 Jianhui Chen
Jianhui Chen Hongping Ma
Hongping Ma Fan Lin
Fan Lin Tuanyao Chai
Tuanyao Chai Hong Wang
Hong Wang- College of Life Sciences, University of Chinese Academy of Sciences, Beijing, China
Reynoutria japonica Houtt. is an important medical plant with a long history of thousands of years in China, however, its mitochondrial genome (mitogenome) has not been reported yet. In this work, we reported and analyzed the R. japonica mitogenome. The main results include: The R. japonica mitogenome was 302,229 bp in length and encoded 48 genes, including 27 protein-coding genes (PCGs), 3 rRNA genes, and 18 tRNA genes. Repeat sequence analysis revealed that there were 54 repeat sequences ranging from 193 bp to 1,983 bp in the R. japonica mitogenome. Relative synonymous codon usage (RSCU) analysis showed that leucine (900, 11.01%) and serine (732, 8.96%) were the two most abundant amino acids, and the codons with RSCU values showed the preference of A or T ending when greater than 1. The RNA editing sites of PCGs in the R. japonica mitogenome were characterized, and 299 RNA editing sites were found. Extensive sequences transfer between mitochondrion and chloroplast were found in R. japonica, where 11 complete plastid-derived tRNA genes stayed intact in the R. japonica mitogenome. Three genes (ccmFC, cox1, and nad1) were seen to play essential roles in the evolution through selection pressure analysis. The phylogenetic analysis showed that Fallopia multiflora was the closest species with R. japonica, in consistency with the results of chloroplast genome. Overall, the current work presents the first mitogenome of R. japonica and could contribute to the phylogenetic analysis of the family Polygonaceae.
1 Introduction
Plant mitochondria are semi-autonomous organelles, possessing relatively independent genetic systems and contributing to metabolism, energy production and cell homeostasis (Gualberto et al., 2014). It is generally believed that plant mitochondria were evolved from free-living bacteria according the endosymbiotic theory (Dyall et al., 2004). The mitochondria genome (mitogenome) in higher plants are very diversified in size, ranging from 22 Kb in Avicennia marina to 11.7 Mb in Larix sibitia (Putintseva et al., 2020; Friis et al., 2021) with distant genetic relationships, even between closely related species (Sloan et al., 2012; Cole et al., 2018). Due to the high frequency homologous recombination with foreign DNA, the mitogenomes in plants are often subject to rearrangement and more complex in size, structure and genes order (Wu et al., 2020). Note also that the homologous sequences in the seed plant mitogenomes are mainly derived from the chloroplast and nucleus (Drouin et al., 2008). Unlike chloroplast genome which is usually a double-stranded and circular molecule, plant mitogenome was found in multiple structural forms rather than the single ring form (Kozik et al., 2019). It is reported that many plant mitogenomes possess linear and branch structures, and a lot of smaller circular molecules (Sloan, 2013; Gualberto et al., 2014). For example, three loops were found in the mitogenome of Hemerocallis citrina and Populus simonii (Bi et al., 2022; Zhang et al., 2022). The repeat sequences are the main reasons that confused the ultimate conformation of mitogenome (Li et al., 2022; Yang et al., 2022). Overall, the plant mitogenome experienced sophisticated changes in size and structure during evolution, hence recovering the conformation of plant mitogenome is a both challenging and rewarding task.
Reynoutria japonica Houtt. (Polygonaceae), a well known traditional Chinese herbal medicine, has been used since ancient time in China (Peng et al., 2013). The dried root of R. japonica in combination with other traditional Chinese medicine herbs have multiple therapeutic uses (Zhang et al., 2013). In this ancient traditional Chinese medicine plant, the chloroplast genome of different regions R. japonica has been systematically analyzed (Chen et al., 2022). But its nuclear genome or mitogenome has not been published yet. Currently, the mitogenomes of the family Polygonaceae still remain largely unknown. Although the mitogenomes of Fallopia multiflora were assembled into two circular chromosomes via Illumina platform (Kim and Kim, 2018), few species mitogenomes in the family Polygonaceae are available in NCBI (National Center for Biotechnology Information) database. Therefore, it is necessary and desirable to attain the R. japonica mitogenome to enrich the Polygonaceae species mitogenome for further evolutionary studies. In recent years, an increasing interest of plant mitogenome is observed largely due to the advancement of sequencing technology and the reduction of sequencing costs, in particular, the application of Oxford Nanopore sequencing technology which has the advantage of long reading sequences to reduce the hassle caused by repetitive sequences compared to Illumina reads. Note that the combination of Illumina and Oxford Nanopore reads, several species mitogenome were obtained, including Mesona chinensis Benth (Tang et al., 2023), Abelmoschus esculentus (Li et al., 2022), Hemerocallis citrina (Zhang et al., 2022), Photinia serratifolia (Wang et al., 2023a), etc. These results demonstrate it is possible to assemble a complete mitogenome via the combination of short reads and long reads.
In this study, we sequenced and assembled the mitogenome of R. japonica via the Illumina short-read and Nanopore long-read integrated pipeline. The characteristic features of the R. japonica mitogenome were compared with those published related species. To our knowledge, this is the first assembly of the R. japonica mitogenome, which could be used for understanding the evolution of R. japonica, as well as the molecular biology research of this medicinal plant.
2 Materials and methods
2.1 Plant materials and genome sequencing
The seeds of R. japonica were collected from the medicinal plant garden of the Institute of Botany, Chinese Academy of Sciences (Beijing, China), planted and germinated in the lab and grown in a climate chamber at the temperature of 24°C ± 2°C with light/dark cycle of 16h/8 h. The well-grown young leaves were collected for DNA extraction. Total genomic DNA was isolated using the modified CTAB method (Arseneau et al., 2017). Then the quality of the extracted DNA was examined by NanoDrop (Thermo Scientific, United States), a Qubit fluorometer (Thermo Scientific, United States), and 0.75% agarose gel electrophoresis, respectively. The BluePippin system (Sage Science, United States) was used to recover large DNA fragments. Then the DNA fragments were treated using damage repair, end preparation, A-tailing, adapter ligation and the purification of DNA from the previous reaction using magnetic beads. The purified library was constructed following the SQK-LSK109 (Oxford, United Kingdom) sequencing kit protocol and loaded into a Nanopore GridION Sequencer (ONT, United Kingdom), which carried out at GrandOmics (Wuhan, China). Effective data were obtained by filtering adapter and removing low-quality reads. In total, 11.53 Gb of data were generated form 577,047 reads (SRA accession SRR24988768).
