 Sok-Kun Tae
Sok-Kun Tae Mazlan RA
Mazlan RA Meow-Keong Thong
Meow-Keong Thong- 1Genetics and Metabolism Unit, Department of Paediatrics, Faculty of Medicine, Kuala Lumpur, Malaysia
- 2Genetic Medicine Unit, University of Malaya Medical Centre, Kuala Lumpur, Malaysia
ESCO2 spectrum disorder is an autosomal recessive developmental disorder characterized by growth retardation, symmetrical mesomelic limb malformation, and distinctive facies with microcephaly, with a wide phenotypic continuum that ranges from Roberts syndrome (MIM #268300) at the severe end to SC phocomelia (MIM #269000) at the milder end. ESCO2 encodes a 601-amino acid protein belonging to the Eco1/Ctf7 family of acetyltransferases that is involved in the establishment of sister chromatid cohesion, which is essential for accurate chromosome segregation and genomic stability and thus belongs to a group of disorders called “cohesinopathies”. We describe a 15-year-old Malaysian female who presented with the characteristic triad of ESCO2 spectrum disorder, with an equivocal chromosomal breakage study and normal karyotyping findings. She was initially suspected to have mosaic Fanconi anemia but whole exome sequencing (WES) showed a likely pathogenic homozygous splice variant c.955 + 2_955+5del in the ESCO2 gene. During the 15-year diagnostic odyssey, she developed type 2 diabetes mellitus, primary ovarian insufficiency, increased optic cup-to-disc ratio with tortuous vessels bilaterally, and an evolving but distinct facial and skin hypopigmentation phenotype. Of note, there was an absence of learning disabilities. Our findings provide further evidence for ESCO2 spectrum disorder in an Asian child and contribute to defining the clinical and radiographic spectrum.
Introduction
ESCO2 spectrum disorder is a rare, autosomal recessive, genetic disorder that has a broad range of clinical phenotypes from a severe type known as Roberts syndrome to a milder type known as SC phocomelia (Vega et al., 2006). Roberts syndrome (RBS, MIM #268300) was first described in 1919 in a baby boy with tetraphocomelia and cleft lip/palate (Roberts, 1919). Vega et al. (2010) established the clinical criteria for RBS, which were based on a cohort of 49 patients: growth retardation, symmetric mesomelic shortening of the limbs predominantly affecting the upper limbs, and characteristic facies with microcephaly (Vega et al., 2010). Fifty years later, Herrmann et al. reported a milder form that was described as pseudothalidomide syndrome or SC phocomelia (MIM #269000) (Herrmann et al., 1969). The prevalence of ESCO2 spectrum disorder is unclear. Approximately 150 individuals of diverse ethnic backgrounds have been reported in the literature (Vega et al., 2006). The clinical description of this condition is rarely reported in patients of Asian ancestry. Kantaputra et al. first reported two siblings of the Lisu tribe from Thailand who were initially diagnosed with Juberg-Hayward syndrome with ESCO2 mutations (Kantaputra et al., 2020).
ESCO2 spectrum disorder belongs to a group of developmental disorders termed “cohesinopathies,” which are associated with biallelic pathogenic variants in the ESCO2 gene (MIM 609353) on chromosome 8p21.1, resulting in complete or partial loss of the acetyltransferase domain (Zakari et al., 2015). Cohesin consists of a ring-shaped multiprotein complex with four subunits (SMC1A, SMC3, RAD21, and STAG1/2) and, together with regulatory factors, is essential for sister chromatid cohesion, genome organization, gene expression regulation, DNA repair, and genome safeguarding (Cucco and Musio, 2016). ESCO2 is one of the regulatory factors in the Cohesin pathway; it encodes a 601-amino acid protein belonging to the Eco1/Ctf7 family of acetyltransferases that is involved in the establishment of sister chromatid cohesion (Faramarz et al., 2020). The cohesion of sister chromatids is essential for accurate chromosome segregation and genomic stability (Tomkins et al., 1979). A cytogenetic study of RBS revealed a rod-like chromosome morphology, resulting in a “railroad-track” appearance and characteristic premature chromatid separation (PCS), otherwise known as heterochromatin repulsion (HR) or puffing (Vega et al., 2005).
