 Yizhe Cheng
Yizhe Cheng Xinyu Liu†
Xinyu Liu† Limei Sun
Limei Sun Xiaoyan Ding
Xiaoyan Ding- State Key Laboratory of Ophthalmology, Zhongshan Ophthalmic Center, Sun Yat-sen University, Guangzhou, China
Background: Leukoencephalopathy and visual impairment have been linked to loss-of-function mutations in the CLCN2 gene (MIM #600570). However, the ocular features caused by the CLCN2 mutations remain poorly understood and seldom reported. This study aims to present a novel mutation and characterize the ocular phenotype in a Chinese female diagnosed with CLCN2-related leukoencephalopathy (CC2L), also known as leukoencephalopathy with ataxia (LKPAT; MIM #615651).
Case presentation: A 20-year-old Chinese female presented with bilateral blurred vision persisting for 2 years, which had worsened over the past 6 months. Ophthalmologic examination revealed bilateral post-capsular cataracts, macular retinal atrophy, and peripheral retinal pigmentation. Swept-source optical coherence tomography (SS-OCT) showed bilateral choroidal capillary atrophy, loss of the outer retinal layer, and a novel noteworthy sign of vacuole-like vitreoretinopathy. Cranial magnetic resonance imaging confirmed leukoencephalopathy. Genetic testing identified a novel homozygous pathogenic c.1382_1386del (p.P461Lfs*13) mutation in exon 13 of the CLCN2 gene.
Conclusion: This case report expands the knowledge of CLCN2 mutations and their associated ocular manifestations in patients with CC2L. The identified ophthalmic features may serve as crucial indicators for early diagnosis in individuals with CC2L, especially in the absence of evident neurological symptoms.
Introduction
CLCN2-related leukoencephalopathy (CC2L), also known as leukoencephalopathy with ataxia (LKPAT; MIM #615651), is a rare autosomal recessively inherited disease caused by CLCN2 mutations. First reported by Depienne in 2013, CC2L is characterized by cerebellar ataxia, recurrent headache, intention tremor, speech impediments and memory decline (Depienne et al., 2013). The CLCN2 gene (MIM #600570) encodes for the chloride channel 2 (ClC-2) protein, a two-pore homodimeric, voltage-gated Cl− channel found in various organs and tissues, including the brain, testis, colon, and retina (Zúñiga et al., 2004).
Disruption of ClC-2 could lead to fluid accumulation, resulting in intramyelinic edema (Jeworutzki et al., 2012). Of note, patients with CC2L not only exhibit neurologic symptoms but also manifest visual impairment, including retinal degeneration, optic neuropathy, retinoschisis and other abnormal changes. However, reports of CC2L patients with ocular manifestations remain limited, with only five cases documented in the literature (Depienne et al., 2013; Di Bella et al., 2014; Giorgio et al., 2017; Guo et al., 2019; Xu et al., 2023). In this report, we present a novel homozygous pathogenic c.1382_1386del (p.P461Lfs*13) mutation identified in exon 13 of the CLCN2 gene in a patient with CC2L. Additionally, a new and noteworthy sign, vacuole-like vitreoretinopathy, was observed in this patient. These findings expanded the genetic and clinical spectrum of CC2L, shedding further light on the ocular manifestations associated with this condition.
Case presentation
A 20-year-old woman presented with bilateral blurred vision persisting for 2 years, worsening over the last 6 months. Initial suspicion of retinitis pigmentosa (RP) was noted 2 years ago. Her medical history was unremarkable, and there was no consanguinity between her parents. She had high myopia for 8 years, with -9D in the right eye and -7D in the left eye. Best corrected visual acuity was 0.82 logMAR in the right eye and 0.70 logMAR in the left eye. The intraocular pressure was unremarkable. Bilateral post-capsular cataract, pepper-like retinal pigmentation was noted in both eyes, with white retinal vascular sheathing in the sub-temporal regions of the right eye (Figures 1A, B). Swept-source optical coherence tomography (OCT) showed bilateral choroidal capillary atrophy, loss of the outer retinal layer, especially in the ellipsoid zone. (Figures 1C–H). Fundus fluorescence angiography (FFA) showed intense macular fluorescence and increased background fluorescence. Full-field electroretinography showed moderately reduced rod and cone responses. Humphrey perimetry revealed bilateral central and temporal field defects.