2.2 Genome assembly and annotation
The Oxford Nanopore long reads were assembled into contigs via NextDenovo v2.5.0 (https://github.com/Nextomics/NextDenovo). Mitochondrial contigs were identified by the BLASTn program (Chen et al., 2015) with Fallopia multiflora (accession number: MF611850, MF611851) mitogenome as references. And the self-loop candidate contigs were found and polished by Pilon v1.23 (Walker et al., 2014) using Illumina Novaseq sequencing reads, which has been used to assemble the R. japonica chloroplast genome before (Chen et al., 2022). Finally, one circular structure of the R. japonica mitogenome was obtained. The self-loop mitogenome of R. japonica was annotated via online tool GeSeq (Tillich et al., 2017) with the mitogenome of F. multiflora (accession number: MF611850, MF611851), and the preliminary annotation was further redressed with the mitogenome of Fallopia aubertii (accession number: MW664926). In order to test the credibility of the mitogenome, BWA 0.7.17-r1188 (Li and Durbin, 2009) and samtools v1.9 (Danecek et al., 2021) were used to calculate the sequencing depth of each locus. Finally, the mitogenome map of R. japonica was drawn using OGDRAW (Greiner et al., 2019).
The dispersed repeat sequences were analyzed by the online REPuter software (https://bibiserv.cebitec.uni-bielefeld.de/reputer) with the parameter of minimal repeats set to 50 bp, and hamming Distance to 3 (Kurtz et al., 2001). The repeat sequences in the R. japonica mitogenome were visualized via Circos v0.69-8 (Krzywinski et al., 2009). The relative synonymous codon usage (RSCU) of the unique protein coding genes (PCGs) of R. japonica mitogenome was calculated by CodonW v1.4.4. For the RNA editing sites analysis of the unique PCGs, the RNA-seq data released by our laboratory (PRJNA626400 and RPJNA623335) in the early stage were first filtered via fastp v0.23.2 (Chen et al., 2018), and then mapped to the PCGs of R. japonica mitogenome via Bowtie2 v2.3.5.1 (Langmead and Salzberg, 2012). And the possible RNA editing sites were identified via bcftools v1.9 (Danecek et al., 2021) according to the mapping results, and the locations with a coverage depth of more than 10× were selected.
2.3 Plastid-like sequences in the mitogenome
The R. japonica mitogenome was compared with its chloroplast genome (GenBank accession number: OP583946) by BLASTn with E value less that 1e-5, and visualized via Circos v0.69-8 (Krzywinski et al., 2009).
2.4 Selection pressure analysis of PCGs
The PCGs were selected to estimate the selection pressure during the evolution of R. japonica. Nonsynonymous (Ka) and synonymous (Ks) substitution rates of the 25 unique PCGs were calculated for R. japonica and other three species (Polygonum aviculare, Fallopia aubertii, and Fallopia multiflora). ParaAT2.0 was used to align and format the PCGs with default parameters (Zhang et al., 2012). The Ka, Ks, and Ka/Ks values were calculated via KaKs_Calculator v3.0 following the YN method (Zhang, 2022).
2.5 Phylogenetic analysis
The conserved mitogenomes PCGs of R. japonica and other ten plant species were identified by BLAT (Kent, 2002). The ten plants, including F. multiflora (MF611850 and MF611851), F. aubertii (MW664926), P. aviculare (OW204033), Nepenthes X ventrata (MH798871), Chenopodium quinoa (MK182703), Beta vulgaris (NC_002511), Silene vulgaris (JF750427), Arachis hypogaea (MW448460), Vitis vinifera (NC_012119), and Arabidopsis thaliana (NC_037304) were downloaded from the NCBI. Twelve PCGs (atp8, atp9, cox1, cox2, nad1, nad3, nad4, nad4L, nad5, nad6, nad7, and nad9) were aligned by MAFFT v7.520 (Katoh and Standley, 2013). And GTR + I + G4 model was selected by ModelTest-NG (Darriba et al., 2020) according to Bayesian information criterion scores. Then maximum likelihood (ML) phylogenetic tree was constructed by RAxML-NG v0.9.0 (Kozlov et al., 2019) with 1000 bootstrap replicates.
3 Results and discussion
3.1 Mitogenome assembly and genomic features
The R. japonica mitochondrial genome (mitogenome) was first assembled with Oxford Nanopore reads, and then polished with Illumina reads due to the short reads possessing higher base recognition accuracy than long-read sequencing (Delahaye and Nicolas, 2021). This is a common strategy when combining short reads and long reads. By this way, 112 contigs were assembled via NextDenovo. And a self-loop contig was obtained with length of 335,479 bp that can be mapped with the mitogenome of F. multiflora. To detect whether it is a circular one, the alignment against itself was performed. Surprisingly, a large fragment was found at the beginning and end, with 99.10% similarity (Supplementary Figure S1) which verifies its circular nature. By removing the tail almost identical sequence, a finally circular structure with 302,229 bp in size was obtained and submitted to NCBI under accession OR228435 (Figure 1). And the average depth was 253× (long reads) and 570× (short reads), respectively (Supplementary Figure S2). The depth of long reads ranging from 3× to 5,212×, achieved all sites of the R. japonica mitogenome and made up for the shortcomings of Illumina reads (Supplementary Table S1; Supplementary Figure S2), indicating that the gap-free R. japonica mitogenome was obtained. In addition, other 4 contigs, including 3 linear and 1 loop molecular, were also mapped to the F. multiflora mitogenome. However, the 3 linear contigs failed to be annotated as mitogenome, and the loop contig was more like a plastid genome than a mitogenome (Supplementary Figure S3).
