 Ayça Şahin
Ayça Şahin Esmer Zeynep Duru Badakal
Esmer Zeynep Duru Badakal Müge Kovancılar Koç
Müge Kovancılar Koç Hilmi Uysal
Hilmi Uysal Ayşe Nazlı Başak
Ayşe Nazlı Başak- 1Suna and İnan Kıraç Foundation, Neurodegeneration Research Laboratory, Koç University Research Center for Translational Medicine (KUTTAM), School of Medicine, Koç University, İstanbul, Türkiye
- 2Department of Neurology, Faculty of Medicine, Akdeniz University, Antalya, Türkiye
Brody Disease is an exceptionally rare, autosomal recessive myopathy attributed to the pathogenic variants in the ATP2A1, which encodes the sarcoplasmic/endoplasmic reticulum Ca (2+) ATPase type 1 protein SERCA1. It was first described by Brody IA in 1969. To date, only thirty-three Brody families with forty-seven patients have been reported in the literature, and the disease prevalence is considered as 1 in 10 million, demonstrating the peculiarity of the disease. Clinical characteristics of Brody Disease include muscle stiffness after exercise, myalgia, and muscle cramps. Brody Disease patients generally have disease onset in the first decade, and genetic diagnosis is delayed as a consequence of both the rareness and the mild course of the disease. Here, we report a Turkish Brody Disease patient with a homozygous c.428G>A p.Arg143Gln (NM_004320.4) missense mutation in the ATP2A1. The male patient, whose symptoms started at the age of 14–15, is now 36 years old. His clinical manifestations are athletic appearance, exotropia, slightly elevated creatine kinase (CK), mild progressive proximal muscle weakness in the lower extremities, muscle cramps, pain and stiffness. The patient described here has a very mild progression with an onset in the second decade, expanding the Brody Disease phenotype. The study also implies that in the era of emerging genetic therapies, the routine testing of patients with myopathies is a prerequisite since not only future therapies will be designed on molecular findings, but also currently available symptomatic and palliative treatment options will be more precisely applied.
1 Introduction
While rare diseases are classified as diseases affecting less than 1 in 2000 people, three major classifications exist: more common, less common, and ultra-rare ones. With a prevalence, estimated as 1 in 10 million, Brody Disease is one of the ultra-rare diseases. To our knowledge, only 33 Brody families with 47 patients have been reported in the literature so far (Braz et al., 2019; Bruels et al., 2019; Molenaar et al., 2020; Brugnoni et al., 2021; Saat and Sahin, 2021; Velardo et al., 2023; Walia and Su, 2023).
Brody Disease is an autosomal recessive myopathy caused by pathogenic variants in the ATP2A1 (ATPase sarcoplasmic/endoplasmic reticulum Ca2+ transporting 1) (OMIM: # 108730) in which patients experience muscle stiffness after exercise, myalgia, and muscle cramps (Molenaar et al., 2020).
The underlying pathology of the disease arises from the difficulty in removing Ca2+ from intracellular space for muscle relaxation. The motor unit, composed of motor neurons and muscle fibers, is the core region of muscle movement, and skeletal muscles are innervated by alpha motor neurons. For a healthy movement, there is a well-established balance between the contraction and relaxation of the muscles. At the physiological level, the synaptic connection between the motor neuron and muscle is called the neuromuscular junction, in which the neurotransmitter acetylcholine is released from the alpha motor neuron, leading to the formation of an action potential that initiates muscle contraction. At the cellular level, upon the transmission of the action potential, Ca2+ is released from the sarcoplasmic reticulum, increasing the [Ca2+] level in the intracellular space, leading to muscle contraction. Relaxation, on the other hand, occurs once the Ca2+ is pumped back to the sarcoplasmic reticulum. Transportation of Ca2+ to the sarcoplasmic reticulum is performed by a calcium pump named SERCA (sarcoplasmic endoplasmic reticulum calcium ATPase). SERCA, which is the most abundant protein in the sarcoplasmic reticulum, hydrolyzes ATP per two molecules of Ca2+ transported (Koeppen and Stanton, 2023).