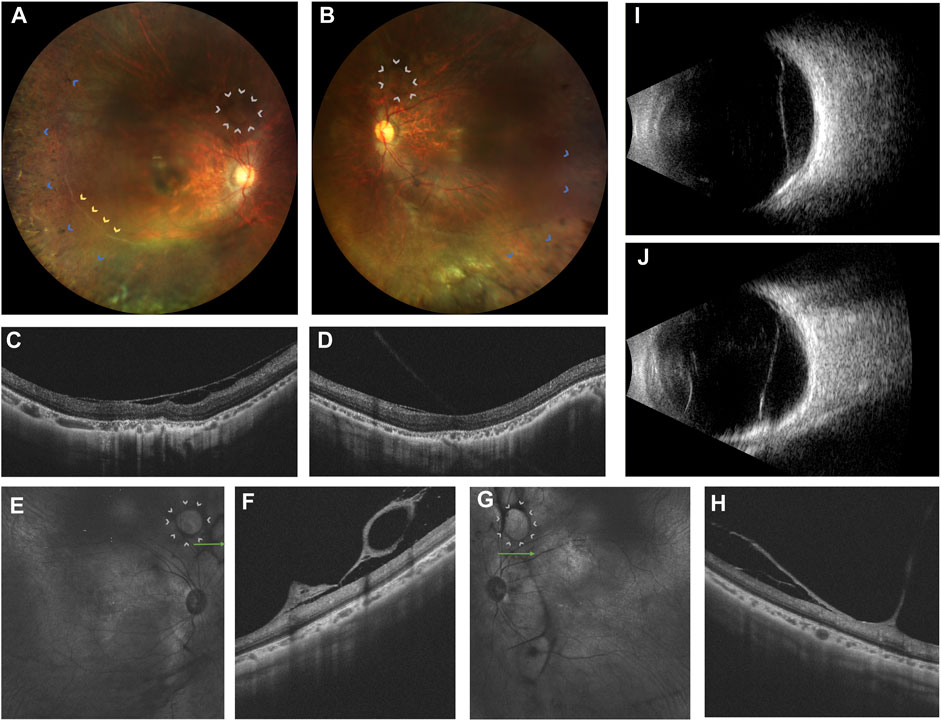
FIGURE 1. Ophthalmic multimodal imaging of the patient. (A,B) showed vacuole-like vitreoretinal fibro-cellular membrane (white arrows), vascular sheathing (yellow arrows), choroiretinal atrophy and bone spicule pigmentation (blue arrows) on fundus photograph. (C,D) showed epiretinal membrane, loss and disruption of the photoreceptor layer and choroidal atrophy. (E–H) showed hyperreflective vacuole-like vitreoretinal abnormalities B-scan optical coherence tomography and low reflectance of vacuole-like membranes on the near-infrared imaging. (I,J) showed a strong echo of a flat notch in the B-ultrasound.
Furthermore, grayish-white vacuole-like or linear fibrous epiretinal thick membranes, adhered with the retina, predominantly in the periphery, were noted. OCT revealed it as a vacuole-like vitreoretinal adhesion. Magnified OCT showed the boundary and shape of this lesion clearly. Near-infrared imaging revealed low reflectance of the vacuole-like epiretinal membrane (Figures 1E–H). Thus, we name it as vacuole-like vitreoretinopathy.