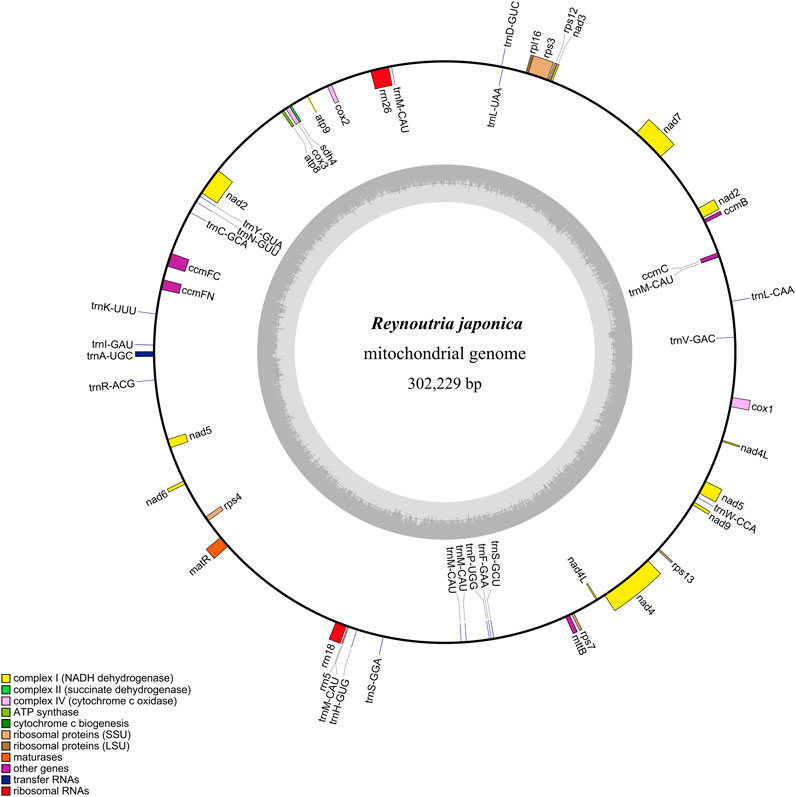
FIGURE 1. Circular map of the R. japonica mitogenome.
The base composition of the R. japonica mitogenome was A (27.80%), T (27.63%), G (22.32%), and C (22.25%). The mitogenome contained 53 genes, representing 48 unique genes, including 27 protein-coding genes (PCGs), 18 tRNA genes, and 3 rRNA genes (Table 1). The 27 PCGs consisted of 9 NADH dehydrogenase genes (nad1, nad2, nad3, nad4, nad4L, nad5, nad6, nad7, and nad9), a transport membrane protein (sdh4), 3 cytochrome C oxidase genes (cox1, cox2, and cox3), 2 ATP synthase genes (atp8 and atp9), a maturase (matR), 4 cytochrome C biogenesis genes (ccmB, ccmC, ccmFc, and ccmFn), a large ribosomal protein (LSU) gene (rpl16) and 5 small ribosomal proteins (SSU) genes (rps3, rps4, rps7, rps12, and rps13). The total length of these 27 PCGs was 24,678 bp, accounting for 8.17% of the R. japonica mitogenome. And the length of tRNA and rRNA genes accounted for 0.56% and 1.71%, respectively. The intergenic region reached a proportion of 89.47%. In the R. japonica mitogenome PCGs, only nad4L had two copies, and the rest are all single copy. There were 7 unique PCGs containing introns in the R. japonica mitogenome (nad1, nad2, nad4, nad5, nad7, ccmFC, and rps3). Additionally, three trans-spliced introns were found in nad1, nad2, and nad5. Four genes, namely, mttB, rpl16, nad1, and nad4L were observed with RNA editing in start codon. A total of 22 tRNAs represented by 18 tRNAs were found, specifying 16 amino acids. And trnL and trnS possessed two copies, while trnM contained five copies (Table 1). By performing the syntenic regions between R. japonica and F. multiflora, the final mitogenome of R. japonica appeared of high similarity to F. multiflora (Supplementary Figure S4). Compared with the mitogenome of F. multiflora, the R. japonica mitogenome has lost several PCGs (namely, atp1, atp4, atp6, cob1, rpl5, rps1, and rps14). It was reported that some mitochondrial genes were lost or transferred to the nucleus during the evolution (Adams et al., 2002). The PCGs failed to be screened in the R. japonica mitogenome and the 112 assembly contigs might be either lost during the evolution or transferred to the nuclear genome as its nuclear genome is not published yet.
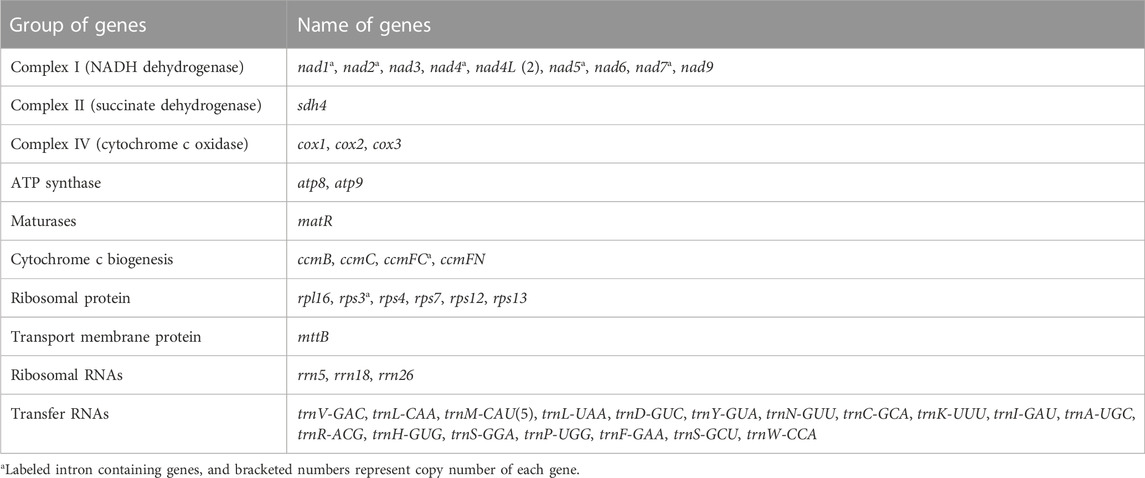
TABLE 1. Gene composition of the R. japonica mitogenome.