SERCA1, a muscle-specific protein, has two splice variants, SERCA1a, present in adults, and neonatal SERCA1b, containing 994 and 1,001 aminoacids, respectively (Wuytack et al., 2002; Xu and Van Remmen, 2021). It is mainly expressed in fast-twitch muscles (Xu and Van Remmen, 2021), also called type 2 muscle fibers (Molenaar et al., 2020). ATP2A1 encodes SERCA1 protein, which plays a role in muscle relaxation by removing Ca2+ from the cytosol (Guglielmi et al., 2013).
The first patient described by Brody in 1969 was a 26-year-old male patient with the prominent symptom of muscle stiffness, especially after strenuous and rapid activities. The patient had no family history of the disease; his first memory was an unexplained fall at the age of five as his muscles got stiffened during a foot race (Brody, 1969).
Here, we report a Turkish male patient with Brody Disease whose parents are first-degree cousins.
2 Case presentation
The patient is a 36-year-old male, referred to the Neurodegeneration Research Laboratory, KUTTAM, Koç University Hospital for genetic analysis from the Neurology Department of Akdeniz University Hospital School of Medicine. He is the second child of first-degree consanguineous parents. His initial complaints started at the age of 14–15, with severe muscle pain while playing football. The current symptoms of the patient with an athletic appearance include mild progressive muscle weakness in lower extremities, left prominent bilateral exotropia, cramps, pain and stiffness in muscles, cold-induced myalgias, and an elevated creatine kinase (CK: 630 IU/L). The MRC sum score is 54 (N = 60). Moderate myopathic changes were observed in the lower extremity proximal muscles on EMG. Silent cramps were not observed, and there was not a significant change in repetitive nerve stimulation. Spontaneous denervation potentials or myotonic discharges were not observed. Sensory and motor conduction velocities were within normal limits. The alpha-glucosidase level was found to be normal. The patient was started on steroids 2 years before the genetic diagnosis. Although the treatment provided some relief from his symptoms, a hip prosthesis was implanted due to aseptic necrosis of the femoral head.
3 Methods
Informed written consent was obtained from the patient as well as from his family members. Genomic DNA was isolated from peripheral blood using Qiagen EZ1 Advanced XL. Whole exome sequencing was performed (Macrogen, Amsterdam) for the index case. WES raw data was processed using the SEQ Platform (Genomize, Istanbul, Turkey), and the data was analyzed at NDAL. The following filtering criteria were applied: OMIM-related genes, minor allele frequency (MAF) < 1%, destructive, missense, and splice region variants. The pathogenicity of the homozygous ATP2A1 variant detected was evaluated by in silico tools. Subsequently, segregation analysis using Sanger sequencing was performed. The pathogenic variant was numbered according to the transcript NM_004320.4, and was submitted to ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/variation/464089/Accession: SCV004023391.1).
4 Results
4.1 Genetic analysis
WES analysis revealed a homozygous c.428G>A p.Arg143Gln variant in the ATP2A1. This variant was earlier reported in a patient with Brody Disease in a compound heterozygous state with c.1317_1318del p.Glu439Aspfs*80 in trans (Molenaar et al., 2020) (Table 1). Sanger sequencing was performed for the proband and his asymptomatic family members. The variant was validated in the proband in homozygous dosage and in his parents in heterozygous form. Two siblings were also found to be carriers, and one sibling was wild type (Figure 1).