The patient did not exhibit any neurological symptoms such as headache, ataxia, poor motor ability, hearing impairment or loss, or memory decline. However, conventional magnetic resonance imaging (MRI) revealed confluent white matter abnormalities with hypointense T1-weighted and hyperintense T2-weighted signals, symmetrically involving the posterior limbs of the internal capsules, splenium of the corpus callosum, midbrain cerebral peduncles, pons, middle cerebellar peduncles (Figures 2A, B). Fluid-attenuated inversion recovery T2 and diffusion-weight imaging revealed hyperintensity in the affected areas (Figures 2C, D). The patient’s mother, father, and brother showed no abnormalities in their fundus (Supplementary Figure).
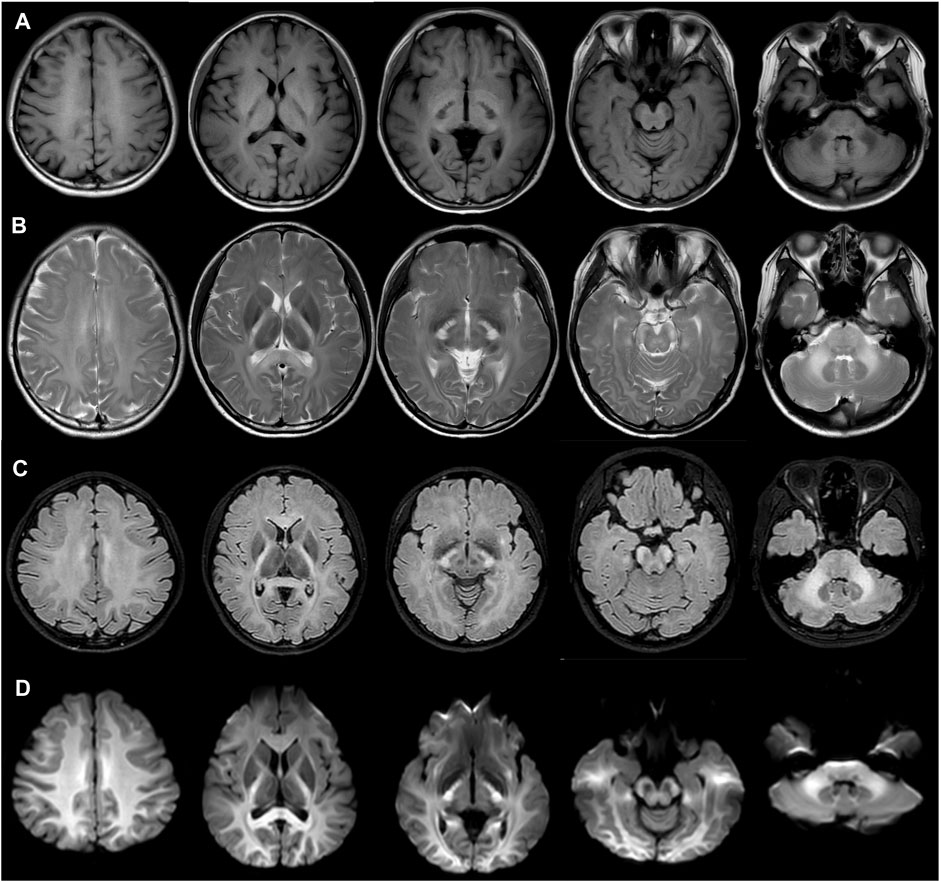
FIGURE 2. Magnetic resonance imaging showed confluent white matter abnormalities with hypointense T1-weighted (A) and hyperintense T2-weighted signals (B), with the symmetrical involvement of the posterior limbs of the internal capsules, splenium of the corpus callosum, midbrain cerebral peduncles, pons, middle cerebellar peduncles. Fluid attenuated inversion recovery T2 (C) and diffusion-weight imaging revealed hyperintensity in the involved areas. (D).
