 Nkechi Martina Odogwu
Nkechi Martina Odogwu Clinton Hagen1
Clinton Hagen1- 1Program for Hypoplastic Left Heart Syndrome, Mayo Clinic, Rochester, MN, United States
- 2Center for Regenerative Medicine, Mayo Clinic, Rochester, MN, United States
- 3Division of General Internal Medicine, Mayo Clinic, Rochester, MN, United States
- 4Department of Molecular Pharmacology and Experimental Therapeutics, Mayo Clinic, Rochester, MN, United States
- 5Division of Pediatric Cardiology, Department of Pediatric and Adolescent Medicine, Mayo Clinic, Rochester, MN, United States
Congenital heart disease (CHD) are genetically complex and comprise a wide range of structural defects that often predispose to - early heart failure, a common cause of neonatal morbidity and mortality. Transcriptome studies of CHD in human pediatric patients indicated a broad spectrum of diverse molecular signatures across various types of CHD. In order to advance research on congenital heart diseases (CHDs), we conducted a detailed review of transcriptome studies on this topic. Our analysis identified gaps in the literature, with a particular focus on the cardiac transcriptome signatures found in various biological specimens across different types of CHDs. In addition to translational studies involving human subjects, we also examined transcriptomic analyses of CHDs in a range of model systems, including iPSCs and animal models. We concluded that RNA-seq technology has revolutionized medical research and many of the discoveries from CHD transcriptome studies draw attention to biological pathways that concurrently open the door to a better understanding of cardiac development and related therapeutic avenue. While some crucial impediments to perfectly studying CHDs in this context remain obtaining pediatric cardiac tissue samples, phenotypic variation, and the lack of anatomical/spatial context with model systems. Combining model systems, RNA-seq technology, and integrating algorithms for analyzing transcriptomic data at both single-cell and high throughput spatial resolution is expected to continue uncovering unique biological pathways that are perturbed in CHDs, thus facilitating the development of novel therapy for congenital heart disease.
1 Introduction
With a prevalence of 6–13 in 1,000 newborn babies and approximately 261,247 infant deaths recorded globally, congenital heart diseases (CHDs) remain the most common and severe birth defect (Hoffman and Kaplan, 2002; Reller et al., 2008; Leirgul et al., 2014; GBD, 2017 Congenital Heart Disease Collaborators, 2020; Bakker et al., 2019; Liu et al., 2019). Based on their clinical manifestation, there are three types of CHDs: left-sided obstruction defects, septation defects, and cyanotic heart disease (Li et al., 2014). Left-sided obstructive lesions include hypoplastic left heart syndrome (HLHS), mitral stenosis, aortic stenosis, aortic coarctation, and IAA. Septation defects mainly affect the separation of the atria (atrial septal defects, ASDs), the ventricles, (ventricular septal defects, VSDs), or both (atrioventricular septal defects, AVSDs). Cyanotic heart diseases include Tetralogy of Fallot (TOF), transposition of the great arteries (TGA), tricuspid atresia, pulmonary atresia, Ebstein’s anomaly of the tricuspid valve, DORV, persistent truncus arteriosus (PTA) and anomalous pulmonary venous connection (Bruneau, 2008).
Diagnostic capabilities and advanced surgical intervention during infancy have dramatically improved over the past decades, improving survival for several patients with CHD who survive into adulthood (McCraken et al., 2018). However, those patients have an increased risk of heart failure and other non-cardiac mortality such as diabetes and cancer through poorly understood mechanisms (Hsu and Pearson, 2009; Ntiloudi et al., 2016; Barrett-Connor et al., 2018; Friedberg and Reddy, 2019).
Next-generation sequencing (NGS) technologies have revolutionized medical research and drastically improved our understanding that genetics has a crucial role in the pathogenesis of CHDs. Whole-genome and exome sequencing analyses facilitated the identification of myriads of genes encoding transcription factors and regulatory genes related to cardiac development (Fahed et al., 2013; Morton et al., 2022). Multiple lines of evidence have also provided a robust indication that a diverse array of genes and genetic aberrations are the main drivers of a majority of CHDs (Iascone et al., 2012; Theis et al., 2015a; Theis et al., 2015b; Zaidi and Brueckner, 2017). The complex anatomy and physiology of CHDs have hindered the development of animal models of CHDs which in turn has hampered progress in the development of novel therapies and explicitly understanding the biological mechanisms of CHDs. Even so, by combining powerful genetic tools with animal models, coupled with the need for more in-depth molecular studies, researchers have and still are uncovering multigenetic backgrounds and complex molecular networks underlying the pathogenesis of CHD. As an illustration, the first study to simulate HLHS, the most severe type of CHD, deployed a mouse-forward genetic screen approach to isolate mutant mice with a hypoplastic LV. In this study, mutations not previously identified were validated by CRISPR–Cas9 genome editing in both mice and zebrafish. Further, RNA-seq analysis of the hypoplastic LV from these mice found changes in gene expression patterns related to metabolism and mitochondrial pathways, calcium signaling, cardiac muscle contraction, and dilated/hypertrophic cardiomyopathy, similar to what has been shown in human SV myocardium-based analysis, advancing our understanding of the complex genetics and mechanisms associated with SV physiology (Liu et al., 2017).
