 Jinghua Li1†
Jinghua Li1† Haipeng Zhu
Haipeng Zhu Limin Feng
Limin Feng- 1Department of Obstetrics and Gynecology, Beijing Tiantan Hospital, Capital Medical University, Beijing, China
- 2Centre for Personalized Cancer Therapy, Ciming Boao International Hospital, Lecheng International Medical Tourism Pilot Zone, Qionghai, Hainan, China
- 3P&A Consulting, Adelaide, SA, Australia
- 4Department of Pathology, Beijing Tiantan Hospital, Capital Medical University, Beijing, China
- 5Department of Radiology, Beijing Tiantan Hospital, Capital Medical University, Beijing, China
Dysgerminoma is a rare occurrence in Turner syndrome patients without Y chromosome mosaicism or hormone therapy during puberty. We present a unique case of a 33-year-old nulliparous Chinese woman with intermittent epilepsy and Mullerian anomalies carrying a double uterus, cervix, and vagina. The patient is also characterized as having Turner syndrome accompanied by 46,X, del(Xp22.33-11.23) and del(2)(q11.1-11.2). MRI exhibited a 17.0 cm × 20.0 cm × 10.5 cm solid ovarian lesion. Radical surgery and pathology revealed dysgerminoma at stage IIIc with lymphatic metastases and a KIT gene mutation identified in exon 13. Furthermore, the tumor microenvironment (TME) displayed robust expression of CD4+ T lymphocytes and PD-1, whereas the distribution of CD8+ T lymphocytes and PDL-1 was sporadic. Despite the administration of enoxaparin to prevent thromboembolism, the patient experienced multiple cerebral infarctions during chemotherapy. Subsequently, the patient chose to decline further treatment and was discharged. This exceptional case imparts several noteworthy lessons. First, the coexistence of Mullerian anomalies, although rare, is not incompatible with Turner syndrome. Second, screening for KIT mutations is imperative to reduce the risk of dysgerminoma in Turner syndrome, especially for patients with Y mosaicism who are recommended for hormone replacement therapy. Lastly, comprehensive anticoagulation therapy is crucial for Turner syndrome patients undergoing cisplatin-based chemotherapy.
Introduction
Turner syndrome encompasses a range of clinical manifestations tightly linked to various X chromosome abnormalities, particularly 45,X, 45,X/46,XY, and 46,X,del(Xp) (Murdock et al., 2017). Recent advances in genetic sequencing techniques have enabled the accurate identification of numerous Turner syndrome karyotypes including 46,X,del(Xp) (Prakash et al., 2014; Murdock et al., 2017). While Turner syndrome occupies many a phenotype, the coexistence of a double vagina, cervix, and uterus is unprecedented (Vaddadi et al., 2013). Furthermore, dysgerminoma occurs in only 1% of Turner syndrome patients without Y mosaicism (Roth et al., 2019), whereas only 2% of Turner syndrome genotypes showed 46,X,del(Xp) (Sybert and McCauley, 2004). In addition, being a tyrosine kinase receptor, the mutant KIT gene plays a pivotal role in carcinogenesis through multiple molecular pathways (Abdellateif et al., 2023). The mutations of the KIT gene are mainly identified in gastrointestinal stromal tumors in exons 8, 9, 11, 13, and 17 (McAuliffe et al., 2008), whereas human germ cell tumors and testicular seminomas contain the mutations in exons 11 and 17 (Hersmus et al., 2012). Currently, there is no evidence showing a mutation of the KIT gene in exon 13 existing in human germ cell tumors (Hersmus et al., 2012).