Whole exome sequencing (WES) was performed to unravel the genetic pathogenesis of this rare disease. Whole-exome sequencing revealed a novel homozygous c.1382_1386del (p.P461Lfs*13) mutation in the exon 13 of the CLCN2 gene, resulting in a frameshift mutation that changed the 461th proline to leucine and caused premature termination of protein synthesis. This mutation was not found in the genetic database and was considered pathogenic. The patient’s mother, father and brother were identified as heterozygous carriers of this mutation (Figures 3A, B).
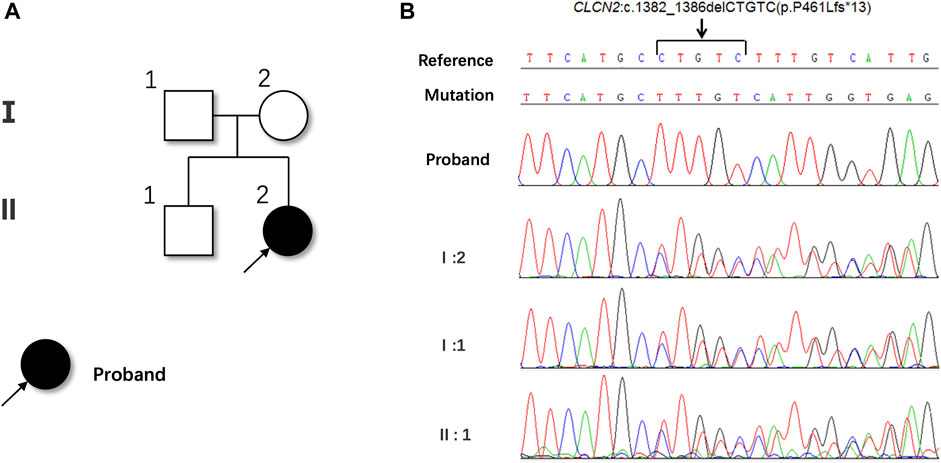
FIGURE 3. Pedigree (A) and sequencing chromatograms (B) of the family.
Material and methods
DNA extraction
Blood samples were collected from the proband and her unaffected family members. Genomic DNA was isolated as mentioned in our previous study (Tang et al., 2016).
Whole exome sequencing
Whole exome sequencing (WES) was performed to unravel the genetic pathogenesis of this rare disease. Sequencing was performed on an Illumina NovaSeq (Illumina, Madison, WI, United States), with a read length of 2 × 150 bp. Single-nucleotide variants (SNVs) and insertions and deletions (InDels) were called by GATK4.0 HC with sequencing depth set to 100-fold. Strand NGS software version 2.0 (Strand Scientific Intelligence Inc., LA, United States) was used to set the sequencing reads to University of California Santa Cruz hg19 (2009.2). The minor allele frequency was defined as less than 0.03% in the public database Genome Aggregation Database (gnomAD, http://www.gnomad-sg.org, version 2.1). The Human Gene Mutation Database (HGMD; http://www.hgmd.cf.ac.uk/ac/index.php, 2020.4), dbSNP151 (http://www.ncbi.nlm.nih.gov/SNP/, build 151), ClinVar (https://www.clinicalgenome.org/data-sharing/clinvar/) and Exome Aggregation Consortium (ExAC, http://exac.broadinstitute.org/) were also used to identify the reported pathogenic variants. The pathogenicity of missense variants was further estimated by using SIFT (ensembl 66, released 2015.01 http://sift.jcvi.org), PolyPhen-2 (version 2.2.2, released v2.2.2, released 2012.02, http://genetics.bwh.harvard.edu/pph2/) (Adzhubei et al., 2010), Mutation Taster (data retrieved in 2015, http://www.mutationtaster.org) (Steinhaus et al., 2021), PROVEAN (version 1.1, ensembl 66, released 2015.01 http://provean.jcvi.org/index.php) (Choi and Chan, 2015) and CADD (version 1.6, http://cadd.gs.washington.edu/) (Rentzsch et al., 2021) online algorithms and via evolutionary conservation analysis. The classification of sequence variants was evaluated based on the ACMG standards and guidelines (Richards et al., 2015).