With the popularity of NGS technologies, bulk RNA sequencing (bulk RNA-seq) has generated a huge amount of data about transcriptomic alterations in congenital heart diseases (Kaynak et al., 2003; Bittel et al., 2011; Grunert et al., 2016; Zhang et al., 2021). Recently, by deploying a multi-omics approach (single-cell RNA sequencing and single nucleic RNA sequencing), the pioneering study of Hill and others discovered distinct transcriptomes from 10 pediatric CHD donor tissues, providing more insight into understanding cardiac tissue molecular biology (Hill et al., 2022). Despite several efforts in understanding the cardiac transcriptomes even at the cellular level, only a subset of CHDs transcriptomes has so far been characterized, and many particularly, the non-coding transcriptomes exhibit high spatial and temporal expression patterns and are emerging as key drivers, regulators of differentiation, development, and disease pathology. Such important spatial and temporal expression patterns are better explored and delineated by deploying even more advanced transcriptome sequencing technology such as spatial transcriptomic sequencing (Zhu, 2016; Rao et al., 2021).
In this article, we review transcriptome studies of CHDs in humans and a variety of model systems, pinpointing promising transcriptomes that are potential diagnostic biomarkers and therapeutic targets while identifying gaps in the literature for future studies. We highlight the potential of advanced transcriptome sequencing technology in studying key functional biological mechanisms in CHDs and propose perspectives for future multi-omics work that could uncover novel treatment options for CHDs. Delineating the transcriptome in CHDs in finer resolution and defining them would improve our understanding of the morphological consequences of damaging variants and the cellular deficits that contribute to lifelong adverse events in patients with CHD, and also provides the opportunity to select new, specific, and more effective therapy to treat CHDs.
2 Role of genes in the development of CHD
Congenital heart diseases arise from abnormal heart development during embryogenesis. The earliest cardiac progenitors arise from lateral plate mesoderm and are controlled by a cascade of interacting transcription factors (Srivastava, 2006). Some notable genes expressed during heart field development include: NKX2-5, TBX, HAND, NOTCH, and GATA family, and several FOX transcription factors (Figure 1). NKX2-5 is expressed at the earliest stages of cardiogenesis before the onset of myogenic differentiation, regulating cardiomyocyte differentiation and proliferation (Komuro and Izumo, 1993). NKX2-5 mutations are known to be associated with AV block and ASD (Benson et al., 1998). GATA4 and GATA6, are expressed broadly in the primitive streak mesoderm (Heikinheimo et al., 1994), while GATA5 has a more restricted expression pattern in the precardiac mesoderm (Morrisey et al., 1997). Most heart defects associated with impaired conduction are observed in individuals with mutations in GATA6, whereas GATA4 mutations more often result in septal defects and endocardial cushion defects, but not issues with conduction (Ang et al., 2016). The TBX transcription factors are expressed throughout the developing heart, especially in the developing inflow tract, atrial wall, atrial septa, and atrioventricular (AV) endocardial cushions and they play a key role in regulating cardiomyocyte identity (Li et al., 1997). Mutations in TBX20 have also been associated with CHD such as TOF. Several forkhead box (FOX) transcription factors also play important roles in heart development, with mutations leading to vascular and cardiac defects and embryonic lethality in mice and are also commonly associated with the clinical processes of congenital heart in humans (Zhu, 2016). Mutations in FOXF1, FOXC2, and FOXL1 are well-characterized causes of TOF and HLHS (Stankiewicz et al., 2009). HAND1 and HAND2 are basic helix–loop–helix (bHLH) transcription factor genes that are excellent ventricular identity markers; their expression changes correlate with altered cardiac morphogenesis (George and Firulli, 2019). A mutation in HAND2 has been associated with VSD. Many other transcription factors have also been shown to cause CHD when mutated, and the phenotypes resulting from this mutation have been reviewed here (Williams et al., 2019).