Case report
A 33-year-old nulliparous woman presented with severe lower abdominal pain was admitted to the Emergency Department in March 2023. The patient had a history of mild hypertension and intermittent epilepsy since the age of 23 years and was treated with oxcarbazepine for 10 years. The patient experienced menarche at the age of 16 years, followed by irregular periods starting at the age of 19 years, and subsequently developed amenorrhea from the age of 23 years onward. The patient showed normal intelligence and social communication, and no history of hormone therapy was documented. Physical examination showed normal breast development and absence of pubic hair growth. The patient’s height was 150 cm and weight was 45 kg. Moreover, an irregularly palpated mass was found in the lower left abdomen, extending to the umbilicus, accompanied by a double vagina and cervix. Blood tests revealed elevated platelet (380*10E9/L), LDH (300.5 U/L), and β-hCG (24.45 mIU/mL) levels, while AFP, CEA, CA-125, and CA-19-9 levels were within the normal range. Additionally, the sexual hormone panel and coagulative file were unremarkable. Except for hyperlipidemia, functional tests of the thyroid, liver, and kidneys were normal. After radical surgery, LDH and β-hCG levels returned to the normal levels at 194.4 U/L and 1.32 mIU/mL, respectively.
Genetic testing in March 2023 revealed the presence of 46,X,del(Xp22.33-11.23), involving a 47.6-Mb deletion, along with del(2)(q11.1-11.2), characterized by a 1.5-Mb deletion (Figure 1). An MRI investigation also exhibited a 17.0 cm × 20.0 cm × 10.5 cm solid ovarian lesion and uterus didelphys (Figure 2), whereas cranial and pulmonary CT examinations yielded unremarkable findings. Echocardiography confirmed slight tricuspid and mitral regurgitation.
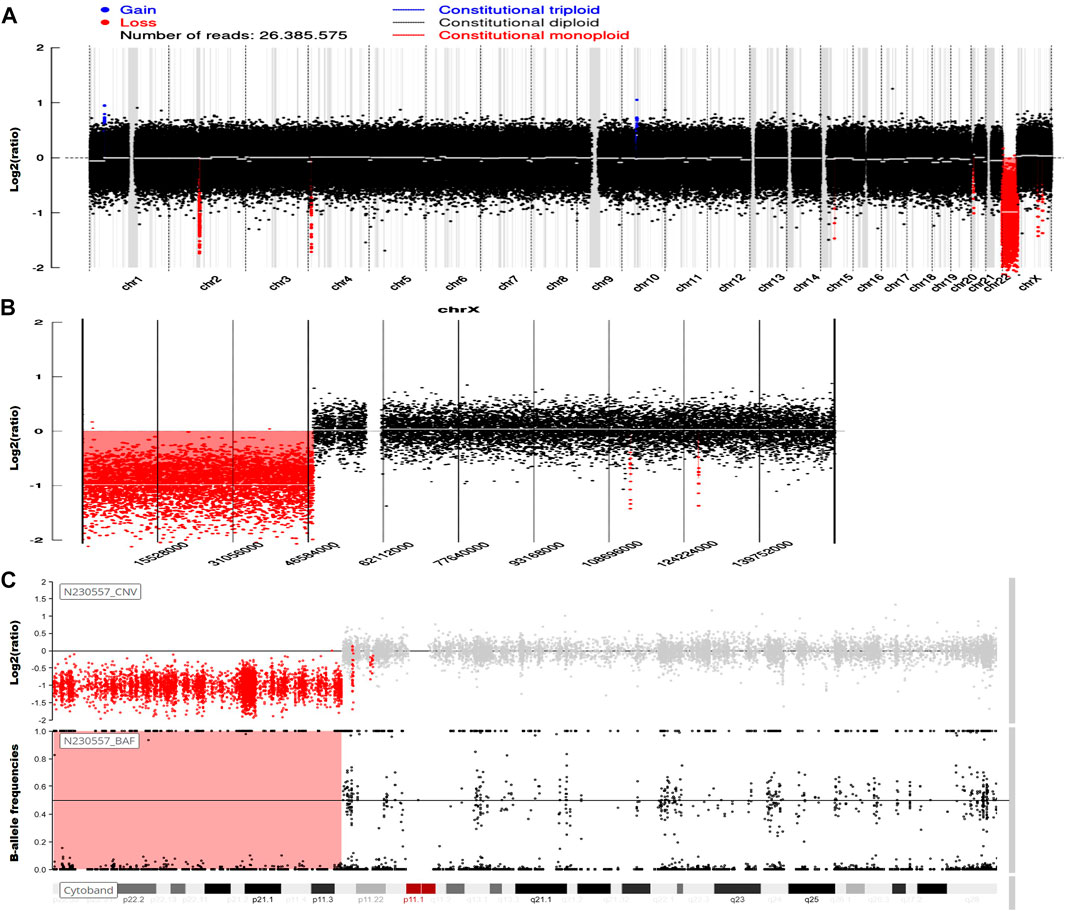
FIGURE 1. Genetic analysis.