A diagnosis of ESCO2 spectrum disorder is established when there are suggestive clinical findings and biallelic pathogenic (or likely pathogenic) variants in the ESCO2 gene as determined by molecular genetic testing, or premature chromatid separation (PCS) as determined by cytogenetic testing. In an analysis of 49 patients with ESCO2 mutations, including 18 previously reported cases, Vega et al. reported no clear genotype/phenotype correlation (Vega et al., 2010). Many patients with ESCO2 spectrum disorder are clinically diagnosed with many overlapping syndromes. Here, we describe a 15-year-old Malaysian female who presented with the characteristic triad of RBS, an equivocal chromosomal breakage study, and normal karyotyping, who was initially diagnosed as mosaic FA for 11 years. The diagnosis was established via whole exome sequencing (WES), where a homozygous, likely pathogenic, variant was identified in the ESCO2 gene.
Case description
A 15-year-old female of Indian ancestry was first referred to the Genetics and Metabolism Unit, University of Malaya Medical Centre (UMMC) at birth for multiple skeletal abnormalities and intrauterine growth retardation (IUGR), with distinctive facial features. She was the first child (V:1, see Figure 1) of a consanguineous union, where her parents were first cousins. Her mother and father were 24 and 28 years old, respectively, when she was born. The maternal grandparents were third cousins. She had a younger brother (V:2) who was healthy, with no skeletal abnormality. There was a family history of type 2 diabetes mellitus. Antenatally, her mother had gestational diabetes mellitus with good glycemic control and there was no known teratogenic exposure. Serial antenatal ultrasound scans noted oligohydramnios and intrauterine growth retardation. There was no reduced fetal movement. She was delivered at term via elective lower segment cesarean section for an abnormal lie, with Apgar scores of 7 and 9 at 1 min and 5 min, respectively. She was admitted to the neonatal intensive care unit (NICU) for 2 weeks for weight management.
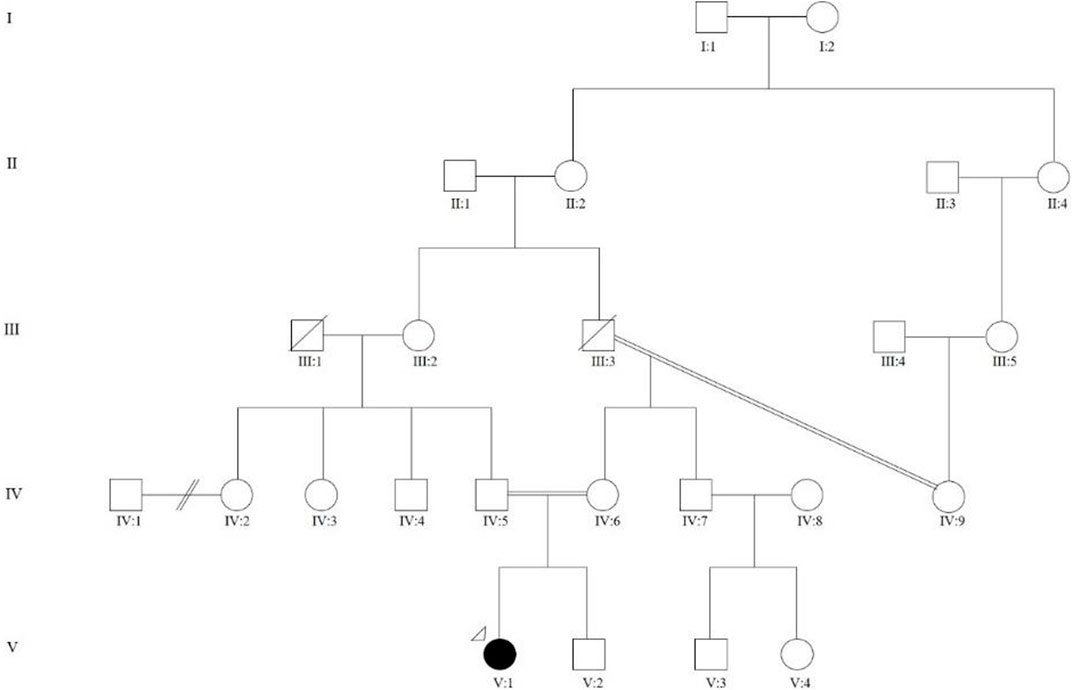
FIGURE 1. Family pedigree showing multi-level consanguinity. V:1 is the proband who was the first child of a consanguineous marriage, where her parents were first cousins (IV:4 and IV:5). The maternal grandparents (III:3 and IV:9) were third cousins.