3.2 Repeat sequence analysis of R. japonica mitogenome
Repeat sequences are the core factor result in the size expansion of plant mitogenome (Alverson et al., 2010). In this study, a total of 54 pairs of repetitive sequences were identified, ranging from 193 bp to 1,983 bp in the R. japonica mitogenome (Supplementary Table S2). Seven large repeat sequences were found bigger than 1 kb, which may participate in intramolecular recombination (Bi et al., 2016). It was reported that the repeat fragments could mediate the homologous recombination in the plant mitogenome, such as sweet potato (Yang et al., 2022) and Scutellaria tsinyunensis (Li et al., 2021). And the repeats in R. japonica mitogenome may also form multiple circular molecules. All the repeats were present primarily as forward or palindromic repeats, and we showed this with different color lines in R. japonica mitogenome. As shown in Figure 2, most of the repeat sequences were located in intergenic region, and mainly between nad7/nad3, atp8/trnY-GUA, matR/rrn18, rps7/nad4L, and nad4L/cox1. The repeat sequences in the R. japonica did not involve any gene, indicating the genes in the R. japonica mitogenome are very conservative.
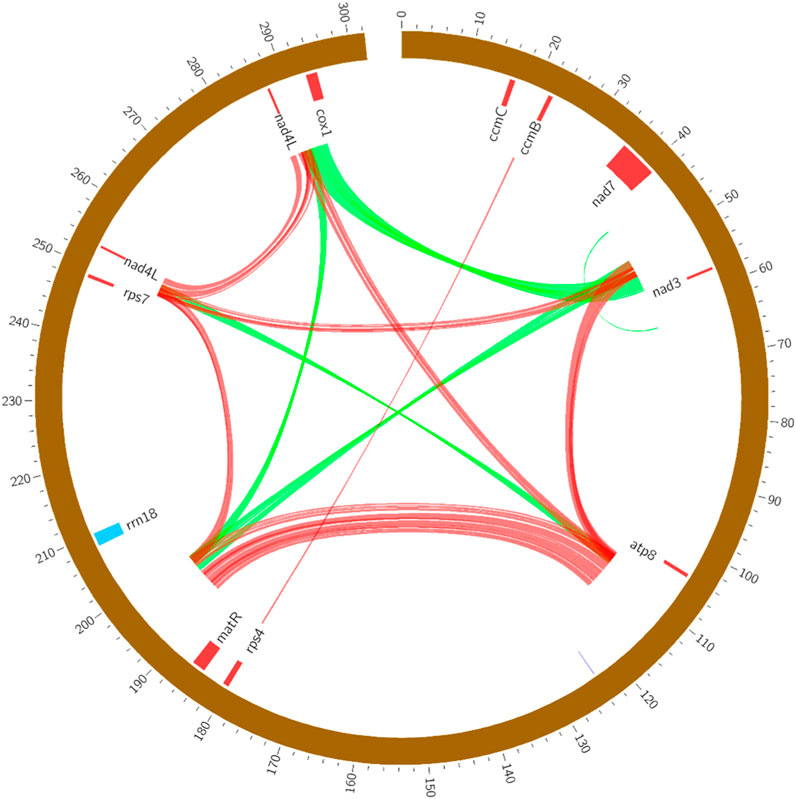
FIGURE 2. The forward (green) and palindromic (red) repeats in the R. japonica mitogenome.
3.3 RSCU and RNA editing sites analysis of PCGs
To investigate the codon preferences of PCGs in the R. japonica mitogenome, the RSCU analysis was performed. Most PCGs used ATG as the start codon, except mttB (ATA), nad1 (ACG), nad4L (ACG), and rpl16 (GTG). By connecting the 27 unique PCGs with only one start codon (ATG) and a stop codon, a total of 8,174 codons were found in the R. japonica mitogenome. Leucine (900, 11.01%) and serine (732, 8.96%) were the two most amino acids, while cystine (124, 1.52%) was the least (Supplementary Table S3). And the most preferentially used codons in the R. japonica mitogenome were A-ended or U-ended codons that have RSCU values greater than 1, being consistent with Hemerocallis citrina (Zhang et al., 2022), with the exception of threonine (ACC) and leucine (UUG) (Supplementary Figure S5; Supplementary Table S3).
RNA editing events are widespread phenomenon in plant mitogenome, and have significant impacts on the changes in amino acids (Maier et al., 1996; Grewe et al., 2009). In this study, 299 RNA editing sites were found in the R. japonica mitogenome (Supplementary Table S4). Briefly, except for the 3 PCGs (atp8, atp9, and cox1), the remaining 24 PCGs possessed RNA editing sites. And nad4 had the most RNA editing sites, followed by nad2, nad5, and nad7 with 34, 29, 25 and 25, respectively (Figure 3A). Of the five types of RNA editing in R. japonica mitogenome, C to T editing had the highest number of occurrences (294 times, 98.33%) and exited in all the 24 PCGs (Figure 3B). C to A editing was only found in rps4, and C to G editing was found in both nad4 and nad5. In addition to C to T editing, nad4 also had C to G, G to T, and G to C editing. As the center of plant energy metabolism, mitochondrion is known as the power house. Base substitution in the sequence of editing key genes may affect plant growth and metabolism. The development of base editor in plant mitogenome (Nakazato et al., 2022) and the mechanism of plant mitogenome editing (Wang et al., 2023b) will help to uncover the mystery of plant mitogenome. The assembly and annotation of R. japonica mitogenome will establish a solid foundation in the field about mitochondrial function research.