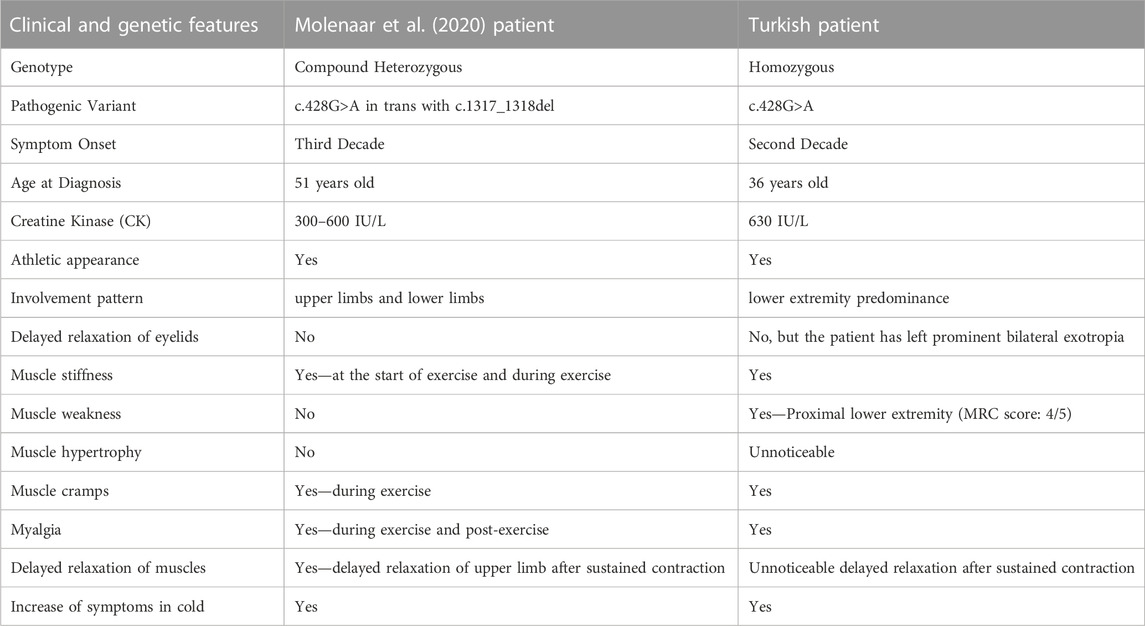
TABLE 1. The comparison between Molenaar’s patient and the Turkish patient under study.
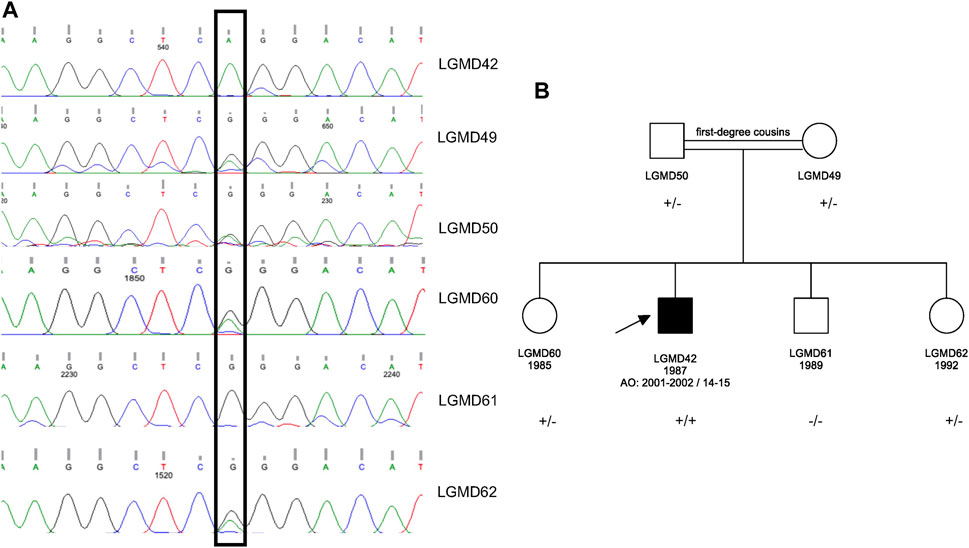
FIGURE 1. (A) Sanger sequencing results for the proband and his asymptomatic family members. (B) Pedigree of the family. The pathogenic variant is ATP2A1 (NM_004320.4) p.Arg143Gln in Exon 5; +: mutant allele and -: wild type allele.
4.2 In silico findings
The homozygous c.428G>A p.Arg143Gln variant identified in the actuator domain of the SERCA1 protein is a missense mutation. While several tools supported the pathogenicity, DANN: 0.999, CADD: 22.7000, FATHMM-MKL: 0.9749, a few remained uncertain or benign-supporting. Conservation scores suggested a conserved site with PhyloP100way: 7.585, PhastCons100way: 1.000, and GERP RS: 4.1799. In our cohort consisting of 1740 WES-subjected individuals from Turkey, the variant is present in five independent cases in heterozygous state.
