenbach, E., Ackermann, A., Baumann, A., Canal, I., et al. (1995). minibrain: a new protein kinase family involved in postembryonic neurogenesis in Drosophila. Neuron 14 (2), 287–301. doi:10.1016/0896-6273(95)90286-4
Uchiyama, Y., Nakashima, M., Watanabe, S., Miyajima, M., Taguri, M., Miyatake, S., et al. (2016). Ultra-sensitive droplet digital PCR for detecting a low-prevalence somatic GNAQ mutation in Sturge-Weber syndrome. Sci. Rep. 6, 22985. doi:10.1038/srep22985
van Bon, B. W., Coe, B. P., Bernier, R., Green, C., Gerdts, J., Witherspoon, K., et al. (2016). Disruptive de novo mutations of DYRK1A lead to a syndromic form of autism and ID. Mol. Psychiatry 21 (1), 126–132. doi:10.1038/mp.2015.5
van Bon, B. W., Hoischen, A., Hehir-Kwa, J., de Brouwer, A. P. M., Ruivenkamp, C., Gijsbers, A. C. J., et al. (2011). Intragenic deletion in DYRK1A leads to mental retardation and primary microcephaly. Clin. Genet. 79 (3), 296–299. doi:10.1111/j.1399-0004.2010.01544.x
Waterhouse, A., Bertoni, M., Bienert, S., Studer, G., Tauriello, G., Gumienny, R., et al. (2018). SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res. 46 (W1), W296–w303. doi:10.1093/nar/gky427
Widowati, E. W., Ernst, S., Hausmann, R., Müller-Newen, G., and Becker, W. (2018). Functional characterization of DYRK1A missense variants associated with a syndromic form of intellectual deficiency and autism. Biol. Open 7 (4), bio032862. doi:10.1242/bio.032862
Wong, K. (2009). Ultrasonographic examination for prevention of fetal birth defect. Beijing Da Xue Xue Bao Yi Xue Ban. 41 (4), 492–499.
Xu, N., Shi, W., Cao, X., Zhou, X., Jin, L., Huang, H. F., et al. (2023). Parental mosaicism detection and preimplantation genetic testing in families with multiple transmissions of de novo mutations. J. Med. Genet. 60 (9), 910–917. doi:10.1136/jmg-2022-108920
Keywords: DYRK1A, MRD7, novel variant, prenatal diagnosis, whole exome sequencing
Citation: Huang C, Luo H, Zeng B, Feng C, Chen J, Yuan H, Huang S, Yang B, Zou Y and Liu Y (2023) Identification of two novel and one rare mutation in DYRK1A and prenatal diagnoses in three Chinese families with intellectual Disability-7. Front. Genet. 14:1290949. doi: 10.3389/fgene.2023.1290949
Received: 08 September 2023; Accepted: 07 December 2023;
Published: 20 December 2023.
Edited by:
Simona Sestito, University “Magna Graecia” of Catanzaro, ItalyReviewed by:
Roberta Polli, University of Padua, ItalyEnrique Medina-Acosta, State University of Northern Rio de Janeiro, Brazil
Copyright © 2023 Huang, Luo, Zeng, Feng, Chen, Yuan, Huang, Yang, Zou and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yanqiu Liu, bHlxMDkxNEAxMjYuY29t; Yongyi Zou, em91eW9uZ3lpQGdtYWlsLmNvbQ==; Bicheng Yang, eWFuZ2JjMTk4NUAxMjYuY29t
†These authors have contributed equally to this work