The patient’s gross and fine motor developmental milestones were delayed due to the skeletal deformities. However, she had normal intellectual development and attended normal school. She was average in her school performance. She underwent multiple corrective surgeries for her lower limbs. For the bilateral congenital talipes equinovarus (CTEV), she was treated with serial casting from birth, followed by a Dennis Brown (DB) splint and shoes until the age of 7 months old. For the fixed flexion deformity of her knees, she underwent bilateral Ilizarov external fixation twice at the age of 3 and 10 years old. She had bilateral distal femur and proximal tibia hemi-epiphysiodesis performed at 6 years of age. These surgeries enabled her to walk for short distances of approximately 50–100 m; however, she developed pain and fatigue while walking for longer distances. Ophthalmology review at birth revealed an increased optic cup-to-disc ratio (CDR) at 0.8, with tortuous vessels bilaterally. There was no increase in intra-ocular pressure, no lens or corneal opacities, nor retinal abnormalities.
She was diagnosed with type 2 diabetes mellitus (T2DM) at the age of 12 years, where she presented with polyuria, polydipsia, significant weight loss, and recurrent skin abscess for a duration of 5 months. The diagnosis was confirmed by a serum blood glucose level of 20.5 mmol/L, with no metabolic acidosis or ketosis and a c-peptide level of 0.9 ng/mL. There were negative islet cell cytoplasmic autoantibodies (ICA), glutamic acid decarboxylase autoantibodies (GADA), insulinoma-associated-2 autoantibodies (IA-2A), and insulin autoantibodies (IAA). She was treated with metformin and subcutaneous insulin. Her T2DM was complicated by diabetic nephropathy, dyslipidemia, and a fatty liver. She attained menarche at 14 years old and was diagnosed with primary ovarian insufficiency when her menses stopped after a few months of irregular menses. A hormonal study revealed low estrogen levels, with high luteinizing hormone (LH) and follicle-stimulating hormone (FSH) levels. Pelvis ultrasonography visualized both ovaries, which were normal in size. Figure 2 shows the clinical course and diagnostic timeline for 15 years.
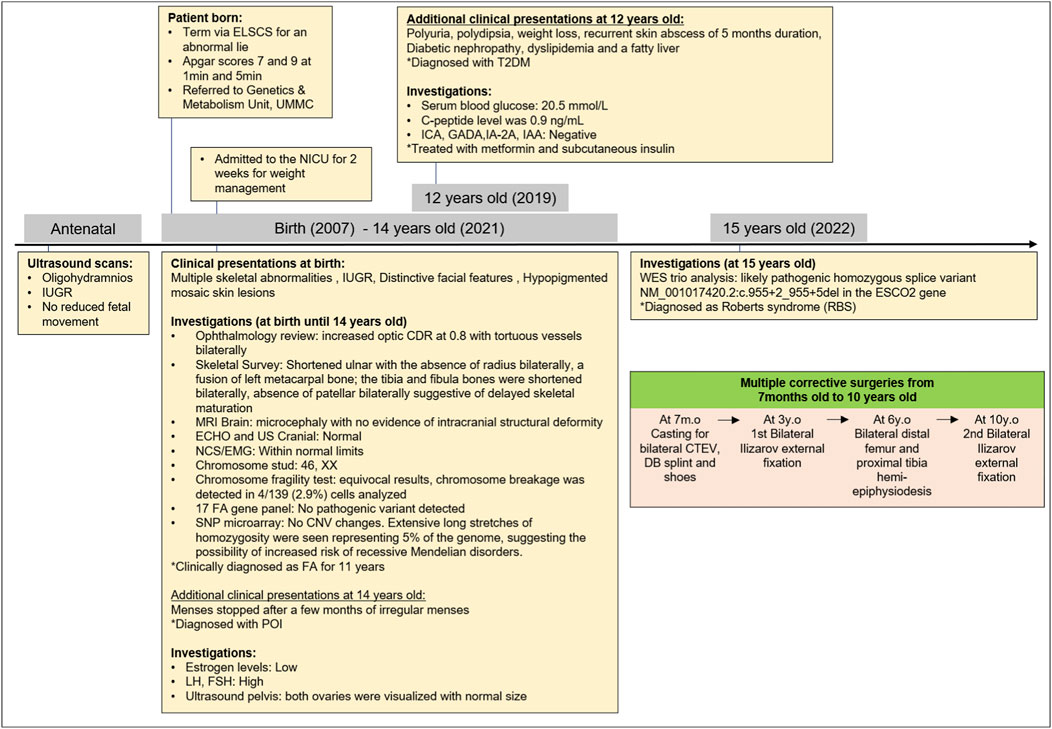
FIGURE 2. Clinical course and diagnostic timeline. *ELCCS, emergency lower C-section; UMMC, University Malaya Medical Centre; NICU, Neonatal Intensive Care Unit; IUGR, intrauterine growth restriction; T2DM, type 2 diabetes mellitus; ICA, islet cell cytoplasmic autoantibodies; GADA, glutamic acid decarboxylase autoantibodies; IA-2A, insulinoma-associated-2 autoantibodies; IAA, insulin autoantibodies; LH, high luteinizing hormone; FH, follicle-stimulating hormone; CDR, cup-to-disc ratio; MRI: magnetic resonance imaging; ECHO, echocardiogram; US, ultrasound; NCS, nerve conduction study; EMG, electromyography; FA, Fanconi anemia; SNP: CNV, copy number of variant; POI, primary ovarian insufficiency; WES, whole exome sequencing; DB, Dennis Brown; CTEV, congenital talipes equino varus; y.o, years old; m.o, month old.