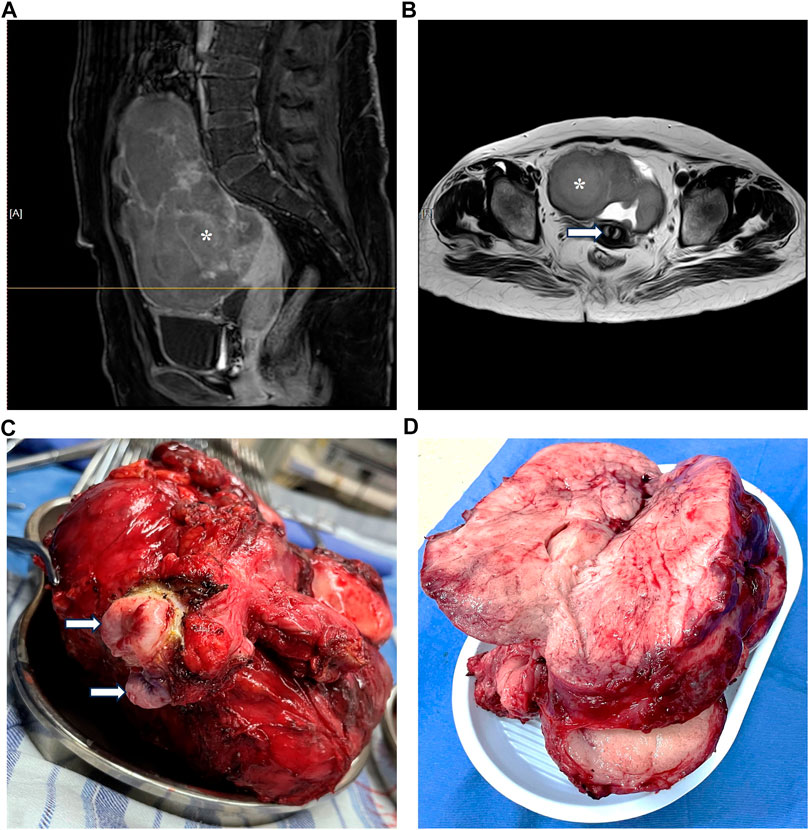
FIGURE 2. Magnetic resonance imaging and radical surgery.
In March 2023, the patient underwent radical surgery, in which pathology verified dysgerminoma at stage IIIc with lymphatic metastases (Figure 2). A cancer-associated mutation screening program utilizing dysgerminoma tissue identified the N655K mutation within exon 13 of the KIT gene. Subsequent immunohistochemistry analysis of sequentially sectioned tumor slides demonstrated the robust expression of CD4+ T cells and programmed cell death protein 1 (PD-1) within the tumor microenvironment (TME), whereas staining of CD8+ T cells and programmed death-ligand 1 (PDL-1) showed sporadic distribution (Figure 3).
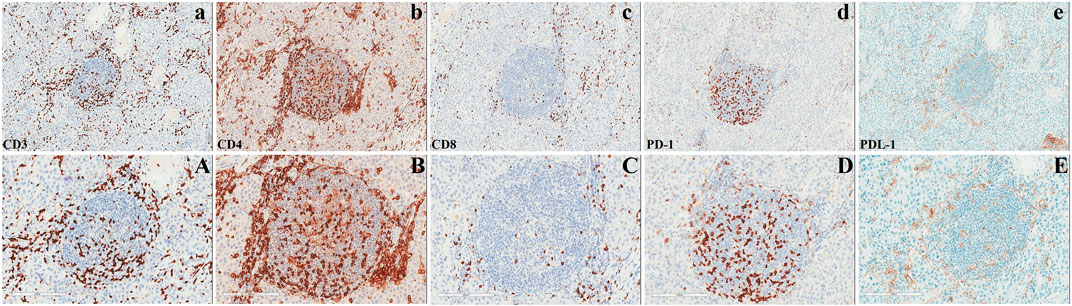
FIGURE 3. Analysis of the microenvironment in dysgerminoma.