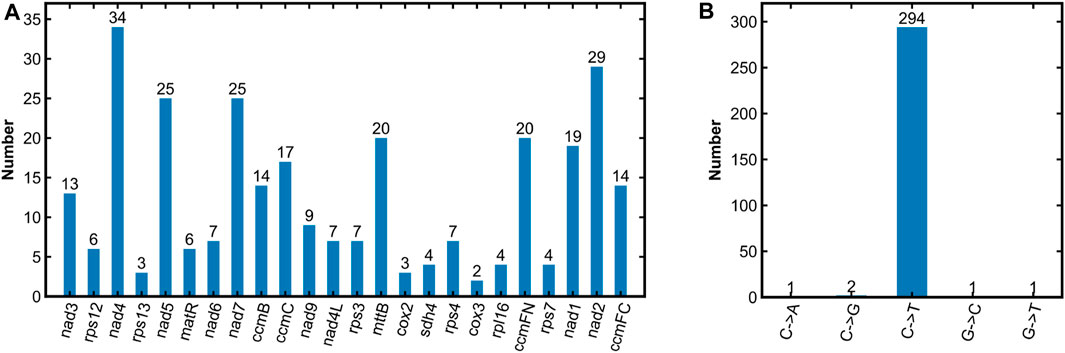
FIGURE 3. Prediction of RNA editing sites in the R. japonica mitogenome. (A) Number of RNA editing sites in PCGs. (B) RNA editing types and their numbers identified in the R. japonica mitogenome.
3.4 Horizontal transfer of sequences from the chloroplast genome
The phenomenon of plant mitogenome containing plastid-like fragments is common in most plant species. It was reported that 23,368 bp in the Salix wilsonii mitogenome (accounting for 3.28%) were derived from the chloroplast genome (Han et al., 2022). A total of 21,542 bp in the Abelmoschus esculentus mitogenome (accounting for 4.07%) were homologous with its chloroplast genome (Li et al., 2022). In the present study, the plastid-like sequences in the R. japonica mitogenome were identified. A total of 17 DNA fragments with a total length of 26,123 bp were similar to chloroplast genome (Supplementary Table S5), accounting for 8.64% of the R. japonica mitogenome. Specifically, the plastid-like fragments ranged from 36 bp to 9,402 bp, containing 11 complete tRNA genes (trnV-GAC, trnL-CAA, trnM-CAU, trnD-GUC, trnN-GUU, trnI-GAU, trnA-UGC, trnR-ACG, trnH-GUG, trnS-GGA, and trnW-CCA) (Figure 4). Additionally, our results demonstrated that the largest plastid-like fragments in the R. japonica mitogenome were derived from the inverted repeat region (IRA and IRB, Figure 4) in chloroplast genome. Apart from tRNA genes, the plastid-like sequences mainly located in non-functional fragments which consistent with most other land plants (Straub et al., 2013). The rrn18 gene in the R. japonica mitogenome may migrate from chloroplast genome due to the partial similarity in sequence, and undergo some integration during the evolutionary process.
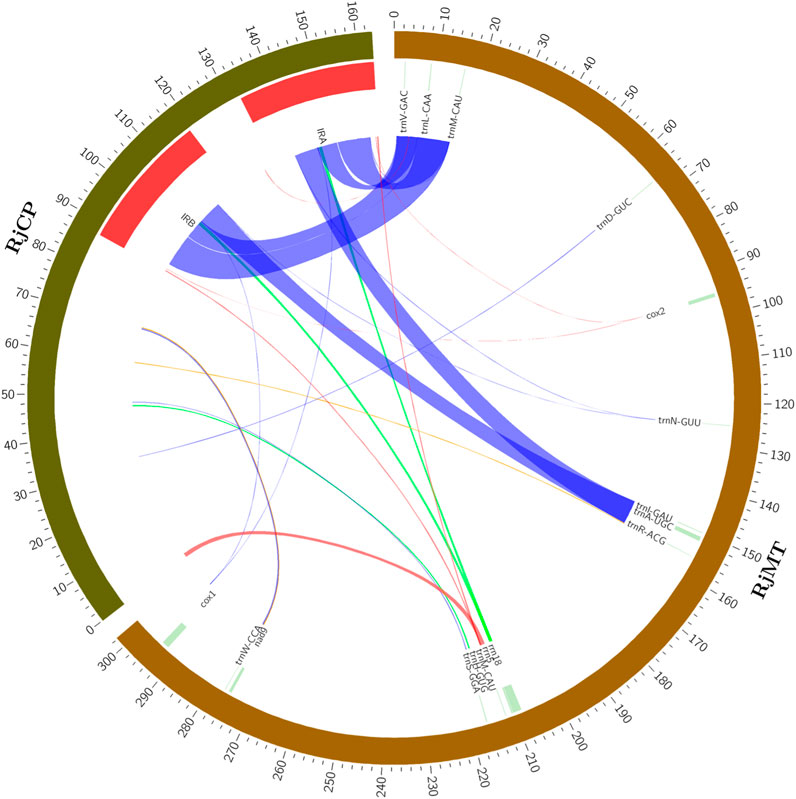
FIGURE 4. Plastid-like sequences in the R. japonica mitogenome. RjCP: the chloroplast genome of R. japonica. RjMT: the mitogenome of R. japonica. The red arcs represent 100% similarity, the blue arcs represent the similarity between 90% and 100%, the orange arcs represent the similarity between 80% and 90%, and the green arcs represent the similarity between 70% and 80%.
3.5 Selection pressure analyses of mitochondrial PCGs
In order to estimate the selection pressure of R. japonica mitochondrial PCGs, a total of 25 shared PCGs were employed to compute the Ka/Ks ratios among the mitogenome of R. japonica, P. aviculaare, F. aubertii, and F. multiflora. The most of the pairwise Ka/Ks ratios were smaller than 1 (Figure 5), indicating that most PCGs were under purifying selection during the evolution of R. japonica. And they may play important roles in stabilizing and maintaining the essential function of mitogenome. However, ccmFC, cox1, and nad1 were found with Ka/Ks ratios bigger than 1, suggesting that these three genes were subject to positive selection during evolution. Note also that the cox1 gene had an extremely high Ka/Ks ratio (R. japonica vs. F. multiflora: 2.83), indicating strong positive selection during the evolution of R. japonica and F. multiflora.
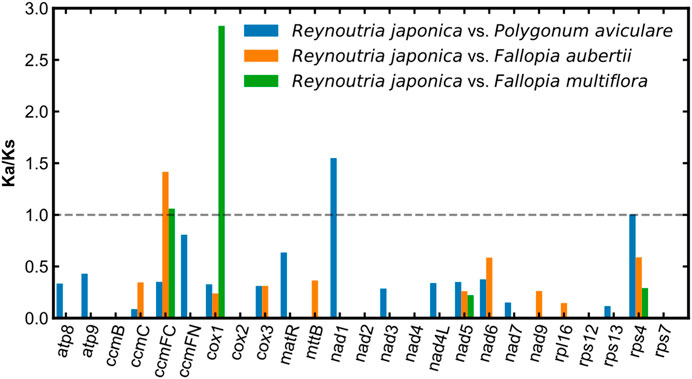
FIGURE 5. Ka/Ks ratio analysis of the R. japonica mitogenome.