Physical examination at birth showed prenatal growth failure, with a birth weight of 1.46 kg (Z = −4.7 SD), length of 35 cm (Z = −8.1 SD), and head circumference of 26 cm (Z = −8.0 SD). Distinctive features included brachymicrocephaly, diffuse alopecia, down-slanting of palpebral fissures, hypertelorism, hypoplastic nasal alae, bilateral low-set and simple ears, malar hypoplasia, and micrognathia (Figure 3). There was no cleft or high-arched palate. Bilateral symmetrical mesomelic shortening of the upper and lower limbs was found, where the upper limbs were more severely affected. There were bilateral absent radii, an absent left thumb with a small, rudimentary right thumb, and bilateral congenital talipes equinovarus. Capillary hemangioma was present at birth over the forehead and extending down to the nose, which gradually faded away when she was a toddler. There were hypopigmented mosaic skin lesions noted since birth over her left cheek, chin, upper limbs, abdomen, and back, which followed Blaschko’s lines. These lesions became more obvious and prominent over time. At 15 years of age, all her growth parameters remained well below the third centile.
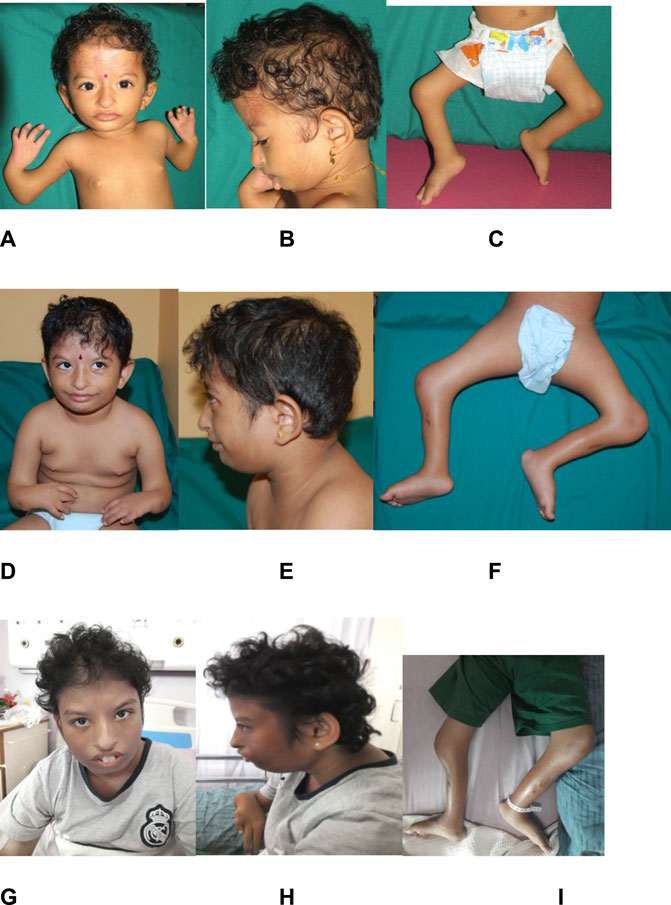
FIGURE 3. Evolving phenotype of the patient over 15 years. Photographs of the patient at 2 years old (images (A–C)), at 6 years old (images (D–F)), and at 14 years old (images (G–I)). Images (A,B), (D,E), and (G,H) show diffuse alopecia, brachymicrocephaly, down-slanting of palpebral fissures, hypertelorism, hypoplastic nasal alae, bilateral low-set and simple ear, malar hypoplasia, and micrognathia. Images (C,F,I) show fixed flexion deformities of both her knees.