Notably, this missense mutation was already reported in trans position with Brody Disease in the compound heterozygous state with a frameshift mutation (Molenaar et al., 2020). In ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/), there were three entries for the variant, classifying it as VUS (variant of uncertain significance). In GnomAD (https://gnomad.broadinstitute.org/), the variant was found in 33 individuals in the heterozygous state, no individual had the variant in homozygous state, and the reported allele frequency was 0.0001168. The variant was reported as likely pathogenic in the LOVD database (https://www.lovd.nl/).
5 Discussion
Since Brody’s identification of the disease 54 years ago, only 47 patients in 33 families have been identified, demonstrating the extreme rareness of the disease. Here, we define the clinical and genetic findings in a Turkish Brody Disease patient.
In general, the first symptoms of Brody Disease manifest in the first decade; however, patients may not feel the urge to consult a physician for several years. In the literature, patients described so far usually have the onset during childhood. Still, their diagnosis is delayed for several years, presumably due to the mild progression of the disease, as well as its rarity and thus ignorance. Also, in our patient, from the first symptoms (describing severe muscle pain at the age of 14–15 while playing football) to the molecular diagnosis, more than 20 years have passed. Owing to the mild course of the disease, the patient describes his complaints as being around for 4–5 years only. However, his first diagnosis was initially put around the age of 18 when he was excluded from the military service because of his muscle-related complaints. Yet, another 16 years had to pass until the patient was referred to our center for molecular testing with the preliminary diagnosis of Limb Girdle Muscular Dystrophy (LGMD). His muscle disease was reported as mildly progressive, with major complaints in his knees, ankles, and hips. The patient has undergone femur surgery, he now has a one-sided hip prosthesis. Although malignant hyperthermia was observed in a few Brody Disease patients (Molenaar et al., 2020), our patient did not manifest malignant hyperthermia during the surgery.
The missense mutation identified (c.428G>A; p.Arg143Gln) is in the actuator domain of the protein. Eight mutations in 10 families comprising 15 patients have been reported in this domain so far (10 index cases and 5 affected siblings). Twelve out of fifteen had an onset in the childhood/first decade, and there was a male predominance. Demographic and clinical characteristics of all patients with a mutation in the actuator domain are compiled in Table 2.
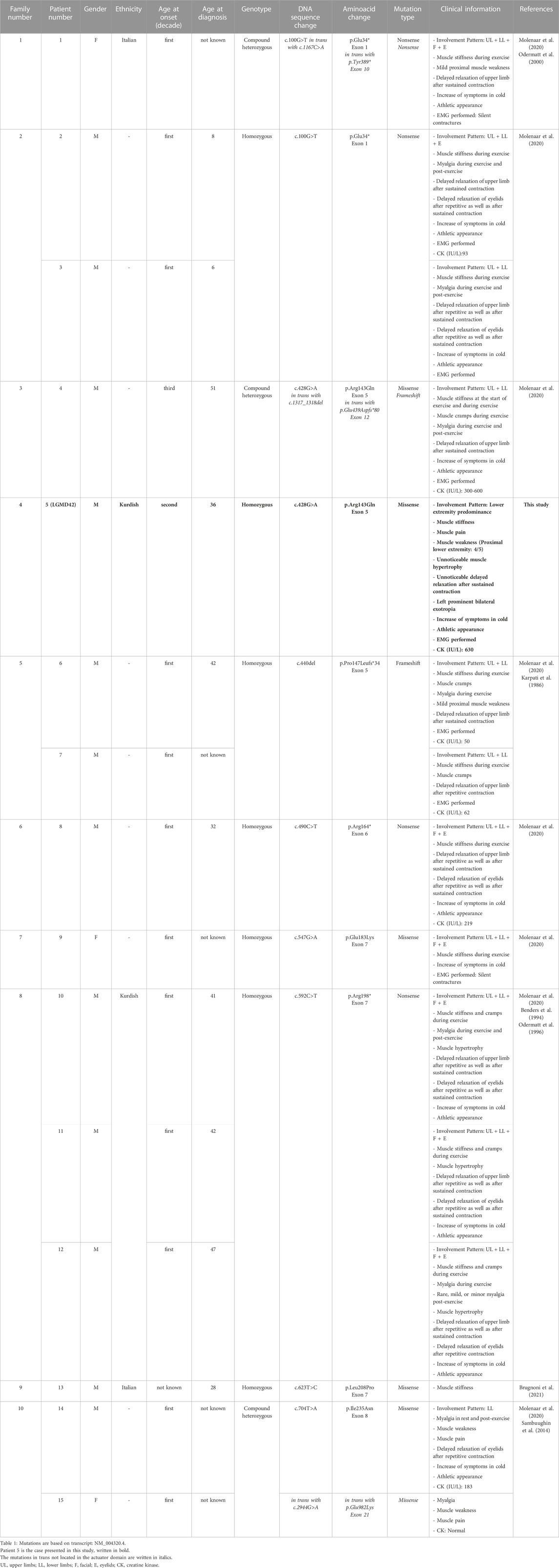
TABLE 2. Genetic and clinical information of patients with mutations in the actuator domain.