3.6 Phylogenetic analysis
The mitogenome is an important tool for developing phylogenetic research. Due to the extensive variations in different plants, the shared conserved PCGs are usually used to conduct phylogenetic analysis. In this study, the maximum likelihood (ML) method was used to construct the phylogenetic tree based on the twelve homologous mitogenome PCGs from eleven species. The results showed that the position of R. japonica stayed closest to F. multiflora, which is consistent with those based on chloroplast genome (Chen et al., 2022). As shown in Figure 6, the ML tree was divided into two clades, one belongs to the order Caryophyllales and others as outgroup. The species in the family Polygonaceae (R. japonica, F. multiflora, F. aubertii, and P. aviculare) were separated from the other families in the order Caryophyllales, indicating that the mitogenome genes are reliable. The low bootstrap value of R. japonica and F. multiflora may be due to the high similarity of the mitogenomes (Supplementary Figure S4), indicating a close kinship. And the R. japonica mitogenome could provide a kind of reference for further phylogenetic studies.
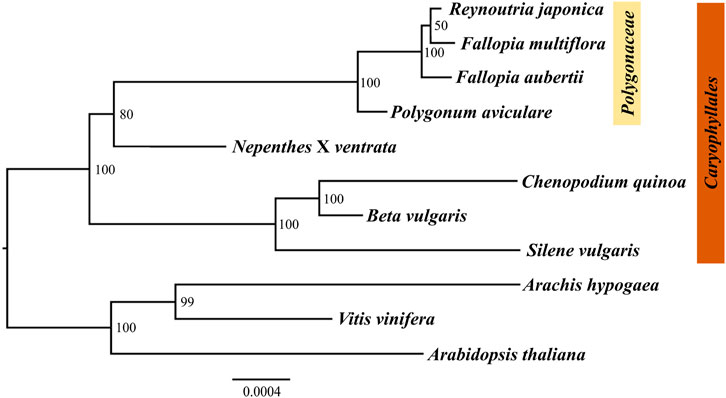
FIGURE 6. Phylogenetic tree based on twelve homologous protein-coding genes in eleven plants’ mitogenomes using ML analysis. Numbers near the nodes are support values with ML bootstrap values.
4 Conclusion
This work presented the first mitogenome assembly and annotation of R. japonica. The main results include: The R. japonica mitogenome was 302,229 bp in length and encoded 48 unique genes, including 27 PCGs, 18 tRNA genes, and 3 rRNA genes. In total, 8,174 codons were encoding the PCGs in the R. japonica mitogenome. The noncoding sequences accounted for 89.47% of the R. japonica mitogenome where the repeat sequences mainly located. In addition, 11 plastid-like tRNA genes were identified in the R. japonica mitogenome, and almost all PCGs were subject to purification selection, except for ccmFC, cox1 and nad1 which were subject to positive selection. In a word, the current study provided valuable genomic resources for further understanding and utilizing R. japonica in the future.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Author contributions
JC: Data curation, Writing–original draft. HM: Investigation, Writing–original draft. HF: Investigation, Writing–original draft. FL: Investigation, Writing–original draft. TC: Writing–review and editing. HW: Writing–review and editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by the National Natural Science Foundation of China (Grant Nos. 61972374 and 32370403) and the Fundamental Research Funds for the Central Universities (Grant No. E1E40605).
Acknowledgments
The authors would like to thank Zhao Su (College of Life Sciences, University of Chinese Academy of Sciences, Beijing 100049, China) for his computing resource assistance during the data analysis of this manuscript.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2023.1289811/full#supplementary-material
References
Adams, K. L., Qiu, Y. L., Stoutemyer, M., and Palmer, J. D. (2002). Punctuated evolution of mitochondrial gene content: high and variable rates of mitochondrial gene loss and transfer to the nucleus during angiosperm evolution. Proc. Natl. Acad. Sci. U. S. A. 99 (15), 9905–9912. doi:10.1073/pnas.042694899
Alverson, A. J., Wei, X., Rice, D. W., Stern, D. B., Barry, K., and Palmer, J. D. (2010). Insights into the evolution of mitochondrial genome size from complete sequences of Citrullus lanatus and Cucurbita pepo (Cucurbitaceae). Mol. Biol. Evol. 27 (6), 1436–1448. doi:10.1093/molbev/msq029
Arseneau, J. R., Steeves, R., and Laflamme, M. (2017). Modified low-salt CTAB extraction of high-quality DNA from contaminant-rich tissues. Mol. Ecol. Resour. 17 (4), 686–693. doi:10.1111/1755-0998.12616
Bi, C., Paterson, A. H., Wang, X., Xu, Y., Wu, D., Qu, Y., et al. (2016). Analysis of the complete mitochondrial genome sequence of the diploid cotton gossypium raimondii by comparative genomics approaches. BioMed Res. Int. 2016, 5040598. doi:10.1155/2016/5040598
Bi, C., Qu, Y., Hou, J., Wu, K., Ye, N., and Yin, T. (2022). Deciphering the multi-chromosomal mitochondrial genome of Populus simonii. Front. plant Sci. 13, 914635. doi:10.3389/fpls.2022.914635
Chen, J., Huang, T., Fan, H., Lin, F., Ma, H., Cao, J., et al. (2022). A comparative phylogenetic analysis on the chloroplast genome in different Reynoutria japonica populations. Genes 13 (11), 1979. doi:10.3390/genes13111979
Chen, S., Zhou, Y., Chen, Y., and Gu, J. (2018). fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinforma. Oxf. Engl. 34 (17), i884–i890. doi:10.1093/bioinformatics/bty560
Chen, Y., Ye, W., Zhang, Y., and Xu, Y. (2015). High speed BLASTN: an accelerated MegaBLAST search tool. Nucleic acids Res. 43 (16), 7762–7768. doi:10.1093/nar/gkv784