Based on her clinical features, a diagnosis of Fanconi anemia (FA) was made. A skeletal survey performed at birth showed a small skull with micrognathia, short ulnae with the absence of radii bilaterally, fusion of the left metacarpal bones, short tibiae and fibulae bones bilaterally, and the absence of patellar bilaterally, suggestive of delayed skeletal maturation. The spine and pelvic bone appeared normal (Figure 4). Cranial MRI showed microcephaly, with no evidence of intracranial structural abnormalities. The echocardiogram was normal. Given multiple contractures, a nerve conduction study and electromyogram were performed, and both were within normal limits. A chromosome study revealed normal results, 46, XX. A chromosome fragility test showed equivocal results, where chromosome breakage was detected in 4/139 (2.9%) cells analyzed. Serial complete blood count showed no evidence of bone marrow failure. A multigene panel sequencing of 17 FA genes showed no pathogenic variant detected, while a chromosomal SNP microarray revealed no clinically significant copy number changes. However, extensive long stretches of homozygosity were seen, representing 5% of the genome, suggesting the possibility of an increased risk of recessive Mendelian disorders.
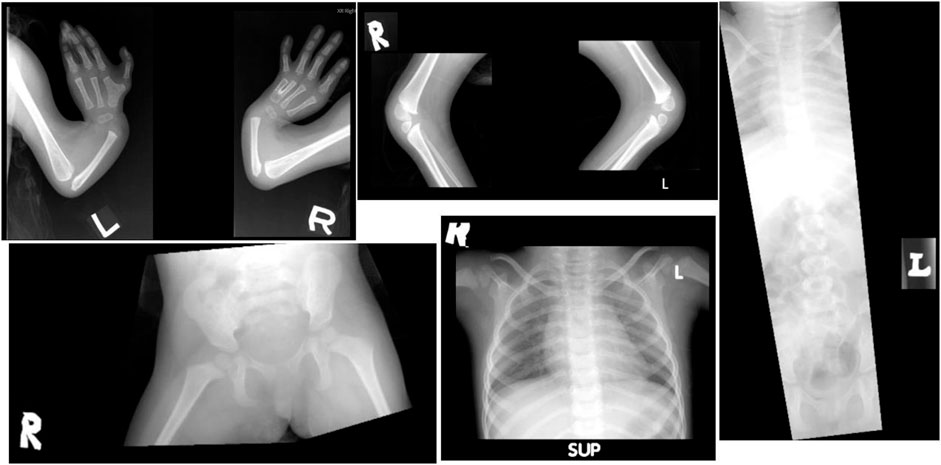
FIGURE 4. Patient’s radiograph at the age of 2 years old. There was a shortened ulnar, with the absence of radius bilaterally, and fusion of the left metacarpal bone. The spine and pelvic bone appeared normal.
The parents were devastated by the negative results. However, they were motivated to pursue further investigations. The patient was treated as mosaic FA and medical surveillance was continued.
The patient was included in the Global Genomic Medicine Collaborative (G2MC) Rare Disease project, in which whole exome sequencing detected a homozygous splice variant NC_000008.10(NM_001017420.3): c.955 + 2_955+5del (GRCh37 (chr8): g.27637786_27637789del) in the ESCO2 gene in the patient. This variant does not present nucleotide change as it is an intronic splicing variant. The frequency allele in gnomAD v2.1.1 is 0.000012 and in the South Asian population is 0.00009827. This sequence change affects a splice site in intron 4 of the ESCO2 gene. It is predicted to alter splicing and results in loss of protein function. Loss of function variants in ESCO2 are known to be pathogenic (Schüle et al., 2005; Vega et al., 2005). Both her parents were heterozygous carriers. This variant was close to the highly conserved splice site. According to HGMD Professional 2021.3, this variant was previously described as disease-causing for Roberts syndrome (Gordillo et al., 2008; Capalbo et al., 2019). ClinVar listed this variant as likely pathogenic (Variation ID: 21252), and homozygosity was confirmed by parental testing. According to the recommendations of the ACMG/AMP variant classification guidelines (Richards et al., 2015), this variant was classified as likely pathogenic.