In total, there are 41 distinct mutations in the ATP2A1 in 34 families worldwide, including our family; 28 mutations located in the different domains of SERCA1 are shown in Figure 2A. The mutations seem to be rare and almost family-private. In the actuator domain, eight variants were identified in 10 index cases; only two of these were redundant. Out of 10 index cases with a mutation in the actuator domain, seven are homozygous, whereas, in three patients, it is in a compound heterozygous state.
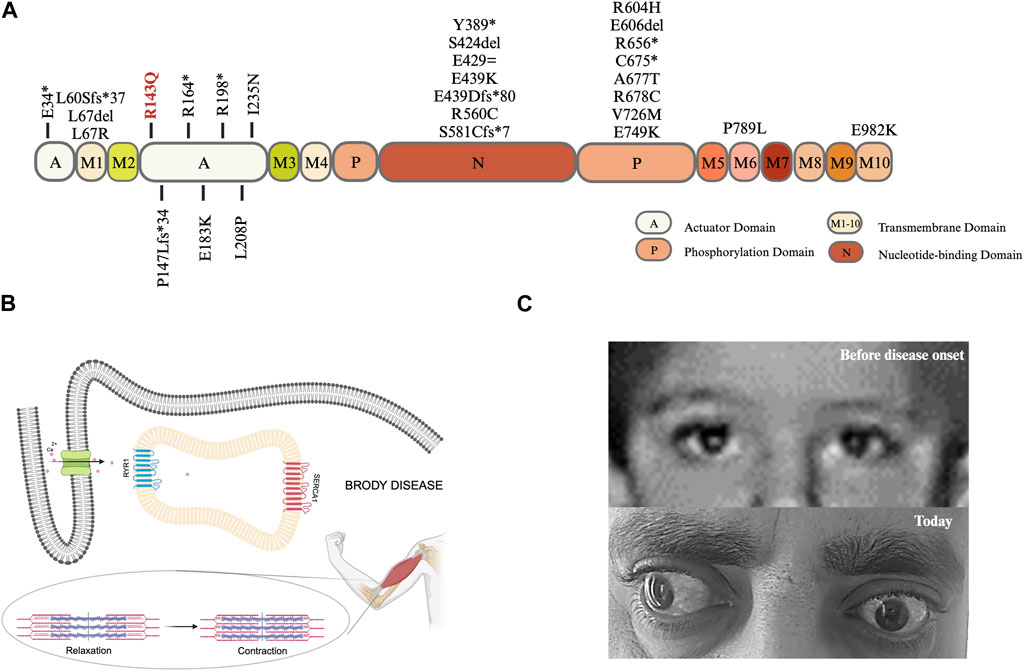
FIGURE 2. (A) Lolliplot diagram: SERCA1a is a transmembrane protein with a molecular weight of 110 kDa. The protein consists of a transmembrane (M) domain and a headpiece within the cytoplasm containing three domains: i) actuator (A) domain accounting for the dephosphorylation ii) phosphorylation (P) domain harboring a site for autophosphorylation, and iii) nucleotide-binding (N) domain which is the ATP binding site (Wuytack et al., 2002; Barbot et al., 2021), created with BioRender.com. (B) Graphical illustration of cellular and physiological mechanisms in Brody Disease. Adapted from Molenaar et al., 2016; Braz et al., 2019, created with BioRender.com. (C) The pictures of patient’s eyes before and after disease onset.