Discussion
In this report, the case clinically and genetically confirmed CC2L, initially with blurred vision, without any neurological symptoms, with a homozygous pathogenic mutation in the CLCN2 gene identified by WES. CC2L, a rare syndrome, manifests with a chronic course, predominantly occurring in females. Most patients with CC2L only have mild neurologic deficits. Specific changes in MRI are important clinical clues to support the diagnosis. Since CLCN2-knocked-out mice were first reported to exhibit brain and retinal abnormalities (Bösl et al., 2001; Blanz et al., 2007), many CLCN2 variants have been identified in individuals with CC2L (Depienne et al., 2013; Di Bella et al., 2014; Hanagasi et al., 2015; Giorgio et al., 2017; Zeydan et al., 2017; Guo et al., 2019; Hoshi et al., 2019; Ngo et al., 2020; Ozaki et al., 2020; Parayil Sankaran et al., 2020). Among these individuals, only five of them exhibited ocular manifestations, including poor vision, optic neuropathy, visual field defects, retinoschisis, retinal atrophy, and normal fundi appearance with abnormal electroretinography and visual evoked potential. To date, we still know little about the ocular manifestation of CCL2.
The underlying mechanisms of CLCN2-related retinal degeneration remained elusive. Depienne et al. believe that the water-electrolyte imbalance of brain tissue caused by the loss of function in the CLCN2 gene, causing osmotic intramyelination edema, is an important cause of this characteristic MRI change (Depienne et al., 2013). The CLCN2-knocked-out mice and CNCL2nmf240 mice homozygotes both develop early-onset and severe photoreceptor degeneration, with only a single layer of photoreceptor cells remaining (Bösl et al., 2001; Edwards et al., 2010). Xu et al. found that the patient induced pluripotent stem cells (iPSC)-derived retinal pigment epithelium (RPE) cells carrying CLCN2 mutations exhibited dysfunctions of ClC-2 chloride channels and outer segment phagocytosis. Repaired by the CRISPR-CaS9 system, the aforementioned dysfunctions were rescued. The findings reported by Xu et al. suggested that the retinal degeneration caused by CLCN2 mutation may be due to RPE dysfunction. Given that, the overaccumulation of metabolic wastes may cause degeneration of retinal photoreceptors (Xu et al., 2023). However, the pathogenic mechanisms should be further investigated, due to the varied phenotypes in different variants of the CLCN2 gene.
We verified that the CLCN2 gene is associated with retinal degeneration, which should be routinely added to the clinical retinopathy genetic sequencing panel. Guo and Xu et al. reported a Chinese 38-year-old female exhibiting a similar ocular appearance to our case, including bilateral cataract, myopia, peripheral retinal pigmentation, retinal atrophy, and loss and disruption of the outer retina on OCT (Xu et al., 2023). Both our case and Guo’s case indicated that CLCN2-related retinal degeneration is an autosomal-recessive inherited retinal dystrophy with systemic abnormalities. Furthermore, we offered detailed descriptions of the novel ocular manifestation of vacuole-like vitreoretinopathy, which is displayed by fundus photography, and infrared- and B-scan OCT. The association between CLCN2 mutation and the occurrence of multiple vacuole-like vitreoretinal adhesion is not clear. We speculate the depletion of the ClC-2 may impair the transport and alter the ionic environment of glial cells and hyalocytes, causing the formation of fibro-cellular proliferation. In addition, inflammation may be another contributor to the formation of vacuole-like vitreoretinopathy (Fujiwara et al., 2016). Since our patient shares some features with RP, the mechanism of the epiretinal membrane formation in RP may hint us the underlying pathway