After radical surgery, the patient experienced a single episode of spontaneous cerebral infarction at home in April 2023, which was treated at another tertiary hospital with complete recovery. Before the onset of chemotherapy, an evaluation at our hospital in May 2023 via intracranial MRI and MRA exhibited previously infarcted foci, highly suspected metastatic loci, and the circle of Willis malformation (Figure 4). Despite the daily administration of enoxaparin at a dose of 4,000 IU (40 mg) to prevent thromboembolism, multiple intracranial infarctions reoccurred (Figure 4) 5 days after commencing the standard chemotherapy regime using bleomycin, etoposide, and cisplatin. Following the comprehensive assessment and subsequent discussion of the situation, the patient chose to decline further treatment and was discharged.
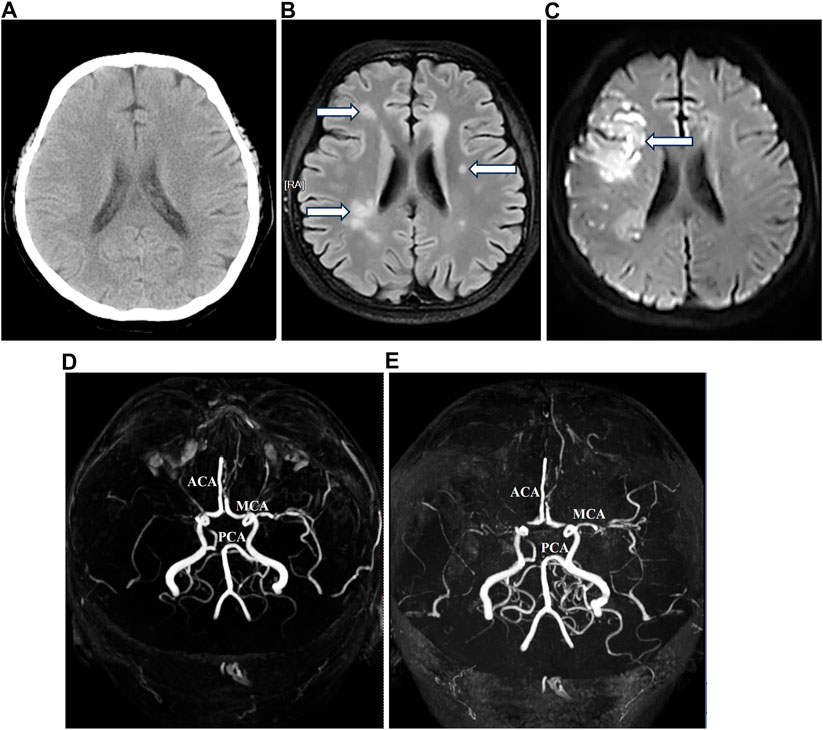
FIGURE 4. Magnetic resonance imaging and magnetic resonance angiography before and after chemotherapy.