So far, three Brody families from Turkey, including our case, with three different mutations have been described. All three families have distinct mutations. The first family is Kurdish, with six children, three of whom are affected. The two older brothers were described by Benders et al. (1994), and their pathogenic variant was identified by Odermatt et al. (1996) (c.592C>T; p.Arg198*). A 5-year-old male patient with muscle weakness and delayed development was reported by Saat and Sahin (2021) in a cohort study with hereditary myopathies (c.2029G>A; p.Ala677Thr). To the best of our knowledge, the present study is the first description of the p.Arg143Gln missense mutation in biallelic state. The patient originating from the Southeastern part of Turkey has Kurdish ethnicity. As in the above two families, the pathogenic variant is present in homozygous form, the result of close consanguinity, very commonly practiced in this part of the country.
The male patient defined by Molenaar et al. (2020) harbored the same variant (c.428G>A; p.Arg143Gln) in a compound heterozygous state in trans with c.1317_1318del; p.Glu439Aspfs*80. Similar to our patient and to other Brody Disease cases, there is a long diagnostic odyssey between symptom onset and definite diagnosis. Table 1 compares the demographic, clinical, and genetic features of both patients. In our patient, the earlier disease onset and higher CK level, compared to Molenaar’s patient, may be the result of the homozygous effect, e.g., double dosage of the variant, or other modifying factors may also have contributed.
Molenaar et al. (2020) reported exercise-induced muscle stiffness in all patients and increase of symptoms when exposed to cold in majority of patients. Our patient’s complaints are consistent with the reported findings, as he frequently expresses exercise-induced muscle stiffness and cold-induced myalgia. Muscle relaxation issues are commonly observed in Brody patients, which are related to the Ca2+ removal difficulty from intracellular space. While RYR1 (ryanodine receptor 1) works by releasing Ca2+, SERCA1 works by removing Ca2+, both are fundamental in muscle contraction and relaxation (Figure 2B).
Eyelid muscle can also be affected in Brody Disease patients as Molenaar et al. (2020) reported delayed relaxation of eyelids i) after repetitive contraction in 61% and ii) after sustained contraction in 59% of total known cases. Although our case does not experience delayed relaxation of eyelids, he has left prominent bilateral exotropia, which developed during the disease course (Figure 2C). Toğrul et al. (2021) conducted a study with the extraocular muscles of patients who had strabismus surgery vs. healthy controls (corneal transplant donors) aiming to compare calcium adenosine 5′-triphosphatase (Ca2+-ATPase) enzyme activity and found that the enzyme activity is lower in the strabismus surgery patients. This finding, as well as the healthy appearance of eyes before disease onset, could indicate that the detected exotropia in our patient might be related to Brody Disease.
Today, there are still no treatment options for most rare diseases, and this also includes Brody Disease. The two drugs, verapamil and dantrolene, with limited therapeutic benefit, work by inhibiting the RYR1 channel and the dihydropyridine receptor, but are not without side effects. Animal models are crucial to enlighten disease-related mechanisms and to discover potential therapeutic candidates. Akyurek et al. (2021) demonstrated Chianina cattle congenital pseudomyotonia as a true counterpart animal model for Brody Disease and a novel pharmacological approach was introduced by the same group.
In the era of gene-based/molecular therapies, a firm differential diagnosis using next-generation technologies as the gold standard is inevitable since not only currently available symptomatic and palliative treatment options will be more precisely applied but also current and future therapies will be designed on molecular findings. In the decade of emerging genetic therapies, the routine testing of patients with myopathies becomes a prerequisite and is not curiosity-driven research anymore. We have all reasons to be optimistic for our rare (disease) patients.
Data availability statement
The datasets for this article are not publicly available due to concerns regarding participant/patient anonymity. Requests to access the datasets should be directed to the corresponding author.