of vacuole-like vitreoretinopathy in CCL2-related retinopathy. Fujiwara et al. retrospectively reviewed a relatively large cohort of RP patients and found that inflammation is a significant cause in association with the presence of epiretinal membrane. Nevertheless, our case showed thicker membranes, more multiple sites of vitreoretinal adhesions and a specifically vacuole-like shape of epiretinal proliferation compared to epiretinal membranes in RP (Fujiwara et al., 2016; Tan et al., 2021). Our case also showed some similarities with Wagner vitreoretinopathy. Wagner vitreoretinopathy, associated with VCAN gene, is characterized by an optically empty vitreous with avascular pre-retinal vitreous strands and veils (Meredith et al., 2007; Li et al., 2020). Progressive chorioretinal atrophy, presenile cataract, mild to moderate myopia, ectopic fovea, and retinal detachment were commonly recorded (Meredith et al., 2007). However, the preretinal vitreous strands and veils were often observed in the mid-peripheral retina (Li et al., 2020), which is different from our patient. In total, both the specific genetic background and inflammation probably contribute to the formation of vacuole-like vitreoretinopathy, though other factors like age and sex cannot be excluded. More experiments and case series studies of CCL2-related retinopathy should be conducted to explore and validate the relationship between the CLCN2 gene and retinal degeneration.
We reported the first CC2L case that initially presented with only ocular symptoms, not exhibiting any neurological symptoms. The ophthalmic findings could provide important evidence for early detection in patients with CC2L but without obvious neurological symptoms. When facing patients initially presented with ocular manifestations, MRI and WES are helpful for early diagnosis of CC2L. However, due to the limited case number and retrospective nature, the diagnostic value of vacuole-like vitreoretinopathy for CC2L needs to be validated in more CC2L patients.
In conclusion, we expanded the genotypic and phenotypic spectrum of the CLCN2 gene. We identified a novel homozygous c.1382_1386del (p.P461Lfs*13) mutation in the CLCN2 gene and found a novel ocular feature of vacuole-like vitreoretinopathy. Fundus complications should be considered during screening, diagnosis, and treatment. Our report verified that CLCN2-related retinopathy may be a kind of inherited retinal dystrophy. The identified ophthalmic features may serve as crucial indicators for early diagnosis in individuals with CC2L, especially in the absence of evident neurological symptoms.
Data availability statement
The datasets for this article are not publicly available due to concerns regarding participant/patient anonymity. Requests to access the datasets should be directed to the corresponding author.
Ethics statement
Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author contributions
YC: Conceptualization, Visualization, Writing–original draft. XL: Data curation, Writing–review and editing. LS: Conceptualization, Writing–review and editing. XD: Funding acquisition, Resources, Supervision, Visualization, Writing–review and editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This study was supported by the Construction Project of High-Level Hospitals in Guangdong Province (303020107 and 303010303058); the National Natural Science Foundation of China (82271092); Guangdong Basic and Applied Basic Research Foundation (2023A1515010430).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2023.1278961/full#supplementary-material
Abbreviations
CC2L, CLCN2-related leukoencephalopathy; FFA, fundus fluorescence angiography, MRI, magnetic resonance imaging; SS-OCT, swept-source optical coherence tomography; WES, whole-exome sequencing.