Discussion
Previously, a large segmental deletion of the Xp22.3 region in Turner syndrome patients was tightly associated with phenotypes of strabismus, ichthyosis, chondrodysplasia punctata, amenorrhea, hyperlipidemia, obesity, short stature, intellectual disability, and Kallmann syndrome due to the loss of ANOS1, GPR143, HDHD1, NLGN4X, PNPLA4, SOX10, STS, and VCX-A genes principally (Nagai et al., 2017; Ma et al., 2020). In this study, including short stature and amenorrhea, our patient also had intermittent epilepsy. Epilepsy in Turner syndrome is less common, with 2%–3% morbidity, which predominantly correlates with structural malformation in the brain and mosaicism karyotypes (Hanew et al., 2018). To the best of our knowledge, only one report of absence of epilepsy was found in a case of 46,X, del(Xp22.33) with a duplication of Xp22.32-22.12 at the age of 10 years (Puusepp et al., 2008). Intriguingly, our patient also carries a 2q11.1-11.2 deletion, which shows an increased propensity for epilepsy (Grosso et al., 2008). These findings indicate that epilepsy in this case is highly likely to manifest in early childhood, as observed in our patient at the age of 23 years after ovarian degeneration. This observation strongly suggests that estrogen plays a pivotal role in suppressing seizure generation. Furthermore, while uterus disappearance due to Mullerian agenesis has been described in the 45,X genotype, actually, most female populations with uterine and cervical malformations caused by Mullerian anomalies possess a normal pattern of sexual chromosomes (Vaddadi et al., 2013). Whether the partial or complete loss of the X chromosome acts as a selective pressure that potentially contributes to Mullerian anomalies during embryonic development, our case, the first in the world, supports an encouraging response to the 46,X,del(Xp22.33-11.23) genotype.
Previously, immunohistochemical studies of dysgerminoma showed abundant infiltrating T cells with reduced or absent expression of the major histocompatibility complex (MHC)-I and -II antigens, respectively (Saito et al., 1989; Stewart et al., 1992). Further studies demonstrated that despite the high expression of CD8+ T cells and PD-1/PDL-1 within the TME, immune checkpoint inhibitors (ICIs) have not yielded significant results in the treatment of refractory dysgerminoma (Adra et al., 2018; Liu et al., 2018; Boldrini et al., 2019). These investigations imply that the loss of specific cytotoxicity due to MHC-I-mediated antigen-processing deficiency in dysgerminoma directly hampers immunotherapy efficacy. Interestingly, the pathology in this case exhibited a tremendously higher expression of CD4+ T cells and PD-1 within the TME, whereas the staining of CD8+ T cells and PDL-1 showed sporadic distribution (Figure 3). Our findings suggest that CD4+ T cells also respond notably to the growing dysgerminoma; however, the absence of MHC-II antigens progressively causes insensitivity of CD4+ T cells to the tumor cells. Considering the role of CD4+ T cells in maintaining tumor-specific cytotoxicity of CD8+ T cells (Ostroumov et al., 2018) and the anticancer activity of CD4+ T cells against self-derived epitopes (Tay et al., 2021), enhancing MHC-II expression in dysgerminoma or infusing specific antigen-activated CD8+ T cells sensitized through lysates of dysgerminoma tissue in vitro may significantly amplify therapeutic efficacy to manage metastatic or refractory dysgerminoma.
Moreover, to the best of our knowledge, we are the first to report a KIT mutation in exon 13 in dysgerminoma (Hersmus et al., 2012). Previous studies demonstrated that imatinib, a KIT gene inhibitor, led to a complete response of stage-IV chemoresistant seminoma (Pectasides et al., 2008), and PD-1 blockers significantly augmented the antitumor efficacy of imatinib (Seifert et al., 2017). Additionally, KIT mutations in exon 13 amplified the sensitivity of tumor cells to imatinib, sunitinib, and regorafenib, leading to prolonged survival in patients (McAuliffe et al., 2008; Napolitano and Vincenzi, 2019). Consequently, after primary radical surgery, combining tyrosine kinase inhibitors with ICIs or dysgerminoma-sensitized adoptive cellular therapy might provide more effective treatments to manage KIT mutation-associated dysgerminoma. Given the significant role of KIT mutations in cancer formation and development and their prevalence in dysgerminoma (Hersmus et al., 2012), it is critical to exclude KIT mutations in Turner syndrome patients, particularly those with Y mosaicism, to reduce the risk of dysgerminoma induced by hormone therapy.