Discussion
To the best of our knowledge, this is the first case report of an individual with ESCO2 spectrum disorder/RBS with Asian Indian ancestry with the classical triad of RBS that consisted of pre-and post-natal growth retardation, bilateral symmetrical mesomelic limb shortening predominantly affecting her upper limbs, and distinctive facial features with microcephaly (Vega et al., 2010). Her limb malformations include bilateral short ulnar and absent radii, thumb defects, oligodactyly, left metacarpal synostosis, flexion contractures over the elbows and knees, bilateral short tibia and fibula, absent patella, and bilateral CTEV. The limb deformities noted in our patient that are rarely reported were flexion contractures (16%), metacarpal synostosis (12%), and absent patella (4%) (Vega et al., 2010). The unique aspect of our case is the 15-year follow-up of the patient’s progress. Whilst she had gross and fine motor impairment due to her skeletal deformities, there was no intellectual impairment. This included the documentation of the absence of learning disabilities and a non-progressive increase in CDR. Interestingly, our patient developed T2DM and primary ovarian insufficiency (POI), which were not reported before in other patient series. The etiology of POI could be autoimmune (4%–30%) (Kirshenbaum and Orvieto, 2019) or genetic (França and Mendonca, 2019), although the majority remains unknown. A few case reports showed a biallelic pathogenic variant in STAG3, a cohesion gene, affected the meiosis-specific cohesion complexes, which might lead to premature ovarian failure (Caburet et al., 2014; Colombo et al., 2017; Akbari et al., 2022). Other meiosis-specific complexes in cohesin, including REC8, RAD21L, and SMC1β, have been shown to be associated with abnormal oocyte development (Caburet et al., 2014; Ward et al., 2016). Thus, more studies will be required to investigate the relationship between the ESCO2 gene, regulatory factors in the cohesin pathway, and human fertility. The evolving facial phenotype and hypopigmented skin lesions documented in our patient will serve as a potential guide for counseling and future dysmorphological studies in the Asian population for RBS/ESCO2 spectrum disorders.
Our patient was initially diagnosed as having FA for 11 years. Some of the clinical characteristics, namely, skeletal deformities, growth retardation, microcephaly, and the extent of MMC-induced chromosomal breakage in patients with cohesinopathies overlapped with those of (mosaic) FA patients (Lo Ten Foe et al., 1997). Patients with mosaic FA may present without bone marrow failure due to spontaneous genetic reversion, which corrects the bone marrow failure (van der Lelij et al., 2010). Chromosomal breakage study, which is the gold standard in diagnosing FA (Hirsch et al., 2020), could be equivocal in both patients with mosaic FA and cohesinopathies. Chromosomal breakage study on skin fibroblasts or conventional karyotyping showed “railroad-track” chromosomes and premature chromatid separation (PCS), which allowed these two conditions to be distinguished. In our patient, the basic karyotyping performed earlier showed normal results, and a chromosomal breakage study on skin fibroblasts was not performed. Due to the highly variable clinical phenotype, a patient with a cohesinopathy such as RBS may exhibit multiple clinical features that overlap with mosaic FA. Cellular hypersensitivity to MMC has also been reported in cohesinopathies, including RBS (van der Lelij et al., 2010). Thus, our patient was treated as mosaic FA initially but the diagnostic odyssey ended with the whole exome sequencing (WES) test, which demonstrated the homozygous splice site variant c.955 + 2_955+5del in the ESCO2 gene. It was important to establish an accurate diagnosis as there were implications for clinical management and surveillance as well as for genetic counseling. When she was initially diagnosed as having Fanconi anemia, the family was informed of an increased risk of tumors, and she received annual ultrasonography of the liver and blood tests such as complete blood count and liver function tests. With the revised diagnosis of ESCO2 disorder, frequent blood tests and liver ultrasonography were no longer required as the cancer risk was not increased. In addition, as both parents were carriers, the family was counseled on the autosomal recessive inheritance and recurrence risks. Cascade screening for extended family members was also performed.
The protracted journey toward the final diagnosis for people living with rare diseases, where up to 50% will remain undiagnosed (Shashi et al., 2014), is not surprising. The clinical implementation of WES has helped in shortening the odyssey and has had significant clinical, genetic counseling, psychosocial, and economic benefits (Sawyer et al., 2016). It was reported that WES had a positive diagnostic yield of approximately 39% in diagnosing disorders related to skeletal malformations (Retterer et al., 2016). There is a need to assess the clinical utility and cost-effectiveness of WES in ending the diagnostic odyssey in low-resourced countries for Asian children with dysmorphic features and multiple congenital malformations. Further discussion with all stakeholders, patient support groups, and governmental intervention is important to ensure equity of care and an appropriate funding model for patients with rare diseases.
Data availability statement
The datasets for this article are not publicly available due to concerns regarding participant/patient anonymity. Requests to access the datasets should be directed to the corresponding author.
Ethics statement
The study involving human participation was approved by the University of Malaya Medical Ethics Research committee and University of Maryland IRB. The studies were conducted in accordance with the local legislation and institutional requirements. The human samples used in this study were acquired from a by-product of routine care or industry. Written informed consent for participation was not required from the participants or the participants’ legal guardians/next of kin in accordance with the national legislation and institutional requirements. Written informed consent was obtained from the minor(s)’ legal guardian/next of kin for the publication of any potentially identifiable images or data included in this article.