References
Adzhubei, I. A., Schmidt, S., Peshkin, L., Ramensky, V. E., Gerasimova, A., Bork, P., et al. (2010). A method and server for predicting damaging missense mutations. Nat. Methods 7, 248–249. doi:10.1038/nmeth0410-248
Blanz, J., Schweizer, M., Auberson, M., Maier, H., Muenscher, A., Hübner, C. A., et al. (2007). Leukoencephalopathy upon disruption of the chloride channel ClC-2. J. Neurosci. 27, 6581–6589. doi:10.1523/JNEUROSCI.0338-07.2007
Bösl, M. R., Stein, V., Hübner, C., Zdebik, A. A., Jordt, S. E., Mukhopadhyay, A. K., et al. (2001). Male germ cells and photoreceptors, both dependent on close cell-cell interactions, degenerate upon ClC-2 Cl(-) channel disruption. Embo J. 20, 1289–1299. doi:10.1093/emboj/20.6.1289
Choi, Y., and Chan, A. P. (2015). PROVEAN web server: a tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics 31, 2745–2747. doi:10.1093/bioinformatics/btv195
Depienne, C., Bugiani, M., Dupuits, C., Galanaud, D., Touitou, V., Postma, N., et al. (2013). Brain white matter oedema due to ClC-2 chloride channel deficiency: an observational analytical study. Lancet Neurol. 12, 659–668. doi:10.1016/S1474-4422(13)70053-X
Di Bella, D., Pareyson, D., Savoiardo, M., Farina, L., Ciano, C., Caldarazzo, S., et al. (2014). Subclinical leukodystrophy and infertility in a man with a novel homozygous CLCN2 mutation. Neurology 83, 1217–1218. doi:10.1212/WNL.0000000000000812
Edwards, M. M., Marín de Evsikova, C., Collin, G. B., Gifford, E., Wu, J., Hicks, W. L., et al. (2010). Photoreceptor degeneration, azoospermia, leukoencephalopathy, and abnormal RPE cell function in mice expressing an early stop mutation in CLCN2. Invest. Ophthalmol. Vis. Sci. 51, 3264–3272. doi:10.1167/iovs.09-4887
Fujiwara, K., Ikeda, Y., Murakami, Y., Nakatake, S., Tachibana, T., Yoshida, N., et al. (2016). Association between aqueous flare and epiretinal membrane in retinitis pigmentosa. Invest. Ophthalmol. Vis. Sci. 57, 4282–4286. doi:10.1167/iovs.16-19686
Giorgio, E., Vaula, G., Benna, P., Lo Buono, N., Eandi, C. M., Dino, D., et al. (2017). A novel homozygous change of CLCN2 (p.His590Pro) is associated with a subclinical form of leukoencephalopathy with ataxia (LKPAT). J. Neurol. Neurosurg. Psychiatry 88, 894–896. doi:10.1136/jnnp-2016-315525
Guo, Z., Lu, T., Peng, L., Cheng, H., Peng, F., Li, J., et al. (2019). CLCN2-related leukoencephalopathy: a case report and review of the literature. BMC Neurol. 19, 156. doi:10.1186/s12883-019-1390-7
Hanagasi, H. A., Bilgiç, B., Abbink, T. E., Hanagasi, F., Tüfekçioğlu, Z., Gürvit, H., et al. (2015). Secondary paroxysmal kinesigenic dyskinesia associated with CLCN2 gene mutation. Park. Relat. Disord. 21, 544–546. doi:10.1016/j.parkreldis.2015.02.013
Hoshi, M., Koshimizu, E., Miyatake, S., Matsumoto, N., and Imamura, A. (2019). A novel homozygous mutation of CLCN2 in a patient with characteristic brain MRI images - a first case of CLCN2-related leukoencephalopathy in Japan. Brain Dev. 41, 101–105. doi:10.1016/j.braindev.2018.07.011
Jeworutzki, E., López-Hernández, T., Capdevila-Nortes, X., Sirisi, S., Bengtsson, L., Montolio, M., et al. (2012). GlialCAM, a protein defective in a leukodystrophy, serves as a ClC-2 Cl(-) channel auxiliary subunit. Neuron 73, 951–961. doi:10.1016/j.neuron.2011.12.039