Ethics statement
The studies involving humans were approved by Koç University Institutional Review Board. The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author contributions
AŞ: Writing–original draft. EB: Writing–review and editing. MK: Writing–review and editing. HU: Writing–review and editing. AB: Conceptualization, Supervision, Writing–original draft, Writing–review and editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This study was supported by Suna and Inan Kirac Foundation Grant 2023-2025.
Acknowledgments
We would like to express our heartfelt thanks to Suna, İnan, and İpek Kıraç; none of our efforts would be possible without their vision, devotion, dedicated mentorship, and sustained support. We cordially thank Koç University-KUTTAM for the inspiring research environment created and Irmak Şahbaz for her excellent technical assistance. We thank with gratitude to the family for their participation in this study. The authors gratefully acknowledge the use of the services and facilities of the Koç University Research Center for Translational Medicine (KUTTAM), funded by the Presidency of Turkey, Presidency of Strategy and Budget. The content is solely the responsibility of the authors and does not necessarily represent the official views of the Presidency of Strategy and Budget.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
ATP2A1, ATPase sarcoplasmic/endoplasmic reticulum Ca2+ transporting 1; SERCA1, Sarcoplasmic/endoplasmic reticulum Ca (2+) ATPase type 1; CK, Creatine kinase; EMG, Electromyography; WES, whole-exome sequencing; LGMD, Limb Girdle Muscular Dystrophy; RYR1, ryanodine receptor 1; CFTR, Cystic Fibrosis Transmembrane Regulator.
References
Akyurek, E. E., Joana, G., Marilena, B., Angelo, P., Sandona, D., Arcangelo, G., et al. (2021). Frontiers in metabolic research, 37. https://hdl.handle.net/11577/3478066.Human Brody disease and its animal model cattle pseudomyotonia: from understanding the pathogenetic mechanism to identification of novel therapeutic approaches
Barbot, T., Beswick, V., Montigny, C., Quiniou, É., Jamin, N., and Mouawad, L. (2021). Deciphering the mechanism of inhibition of SERCA1a by sarcolipin using molecular simulations. Front. Mol. Biosci. 7, 606254. doi:10.3389/fmolb.2020.606254
Benders, A. A., Veerkamp, J. H., Oosterhof, A., Jongen, P. J., Bindels, R. J., Smit, L. M., et al. (1994). Ca2+ homeostasis in Brody’s disease. A study in skeletal muscle and cultured muscle cells and the effects of dantrolene an verapamil. J. Clin. Investig. 94, 741–748. doi:10.1172/JCI117393
Braz, L., Soares-dos-Reis, R., Seabra, M., Silveira, F., and Guimarães, J. (2019). Brody disease: when myotonia is not myotonia. Pract. Neurol. 19, 417–419. doi:10.1136/practneurol-2019-002224
Brody, I. A. (1969). Muscle contracture induced by exercise. A syndrome attributable to decreased relaxing factor. N. Engl. J. Med. 281, 187–192. doi:10.1056/NEJM196907242810403
Bruels, C. C., Li, C., Mendoza, T., Khan, J., Reddy, H. M., Estrella, E. A., et al. (2019). Identification of a pathogenic mutation in ATP2A1 via in silico analysis of exome data for cryptic aberrant splice sites. Mol. Genet. Genomic Med. 7, e552. doi:10.1002/mgg3.552
Brugnoni, R., Maggi, L., Canioni, E., Verde, F., Gallone, A., Ariatti, A., et al. (2021). Next-generation sequencing application to investigate skeletal muscle channelopathies in a large cohort of Italian patients. Neuromuscul. Disord. 31, 336–347. doi:10.1016/j.nmd.2020.12.003
Guglielmi, V., Voermans, N. C., Gualandi, F., Van Engelen, B. G., Ferlini, A., Tomelleri, G., et al. (2013). Fourty-four years of Brody disease: it is time to Review. J. Genet. Syndr. Gene Ther. 04. doi:10.4172/2157-7412.1000181
Karpati, G., Charuk, J., Carpenter, S., Jablecki, C., and Holland, P. (1986). Myopathy caused by a deficiency of Ca2+-adenosine triphosphatase in sarcoplasmic reticulum (Brody’s disease). Ann. Neurol. 20, 38–49. doi:10.1002/ana.410200108
Koeppen, B. M., and Stanton, B. A. (2023). Berne and levy physiology e-book. Elsevier Health Sciences.