Lastly, dysfunction in blood dynamics and abnormality of intracranial arteries extensively exist in Turner syndrome patients (Silberbach et al., 2018; Kruszka et al., 2019). Our patient also showed malformation of the circle of Willis (Figure 4). Considering that cisplatin-based chemotherapy is the standard treatment for dysgerminoma and is prone to induce hypercoagulability (De Giorgi et al., 2019), intracranial MRA becomes a crucial procedure in the evaluation of prognosis due to the high risk of thromboembolism. In this case, our patient exhibited normal coagulative parameters before chemotherapy, despite experiencing spontaneous intracranial embolism once. Following prophylactic anticoagulation with enoxaparin at a dose of 4,000 IU (40 mg) daily, multiple cerebral infarctions after chemotherapy were larger and more widespread than before (Figure 4). This indicates that selecting a moderate and sustained anticoagulation strategy, such as combining enoxaparin with Ginkgo leaf extract and dipyridamole (Liu et al., 2019), may provide more advantages to Turner syndrome patients undergoing cisplatin-based chemotherapy for dysgerminoma.
Conclusion
In summary, Mullerian anomalies are not incompatible with Turner syndrome. Assessing KIT mutations is critical to Turner syndrome patients prior to considering further treatments. Comprehensive anticoagulation therapy is a pivotal procedure in the management of Turner syndrome patients undergoing cisplatin-based chemotherapy.
Data availability statement
The datasets presented in this article are not readily available because of ethical/privacy restrictions. Requests to access the datasets should be directed to the corresponding authors.
Ethics statement
The studies involving humans were approved by the Medical Ethics Committee of Beijing Tiantan Hospital, Capital Medical University. The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article. Written informed consent was obtained from the participant/patient(s) for the publication of this case report.
Author contributions
JHL: data curation, formal analysis, investigation, methodology, project administration, and writing–review and editing. HZ: conceptualization, formal analysis, methodology, validation, writing–original draft, writing–review and editing, and data curation. XM: data curation, investigation, and writing–review and editing. JL: data curation, investigation, and writing–review and editing. JX: data curation and writing–review and editing. LF: data curation, formal analysis, funding acquisition, investigation, methodology, project administration, resources, supervision, and writing–review and editing.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This study was supported by the Natural Science Foundation of Beijing Municipality (J200002) awarded to LF who is the principal investigator.
Conflict of interest
Author HZ was employed by P&A Consulting.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
AFP, alpha fetal protein; CA-125, carbohydrate antigen 125; CA-19-9, carbohydrate antigen 19-9; CEA, carcinoembryonic antigen; CTL, cytotoxic T lymphocyte; ICI, immune checkpoint inhibitor; LDH, lactic dehydrogenase; LMWH, low-molecular-weight heparin; MRA, magnetic resonance angiography; MRI, magnetic resonance imaging; PD-1, programmed cell death protein 1; PDL-1, programmed death-ligand 1; TME, tumor microenvironment; β-hCG, beta human choriogonadotropin.
References
Abdellateif, M. S., Bayoumi, A. K., and Mohammed, M. A. (2023). c-Kit receptors as a therapeutic target in cancer: current insights. Onco Targets Ther. 16, 785–799. doi:10.2147/OTT.S404648
Adra, N., Einhorn, L. H., Althouse, S. K., Ammakkanavar, N. R., Musapatika, D., Albany, C., et al. (2018). Phase II trial of pembrolizumab in patients with platinum refractory germ-cell tumors: a Hoosier Cancer Research Network Study GU14-206. Ann. Oncol. 29 (1), 209–214. doi:10.1093/annonc/mdx680