Li, H., Li, H., Yang, L., Sun, Z., Wu, S., and Sui, R. (2020). Clinical and genetic study on two Chinese families with Wagner vitreoretinopathy. Ophthalmic Genet. 41, 432–439. doi:10.1080/13816810.2020.1786843
Meredith, S. P., Richards, A. J., Flanagan, D. W., Scott, J. D., Poulson, A. V., and Snead, M. P. (2007). Clinical characterisation and molecular analysis of Wagner syndrome. Br. J. Ophthalmol. 91, 655–659. doi:10.1136/bjo.2006.104406
Ngo, K. J., Rexach, J. E., Lee, H., Petty, L. E., Perlman, S., Valera, J. M., et al. (2020). A diagnostic ceiling for exome sequencing in cerebellar ataxia and related neurological disorders. Hum. Mutat. 41, 487–501. doi:10.1002/humu.23946
Ozaki, A., Sasaki, M., Hiraide, T., Sumitomo, N., Takeshita, E., Shimizu-Motohashi, Y., et al. (2020). A case of CLCN2-related leukoencephalopathy with bright tree appearance during aseptic meningitis. Brain Dev. 42, 462–467. doi:10.1016/j.braindev.2020.02.008
Parayil Sankaran, B., Nagappa, M., Chiplunkar, S., Kothari, S., Govindaraj, P., Sinha, S., et al. (2020). Leukodystrophies and genetic leukoencephalopathies in children specified by exome sequencing in an expanded gene panel. J. Child. Neurol. 35, 433–441. doi:10.1177/0883073820904294
Rentzsch, P., Schubach, M., Shendure, J., and Kircher, M. (2021). CADD-Splice-improving genome-wide variant effect prediction using deep learning-derived splice scores. Genome Med. 13, 31. doi:10.1186/s13073-021-00835-9
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American college of medical genetics and Genomics and the association for molecular pathology. Genet. Med. 17, 405–424. doi:10.1038/gim.2015.30
Steinhaus, R., Proft, S., Schuelke, M., Cooper, D. N., Schwarz, J. M., and Seelow, D. (2021). MutationTaster. Nucleic Acids Res. 49, W446–W451. doi:10.1093/nar/gkab266
Tan, L., Long, Y., Li, Z., Ying, X., Ren, J., Sun, C., et al. (2021). Ocular abnormalities in a large patient cohort with retinitis pigmentosa in Western China. BMC Ophthalmol. 21, 43. doi:10.1186/s12886-020-01797-z
Tang, M., Ding, X., Li, J., Hu, A., Yuan, M., Yang, Y., et al. (2016). Novel mutations in FZD4 and phenotype-genotype correlation in Chinese patients with familial exudative vitreoretinopathy. Mol. Vis. 22, 917–932.
Xu, P., Chen, Z., Ma, J., Shan, Y., Wang, Y., Xie, B., et al. (2023). Biallelic CLCN2 mutations cause retinal degeneration by impairing retinal pigment epithelium phagocytosis and chloride channel function. Hum. Genet. 142, 577–593. doi:10.1007/s00439-023-02531-7
Zeydan, B., Uygunoglu, U., Altintas, A., Saip, S., Siva, A., Abbink, T. E. M., et al. (2017). Identification of 3 novel patients with CLCN2-related leukoencephalopathy due to CLCN2 mutations. Eur. Neurol. 78, 125–127. doi:10.1159/000478089
Keywords: CLCN2 gene, leukoencephalopathies, retinal degeneration, magnetic resonance imaging, optical coherence tomography
Citation: Cheng Y, Liu X, Sun L and Ding X (2023) Case report: A frameshift mutation in CLCN2-related leukoencephalopathy and retinopathy. Front. Genet. 14:1278961. doi: 10.3389/fgene.2023.1278961
Received: 05 September 2023; Accepted: 26 October 2023;
Published: 09 November 2023.
Edited by:
Stephen J. Bush, Xi’an Jiaotong University, ChinaCopyright © 2023 Cheng, Liu, Sun and Ding. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiaoyan Ding, ZGluZ3hpYW95YW5AZ3p6b2MuY29t
†These authors have contributed equally to this work and shared the first authorship