Author contributions
S-KT: Conceptualization, Data curation, Investigation, Methodology, Software, Writing–original draft, Writing–review and editing. MR: Investigation, Writing–original draft, Writing–review and editing. M-KT: Conceptualization, Formal Analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Supervision, Visualization, Writing–original draft, Writing–review and editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This study was supported by the Global Genomic Medicine Consortium (G2MC) and the Genetic Medicine Unit, University of Malaya Medical Centre, Kuala Lumpur, Malaysia.
Acknowledgments
The authors thank the patient and family for cooperating in the study. We thank the Global Genomic Medicine Consortium (G2MC) and Centogene for helping with the whole exome sequencing, analysis, and variant interpretation.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Akbari, A., Zoha Tabatabaei, S., Salehi, N., Padidar, K., Almadani, N., Ali Sadighi Gilani, M., et al. (2022). Novel STAG3 variant associated with primary ovarian insufficiency and non-obstructive azoospermia in an Iranian consanguineous family. Gene 821, 146281. doi:10.1016/j.gene.2022.146281
Caburet, S., Arboleda, V. A., Llano, E., Overbeek, P. A., Barbero, J. L., Oka, K., et al. (2014). Mutant cohesin in premature ovarian failure. N. Engl. J. Med. 370 (10), 943–949. doi:10.1056/NEJMoa1309635
Capalbo, A., Valero, R. A., Jimenez-Almazan, J., Pardo, P. M., Fabiani, M., Jiménez, D., et al. (2019). Optimizing clinical exome design and parallel gene-testing for recessive genetic conditions in preconception carrier screening: translational research genomic data from 14,125 exomes. PLoS Genet. 15 (10), e1008409. doi:10.1371/journal.pgen.1008409
Colombo, R., Pontoglio, A., and Bini, M. (2017). A STAG3 missense mutation in two sisters with primary ovarian insufficiency. Eur. J. obstetrics, Gynecol. reproductive Biol. 216, 269–271. doi:10.1016/j.ejogrb.2017.08.005
Cucco, F., and Musio, A. (2016). Genome stability: what we have learned from cohesinopathies. Am. J. Med. Genet. Part C, Seminars Med. Genet. 172 (2), 171–178. doi:10.1002/ajmg.c.31492
Faramarz, A., Balk, J. A., van Schie, J. J. M., Oostra, A. B., Ghandour, C. A., Rooimans, M. A., et al. (2020). Non-redundant roles in sister chromatid cohesion of the DNA helicase DDX11 and the SMC3 acetyl transferases ESCO1 and ESCO2. PloS one 15 (1), e0220348. doi:10.1371/journal.pone.0220348
França, M. M., and Mendonca, B. B. (2019). Genetics of primary ovarian insufficiency in the next-generation sequencing era. J. Endocr. Soc. 4 (2), bvz037. doi:10.1210/jendso/bvz037
Gordillo, M., Vega, H., Trainer, A. H., Hou, F., Sakai, N., Luque, R., et al. (2008). The molecular mechanism underlying Roberts syndrome involves loss of ESCO2 acetyltransferase activity. Hum. Mol. Genet. 17 (14), 2172–2180. doi:10.1093/hmg/ddn116
Herrmann, J., Feingold, M., Tuffli, G. A., and Opitz, J. M. (1969). A familial dysmorphogenetic syndrome of limb deformities, characteristic facial appearance and associated anomalies: the “pseudothalidomide” or “SC-syndrome”. Birth Defects Orig. Artic. Ser. 5, 81e9. doi:10.1055/s-2007-999392
Hirsch, B., McIntyre, K., and Olson, S. (2020). “Diagnosis of Fanconi Anemia: testing and genetic counseling,” in Fanconi Anemia clinical care guideline. Editor I. Sroka 5th ed. Eugene, OR: Fanconi Anemia research fund, 21–52. https://www.fanconi.org/images/uploads/other/Fanconi_Anemia_Clinical_Care_Guidelines_5thEdition_web.pdf (Accessed April 30, 2023).