Molenaar, J. P., Snoeck, M. M., Voermans, N. C., and van Engelen, B. G. (2016). Overactive muscles: it can be more serious than common myalgia or cramp. Ned. Tijdschr. Geneeskd. 160, A9675. https://hdl.handle.net/2066/165866.
Molenaar, J. P., Verhoeven, J. I., Rodenburg, R. J., Kamsteeg, E. J., Erasmus, C. E., Vicart, S., et al. (2020). Clinical, morphological and genetic characterization of Brody disease: an international study of 40 patients. Brain 143, 452–466. doi:10.1093/brain/awz410
Odermatt, A., Barton, K., Khanna, V. K., Mathieu, J., Escolar, D., Kuntzer, T., et al. (2000). The mutation of Pro789 to Leu reduces the activity of the fast-twitch skeletal muscle sarco(endo)plasmic reticulum Ca2+ ATPase (SERCA1) and is associated with Brody disease. Hum. Genet. 106, 482–491. doi:10.1007/s004390000297
Odermatt, A., Taschner, P. E. M., Khanna, V. K., Busch, H. F., Karpati, G., Jablecki, C. K., et al. (1996). Mutations in the gene–encoding SERCA1, the fast–twitch skeletal muscle sarcoplasmic reticulum Ca2+ ATPase, are associated with Brody disease. Nat. Genet. 14, 191–194. doi:10.1038/ng1096-191
Saat, H., and Sahin, I. (2021). Mutation spectrum of hereditary myopathies in Turkish patients and novel variants. Ann. Hum. Genet. 85, 178–185. doi:10.1111/ahg.12429
Sambuughin, N., Zvaritch, E., Kraeva, N., Sizova, O., Sivak, E., Dickson, K., et al. (2014). Exome analysis identifies Brody myopathy in a family diagnosed with malignant hyperthermia susceptibility. Mol. Genet. Genomic Med. 2, 472–483. doi:10.1002/mgg3.91
Toğrul, V., Günaydın, N. T., and Tanyıldız, B. (2021). CALCIUM ADENOSINE 5’ TRIPHOSPHATASE ENZYME ACTIVITY IN EXTRAOCULAR MUSCLES IN STRABISMUS. KTD 22, 93–97. doi:10.18229/kocatepetip.656268
Velardo, D., Antognozzi, S., Rimoldi, M., Pagliarani, S., Cogiamanian, F., Barbieri, S., et al. (2023). Case report: clinical and molecular characterization of two siblings affected by Brody myopathy. Front. Neurol. 14, 1170071. doi:10.3389/fneur.2023.1170071
Walia, S., and Su, X. (2023). Brody myopathy associated with novel compound heterozygous ATP2A1 mutations (P6-8.015). Neurology 100. doi:10.1212/WNL.0000000000202716
Wuytack, F., Raeymaekers, L., and Missiaen, L. (2002). Molecular physiology of the SERCA and SPCA pumps. Cell. Calcium 32, 279–305. doi:10.1016/S0143416002001847
Keywords: Brody Disease, Brody Myopathy, ATP2A1, SERCA1, rare disease, whole-exome sequencing
Citation: Şahin A, Badakal EZD, Kovancılar Koç M, Uysal H and Başak AN (2023) Case report: Revealing the rare—a Brody Disease patient from Turkey expanding the phenotype. Front. Genet. 14:1289312. doi: 10.3389/fgene.2023.1289312
Received: 05 September 2023; Accepted: 16 November 2023;
Published: 30 November 2023.
Edited by:
Mohiuddin Mohammed Taher, Umm Al-Qura University, Saudi ArabiaReviewed by:
Sara Gibertini, IRCCS Carlo Besta Neurological Institute Foundation, ItalyAcary Oliveira, Federal University of São Paulo, Brazil
Copyright © 2023 Şahin, Badakal, Kovancılar Koç, Uysal and Başak. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ayşe Nazlı Başak, bmJhc2FrQGt1LmVkdS50cg==