Boldrini, R., De Pasquale, M. D., Melaiu, O., Chierici, M., Jurman, G., Benedetti, M. C., et al. (2019). Tumor-infiltrating T cells and PD-L1 expression in childhood malignant extracranial germ-cell tumors. Oncoimmunology 8 (2), e1542245. doi:10.1080/2162402X.2018.1542245
De Giorgi, U., Casadei, C., Bergamini, A., Attademo, L., Cormio, G., Lorusso, D., et al. (2019). Therapeutic challenges for cisplatin-resistant ovarian germ cell tumors. Cancers (Basel) 11 (10), 1584. doi:10.3390/cancers11101584
Grosso, S., Pucci, L., Curatolo, P., Coppola, G., Bartalini, G., Di Bartolo, R., et al. (2008). Epilepsy and electroencephalographic anomalies in chromosome 2 aberrations. A review. Epilepsy Res. 79 (1), 63–70. doi:10.1016/j.eplepsyres.2007.12.011
Hanew, K., Tanaka, T., Horikawa, R., Hasegawa, T., and Yokoya, S. (2018). Prevalence of diverse complications and its association with karyotypes in Japanese adult women with Turner syndrome-a questionnaire survey by the Foundation for Growth Science. Endocr. J. 65 (5), 509–519. doi:10.1507/endocrj.EJ17-0401
Hersmus, R., Stoop, H., van de Geijn, G. J., Eini, R., Biermann, K., Oosterhuis, J. W., et al. (2012). Prevalence of c-KIT mutations in gonadoblastoma and dysgerminomas of patients with disorders of sex development (DSD) and ovarian dysgerminomas. PLoS One 7 (8), e43952. doi:10.1371/journal.pone.0043952
Kruszka, P., Buscetta, A., Acosta, M. T., Banks, N., Addissie, Y. A., Toro, C., et al. (2019). Circle of Willis anomalies in Turner syndrome: absent A1 segment of the anterior cerebral artery. Birth Defects Res. 111 (19), 1584–1588. doi:10.1002/bdr2.1609
Liu, B., Arakawa, Y., Yokogawa, R., Tokunaga, S., Terada, Y., Murata, D., et al. (2018). PD-1/PD-L1 expression in a series of intracranial germinoma and its association with Foxp3+ and CD8+ infiltrating lymphocytes. PLoS One 13 (4), e0194594. doi:10.1371/journal.pone.0194594
Liu, Y., Wu, X., and Yu, Z. (2019). Ginkgo leaf extract and dipyridamole injection as adjuvant treatment for acute cerebral infarction: protocol for systemic review and meta-analysis of randomized controlled trials. Med. Baltim. 98 (8), e14643. doi:10.1097/MD.0000000000014643
Ma, W., Mao, J., Wang, X., Duan, L., Song, Y., Lian, X., et al. (2020). Novel microdeletion in the X chromosome leads to Kallmann syndrome, ichthyosis, obesity, and strabismus. Front. Genet. 11, 596. doi:10.3389/fgene.2020.00596
McAuliffe, J. C., Wang, W. L., Pavan, G. M., Pricl, S., Yang, D., Chen, S. S., et al. (2008). Unlucky number 13? Differential effects of KIT exon 13 mutation in gastrointestinal stromal tumors. Mol. Oncol. 2 (2), 161–163. doi:10.1016/j.molonc.2008.05.002
Murdock, D. R., Donovan, F. X., Chandrasekharappa, S. C., Banks, N., Bondy, C., Muenke, M., et al. (2017). Whole-exome sequencing for diagnosis of turner syndrome: toward next-generation sequencing and newborn screening. J. Clin. Endocrinol. Metab. 102 (5), 1529–1537. doi:10.1210/jc.2016-3414
Nagai, K., Shima, H., Kamimura, M., Kanno, J., Suzuki, E., Ishiguro, A., et al. (2017). Xp22.31 microdeletion due to microhomology-mediated break-induced replication in a boy with contiguous gene deletion syndrome. Cytogenet Genome Res. 151 (1), 1–4. doi:10.1159/000458469
Napolitano, A., and Vincenzi, B. (2019). Secondary KIT mutations: the GIST of drug resistance and sensitivity. Br. J. Cancer 120 (6), 577–578. doi:10.1038/s41416-019-0388-7
Ostroumov, D., Fekete-Drimusz, N., Saborowski, M., Kuhnel, F., and Woller, N. (2018). CD4 and CD8 T lymphocyte interplay in controlling tumor growth. Cell Mol. Life Sci. 75 (4), 689–713. doi:10.1007/s00018-017-2686-7
Pectasides, D., Nikolaou, M., Pectasides, E., Koumarianou, A., Valavanis, C., and Economopoulos, T. (2008). Complete response after imatinib mesylate administration in a patient with chemoresistant stage IV seminoma. Anticancer Res. 28 (4C), 2317–2320.