Cole, L. W., Guo, W., Mower, J. P., and Palmer, J. D. (2018). High and variable rates of repeat-mediated mitochondrial genome rearrangement in a genus of plants. Mol. Biol. Evol. 35 (11), 2773–2785. doi:10.1093/molbev/msy176
Danecek, P., Bonfield, J. K., Liddle, J., Marshall, J., Ohan, V., Pollard, M. O., et al. (2021). Twelve years of SAMtools and BCFtools. GigaScience 10 (2), giab008. doi:10.1093/gigascience/giab008
Darriba, D., Posada, D., Kozlov, A. M., Stamatakis, A., Morel, B., and Flouri, T. (2020). ModelTest-NG: a new and scalable tool for the selection of DNA and protein evolutionary models. Mol. Biol. Evol. 37 (1), 291–294. doi:10.1093/molbev/msz189
Delahaye, C., and Nicolas, J. (2021). Sequencing DNA with nanopores: troubles and biases. PloS one 16 (10), e0257521. doi:10.1371/journal.pone.0257521
Drouin, G., Daoud, H., and Xia, J. (2008). Relative rates of synonymous substitutions in the mitochondrial, chloroplast and nuclear genomes of seed plants. Mol. phylogenetics Evol. 49 (3), 827–831. doi:10.1016/j.ympev.2008.09.009
Dyall, S. D., Brown, M. T., and Johnson, P. J. (2004). Ancient invasions: from endosymbionts to organelles. Sci. (New York, N.Y.) 304 (5668), 253–257. doi:10.1126/science.1094884
Friis, G., Vizueta, J., Smith, E. G., Nelson, D. R., Khraiwesh, B., Qudeimat, E., et al. (2021). A high-quality genome assembly and annotation of the gray mangrove, Avicennia marina. Avicennia Mar. 11 (1), jkaa025. doi:10.1093/g3journal/jkaa025
Greiner, S., Lehwark, P., and Bock, R. (2019). OrganellarGenomeDRAW (OGDRAW) version 1.3.1: expanded toolkit for the graphical visualization of organellar genomes. Nucleic acids Res. 47 (W1), W59-W64–W64. doi:10.1093/nar/gkz238
Grewe, F., Viehoever, P., Weisshaar, B., and Knoop, V. (2009). A trans-splicing group I intron and tRNA-hyperediting in the mitochondrial genome of the lycophyte Isoetes engelmannii. Nucleic acids Res. 37 (15), 5093–5104. doi:10.1093/nar/gkp532
Gualberto, J. M., Mileshina, D., Wallet, C., Niazi, A. K., Weber-Lotfi, F., and Dietrich, A. (2014). The plant mitochondrial genome: dynamics and maintenance. Biochimie 100, 107–120. doi:10.1016/j.biochi.2013.09.016
Han, F., Qu, Y., Chen, Y., Xu, L., and Bi, C. (2022). Assembly and comparative analysis of the complete mitochondrial genome of Salix wilsonii using PacBio HiFi sequencing. Front. plant Sci. 13, 1031769. doi:10.3389/fpls.2022.1031769
Katoh, K., and Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30 (4), 772–780. doi:10.1093/molbev/mst010
Kent, W. J. (2002). BLAT--the BLAST-like alignment tool. Genome Res. 12 (4), 656–664. doi:10.1101/gr.229202
Kim, C. K., and Kim, Y. K. (2018). The multipartite mitochondrial genome of Fallopia multiflora (Caryophyllales: Polygonaceae). Mitochondrial DNA. Part B, Resour. 3 (1), 155–156. doi:10.1080/23802359.2018.1437796
Kozik, A., Rowan, B. A., Lavelle, D., Berke, L., Schranz, M. E., Michelmore, R. W., et al. (2019). The alternative reality of plant mitochondrial DNA: one ring does not rule them all. PLoS Genet. 15 (8), e1008373. doi:10.1371/journal.pgen.1008373
Kozlov, A. M., Darriba, D., Flouri, T., Morel, B., and Stamatakis, A. (2019). RAxML-NG: a fast, scalable and user-friendly tool for maximum likelihood phylogenetic inference. Bioinforma. Oxf. Engl. 35 (21), 4453–4455. doi:10.1093/bioinformatics/btz305
Krzywinski, M., Schein, J., Birol, I., Connors, J., Gascoyne, R., Horsman, D., et al. (2009). Circos: an information aesthetic for comparative genomics. Genome Res. 19 (9), 1639–1645. doi:10.1101/gr.092759.109
Kurtz, S., Choudhuri, J. V., Ohlebusch, E., Schleiermacher, C., Stoye, J., and Giegerich, R. (2001). REPuter: the manifold applications of repeat analysis on a genomic scale. Nucleic acids Res. 29 (22), 4633–4642. doi:10.1093/nar/29.22.4633
Langmead, B., and Salzberg, S. L. (2012). Fast gapped-read alignment with Bowtie 2. Nat. methods 9 (4), 357–359. doi:10.1038/nmeth.1923
Li, H., and Durbin, R. (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinforma. Oxf. Engl. 25 (14), 1754–1760. doi:10.1093/bioinformatics/btp324
Li, J., Li, J., Ma, Y., Kou, L., Wei, J., and Wang, W. (2022). The complete mitochondrial genome of okra (Abelmoschus esculentus): using nanopore long reads to investigate gene transfer from chloroplast genomes and rearrangements of mitochondrial DNA molecules. BMC genomics 23 (1), 481. doi:10.1186/s12864-022-08706-2
Li, J., Xu, Y., Shan, Y., Pei, X., Yong, S., Liu, C., et al. (2021). Assembly of the complete mitochondrial genome of an endemic plant, Scutellaria tsinyunensis, revealed the existence of two conformations generated by a repeat-mediated recombination. Planta 254 (2), 36. doi:10.1007/s00425-021-03684-3
Lilly, J. W., and Havey, M. J. (2001). Small, repetitive DNAs contribute significantly to the expanded mitochondrial genome of cucumber. Genetics 159 (1), 317–328. doi:10.1093/genetics/159.1.317
Maier, R. M., Zeltz, P., Kössel, H., Bonnard, G., Gualberto, J. M., and Grienenberger, J. M. (1996). RNA editing in plant mitochondria and chloroplasts. Plant Mol. Biol. 32 (1-2), 343–365. doi:10.1007/BF00039390