Kantaputra, P. N., Dejkhamron, P., Tongsima, S., Ngamphiw, C., Intachai, W., Ngiwsara, L., et al. (2020). Juberg-Hayward syndrome and Roberts syndrome are allelic, caused by mutations in ESCO2. Arch. Oral Biol. 119, 104918. doi:10.1016/j.archoralbio.2020.104918
Kirshenbaum, M., and Orvieto, R. (2019). Premature ovarian insufficiency (POI) and autoimmunity-an update appraisal. J. Assisted Reproduction Genet. 36 (11), 2207–2215. doi:10.1007/s10815-019-01572-0
Lo Ten Foe, J. R., Kwee, M. L., Rooimans, M. A., Oostra, A. B., Veerman, A. J., van Weel, M., et al. (1997). Somatic mosaicism in Fanconi anemia: molecular basis and clinical significance. Eur. J. Hum. Genet. 5 (3), 137–148. doi:10.1159/000484749
Retterer, K., Juusola, J., Cho, M. T., Vitazka, P., Millan, F., Gibellini, F., et al. (2016). Clinical application of whole-exome sequencing across clinical indications. Genet. Med. 18 (7), 696–704. doi:10.1038/gim.2015.148
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genet. Med. 17 (5), 405–424. doi:10.1038/gim.2015.30
Roberts, J. B. (1919). A child with double cleft lip and palate, protrusion of the intermaxillary portion of the upper jaw and imperfect development of the bones of the four extremities. Ann. Surg. 70, 252e3.
Sawyer, S. L., Hartley, T., Dyment, D. A., Beaulieu, C. L., Schwartzentruber, J., Smith, A., et al. (2016). Utility of whole-exome sequencing for those near the end of the diagnostic odyssey: time to address gaps in care. Clin. Genet. 89 (3), 275–284. doi:10.1111/cge.12654
Schüle, B., Oviedo, A., Johnston, K., Pai, S., and Francke, U. (2005). Inactivating mutations in ESCO2 cause SC phocomelia and Roberts syndrome: no phenotype-genotype correlation. Am. J. Hum. Genet. 77 (6), 1117–1128. doi:10.1086/498695
Shashi, V., McConkie-Rosell, A., Rosell, B., Schoch, K., Vellore, K., McDonald, M., et al. (2014). The utility of the traditional medical genetics diagnostic evaluation in the context of next-generation sequencing for undiagnosed genetic disorders. Genet. Med. 16 (2), 176–182. doi:10.1038/gim.2013.99
Tomkins, D., Hunter, A., and Roberts, M. (1979). Cytogenetic findings in Roberts-SC phocomelia syndrome(s). Am. J. Med. Genet. 4 (1), 17–26. doi:10.1002/ajmg.1320040104
van der Lelij, P., Oostra, A. B., Rooimans, M. A., Joenje, H., and de Winter, J. P. (2010). Diagnostic overlap between Fanconi anemia and the cohesinopathies: Roberts syndrome and Warsaw breakage syndrome. Anemia 2010, 565268. doi:10.1155/2010/565268
Vega, H., Gordillo, M., and Jabs, E. W. (2006). “ESCO2 spectrum disorder,” in GeneReviews®. Editor M. P. Adam (Seattle: University of Washington).
Vega, H., Trainer, A. H., Gordillo, M., Crosier, M., Kayserili, H., Skovby, F., et al. (2010). Phenotypic variability in 49 cases of ESCO2 mutations, including novel missense and codon deletion in the acetyltransferase domain, correlates with ESCO2 expression and establishes the clinical criteria for Roberts syndrome. J. Med. Genet. 47 (1), 30–37. doi:10.1136/jmg.2009.068395
Vega, H., Waisfisz, Q., Gordillo, M., Sakai, N., Yanagihara, I., Yamada, M., et al. (2005). Roberts syndrome is caused by mutations in ESCO2, a human homolog of yeast ECO1 that is essential for the establishment of sister chromatid cohesion. Nat. Genet. 37 (5), 468–470. doi:10.1038/ng1548
Ward, A., Hopkins, J., Mckay, M., Murray, S., and Jordan, P. W. (2016). Genetic interactions between the meiosis-specific cohesin components, STAG3, REC8, and RAD21L. G3 (Bethesda) 6 (6), 1713–1724. doi:10.1534/g3.116.029462
Keywords: Roberts syndrome, ESCO2 spectrum disorders, cohesinopathy, ESCO2 gene, Malaysia
Citation: Tae S-K, RA M and Thong M-K (2024) Case report: The evolving phenotype of ESCO2 spectrum disorder in a 15-year-old Malaysian child. Front. Genet. 14:1286489. doi: 10.3389/fgene.2023.1286489
Received: 31 August 2023; Accepted: 26 December 2023;
Published: 15 January 2024.
Edited by:
Claudia Gonzaga-Jauregui, Universidad Nacional Autónoma de México, MexicoReviewed by:
Hiroki Ura, Kanazawa Medical University, JapanVindya Subasinghe, Birmingham Women’s and Children’s Hospital, United Kingdom
Copyright © 2024 Tae, RA and Thong. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Meow-Keong Thong, dGhvbmdta0B1bS5lZHUubXk=