Prakash, S., Guo, D., Maslen, C. L., Silberbach, M., Milewicz, D., Bondy, C. A., et al. (2014). Single-nucleotide polymorphism array genotyping is equivalent to metaphase cytogenetics for diagnosis of Turner syndrome. Genet. Med. 16 (1), 53–59. doi:10.1038/gim.2013.77
Puusepp, H., Zordania, R., Paal, M., Bartsch, O., and Ounap, K. (2008). Girl with partial Turner syndrome and absence epilepsy. Pediatr. Neurol. 38 (4), 289–292. doi:10.1016/j.pediatrneurol.2007.11.008
Roth, L. M., Davis, M. M., and Czernobilsky, B. (2019). Classic and "dissecting" gonadoblastoma in a phenotypic girl with a 46, XX peripheral karyotype and No evidence of a disorder of sex development. Int. J. Gynecol. Pathol. 38 (6), 581–587. doi:10.1097/PGP.0000000000000551
Saito, T., Tanaka, R., Kouno, M., Washiyama, K., Abe, S., and Kumanishi, T. (1989). Tumor-infiltrating lymphocytes and histocompatibility antigens in primary intracranial germinomas. J. Neurosurg. 70 (1), 81–85. doi:10.3171/jns.1989.70.1.0081
Seifert, A. M., Zeng, S., Zhang, J. Q., Kim, T. S., Cohen, N. A., Beckman, M. J., et al. (2017). PD-1/PD-L1 blockade enhances T-cell activity and antitumor efficacy of imatinib in gastrointestinal stromal tumors. Clin. Cancer Res. 23 (2), 454–465. doi:10.1158/1078-0432.CCR-16-1163
Silberbach, M., Roos-Hesselink, J. W., Andersen, N. H., Braverman, A. C., Brown, N., Collins, R. T., et al. (2018). Cardiovascular Health in turner syndrome: a scientific statement from the American heart association. Circ. Genom Precis. Med. 11 (10), e000048. doi:10.1161/HCG.0000000000000048
Stewart, C. J., Farquharson, M. A., and Foulis, A. K. (1992). Characterization of the inflammatory infiltrate in ovarian dysgerminoma: an immunocytochemical study. Histopathology 20 (6), 491–497. doi:10.1111/j.1365-2559.1992.tb01033.x
Sybert, V. P., and McCauley, E. (2004). Turner's syndrome. N. Engl. J. Med. 351 (12), 1227–1238. doi:10.1056/NEJMra030360
Tay, R. E., Richardson, E. K., and Toh, H. C. (2021). Revisiting the role of CD4(+) T cells in cancer immunotherapy-new insights into old paradigms. Cancer Gene Ther. 28 (1-2), 5–17. doi:10.1038/s41417-020-0183-x
Keywords: epilepsy, uterus didelphys, Turner syndrome, dysgerminoma, KIT mutation
Citation: Li J, Zhu H, Ma X, Li J, Xue J and Feng L (2024) Case Report: From epilepsy and uterus didelphys to Turner syndrome-associated dysgerminoma. Front. Genet. 14:1286515. doi: 10.3389/fgene.2023.1286515
Received: 01 September 2023; Accepted: 14 December 2023;
Published: 11 January 2024.
Edited by:
Amit V. Pandey, University of Bern, SwitzerlandReviewed by:
Bicheng Yang, Jiangxi Maternal and Child Health Hospital, ChinaItaru Kushima, Nagoya University, Japan
Copyright © 2024 Li, Zhu, Ma, Li, Xue and Feng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Haipeng Zhu, ZHIuaGFpcGVuZ3podUBob3RtYWlsLmNvbQ==; Limin Feng, bHVjeWZlbmcxOTY2QDE2My5jb20=
†These authors have contributed equally to this work and share first authorship