Nakazato, I., Okuno, M., Zhou, C., Itoh, T., Tsutsumi, N., Takenaka, M., et al. (2022). Targeted base editing in the mitochondrial genome of Arabidopsis thaliana. Proc. Natl. Acad. Sci. U. S. A. 119 (20), e2121177119. doi:10.1073/pnas.2121177119
Peng, W., Qin, R., Li, X., and Zhou, H. (2013). Botany, phytochemistry, pharmacology, and potential application of Polygonum cuspidatum Sieb.et Zucc.: a review. J. Ethnopharmacol. 148 (3), 729–745. doi:10.1016/j.jep.2013.05.007
Putintseva, Y. A., Bondar, E. I., Simonov, E. P., Sharov, V. V., Oreshkova, N. V., Kuzmin, D. A., et al. (2020). Siberian larch (Larix sibirica Ledeb.) mitochondrial genome assembled using both short and long nucleotide sequence reads is currently the largest known mitogenome. BMC genomics 21 (1), 654. doi:10.1186/s12864-020-07061-4
Sloan, D. B. (2013). One ring to rule them all? Genome sequencing provides new insights into the 'master circle' model of plant mitochondrial DNA structure. New phytologist 200 (4), 978–985. doi:10.1111/nph.12395
Sloan, D. B., Alverson, A. J., Chuckalovcak, J. P., Wu, M., McCauley, D. E., Palmer, J. D., et al. (2012). Rapid evolution of enormous, multichromosomal genomes in flowering plant mitochondria with exceptionally high mutation rates. PLoS Biol. 10 (1), e1001241. doi:10.1371/journal.pbio.1001241
Straub, S. C., Cronn, R. C., Edwards, C., Fishbein, M., and Liston, A. (2013). Horizontal transfer of DNA from the mitochondrial to the plastid genome and its subsequent evolution in milkweeds (apocynaceae). Genome Biol. Evol. 5 (10), 1872–1885. doi:10.1093/gbe/evt140
Tang, D., Huang, S., Quan, C., Huang, Y., Miao, J., and Wei, F. (2023). Mitochondrial genome characteristics and phylogenetic analysis of the medicinal and edible plant Mesona chinensis Benth. Front. Genet. 13, 1056389. doi:10.3389/fgene.2022.1056389
Tillich, M., Lehwark, P., Pellizzer, T., Ulbricht-Jones, E. S., Fischer, A., Bock, R., et al. (2017). GeSeq - versatile and accurate annotation of organelle genomes. Nucleic acids Res. 45 (W1), W6-W11. W6–W11. doi:10.1093/nar/gkx391
Walker, B. J., Abeel, T., Shea, T., Priest, M., Abouelliel, A., Sakthikumar, S., et al. (2014). Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PloS one 9 (11), e112963. doi:10.1371/journal.pone.0112963
Wang, Y., Chen, S., Chen, J., Chen, C., Lin, X., Peng, H., et al. (2023a). Characterization and phylogenetic analysis of the complete mitochondrial genome sequence of Photinia serratifolia. Sci. Rep. 13 (1), 770. doi:10.1038/s41598-022-24327-x
Wang, Y., Li, H., Huang, Z. Q., Ma, B., Yang, Y. Z., Xiu, Z. H., et al. (2023b). Maize PPR-E proteins mediate RNA C-to-U editing in mitochondria by recruiting the trans deaminase PCW1. Plant Cell 35 (1), 529–551. doi:10.1093/plcell/koac298
Wu, Z., Liao, X., Zhang, X., Tembrock, L. R., and Broz, A. (2020). Genomic architectural variation of plant mitochondria—a review of multichromosomal structuring. J. Syst. Evol. 60 (1), 160–168. doi:10.1111/jse.12655
Yang, Z., Ni, Y., Lin, Z., Yang, L., Chen, G., Nijiati, N., et al. (2022). De novo assembly of the complete mitochondrial genome of sweet potato (Ipomoea batatas [L.] Lam) revealed the existence of homologous conformations generated by the repeat-mediated recombination. BMC plant Biol. 22 (1), 285. doi:10.1186/s12870-022-03665-y
Zhang, H., Li, C., Kwok, S. T., Zhang, Q. W., and Chan, S. W. (2013). A review of the pharmacological effects of the dried root of polygonum cuspidatum (hu Zhang) and its constituents. Evidence-Based complementary and alternative medicine: eCAM. Evidence-Based Complementary Altern. Med. 2013, 208349. doi:10.1155/2013/208349
Zhang, K., Wang, Y., Zhang, X., Han, Z., and Shan, X. (2022). Deciphering the mitochondrial genome of Hemerocallis citrina (Asphodelaceae) using a combined assembly and comparative genomic strategy. Front. plant Sci. 13, 1051221. doi:10.3389/fpls.2022.1051221
Zhang, Z. (2022). KaKs_Calculator 3.0: calculating selective pressure on coding and non-coding sequences. Genomics, proteomics Bioinforma. 20 (3), 536–540. doi:10.1016/j.gpb.2021.12.002
Keywords: Reynoutria japonica, mitogenome, codon usage, repeat sequences, RNA editing, evolution
Citation: Chen J, Ma H, Fan H, Lin F, Chai T and Wang H (2023) De novo assembly and comparative analysis of the mitochondrial genome of Reynoutria japonica. Front. Genet. 14:1289811. doi: 10.3389/fgene.2023.1289811
Received: 06 September 2023; Accepted: 10 November 2023;
Published: 23 November 2023.
Edited by:
Fatemeh Maghuly, University of Natural Resources and Life Sciences Vienna, AustriaReviewed by:
Jose Pablo Jiménez-Madrigal, Instituto Tecnológico de Costa Rica (ITCR), Costa RicaFederico Albertazzi, Universidad de Costa Rica, Costa Rica
Copyright © 2023 Chen, Ma, Fan, Lin, Chai and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tuanyao Chai, dHljaGFpQHVjYXMuYWMuY24=; Hong Wang, aHdhbmdAdWNhcy5hYy5jbg==