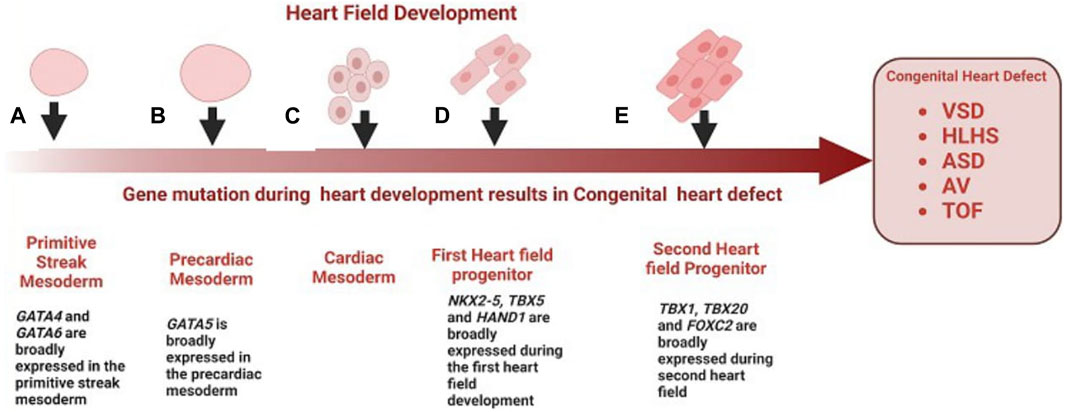
FIGURE 1. Role of genes in the development of congenital heart disease. (A) GATA4 and GATA6 are broadly expressed in the primitive streak mesoderm, mutations of GATA4 and GATA6 are a well-characterized cause of VSD (B) GATA5 is broadly expressed in the precardiac mesoderm, mutation in GATA5 have been associated with BAV and AV block (C) Cardiac mesoderm speciation give rise to first cardiac progenitors (D) First cardiac progenitors, NKX2-5, HAND1 and TBX5 are expressed at the earliest stages of cardiogenesis before the onset of myogenic differentiation, mutation in these genes during early cardiogenesis result is associated with ASV, VSD and TOF respectively (E) FOX transcription factors and TBX20 are expressed in the second heart field progenitor and throughout the developing heart, mutation of FOXF1, FOXC2 and TBX20 is associated with HLHS, and TOF.
3 Model systems for recapitulating human CHD
Following the widespread acceptance of the genetic theory of diseases, the presumption that diseases and specific traits are caused by genetic variations has remained one of the most firmly upheld doctrines of medicine. The central dogma of molecular biology which suggested that genetic information flows primarily from nucleic acids in the form of DNA and RNA to functional proteins has brought light to the mechanisms governing the specification and transmission of genetic traits (Crick, 1970). Over the past decade, there has been an increasing interest in understanding the role of genetic variation in complex traits and human disease. However, the molecular mechanisms by which this genetic variation predisposes individuals to disease are still limited, impeding the development of therapeutic interventions. Modeled after the Human Genome Project, the NIH Genotype-Tissue Expression (The GTEx Consortium Science, 2015) Project was designed to explicitly delineate the molecular biology of tissues including genetic variation, gene expression, and other molecular phenotypes in multiple human tissues thus providing useful insights into understanding the role of transcriptomes across various mammals and pinpointing the functional interpretation and insights into disease etiology in a large scale (GTEx Consortium, 2013). The National Human Genome Research Institute (NHGRI) Mouse Transcriptome Project and Mammalian Gene Collection initiative focused on generating transcriptomes and a reference expression database for the C57BL/6 J mouse further describing the important role of animal model in understanding biological complexities of diseases (Strausberg et al., 1999; Li et al., 2017). Transcriptomic analysis of cardiovascular disease has been described in a variety of model systems ranging from iPSCs, rodents (Rao et al., 2022), pigs, non-human primates, chicken embryos, and zebrafish. In recent years, the development of a variety of model systems for CHD has opened up novel frontiers in understanding the molecular basis of CHDs. Human CHDs can never be perfectly emulated in pre-clinical animal models, however since early heart failure is a common long-term complication of several types of CHDs. Animal models of right heart failure, cardiac hypertrophy, and right ventricle pressure overload are often utilized to study CHD development (Diller et al., 2015; Liu et al., 2017). For instance, murine models of RV hypertrophy and chronic RV volume overload have improved our understanding of RV-specific adaptions and single ventricle-CHDs (Urashima et al., 2008; Reddy et al., 2012; Reddy et al., 2013; Blakeslee et al., 2017). In a pulmonary blood (PBF) shunt ovine model of CHD, Tian, and others traced “angiogenesis burst” between 1 and 4 weeks of age and described how disordered angiogenesis is implicated in pulmonary vascular remodeling secondary to congenital heart diseases (CHD) (Tian et al., 2011). Mouse models of CHD represent many aspects of human cardiac development as these models maintain the anatomical arrangement of the heart, and thus provide incredible insight into the role of specific genes and cell types that contribute to proper cardiogenesis. However, mutations modeled in mice often do not recapitulate many aspects of the human phenotype. Drosophila is another animal model of choice for human cardiac disease gene testing (Schroeder et al., 2019; Vissers et al., 2020; Souidi and Jagla, 2021) because of its low cost and high throughput. However, the classical fly mutants from forward genetic screens are typically loss-of-function alleles therefore fly mutations or some transcriptional factors may not be precise representations of human mutant transcripts (Koon and Chan, 2017). hPSC-based CHD models are highly scalable human cellular models of CHD used to study the mechanisms underlying cellular defects occurring in CHDs. However, while hPSC-based CHD models are highly scalable human cellular models of CHD, which are more likely to capture human-specific biology they lack anatomical structure, these models are two-dimensional and lack the spatial context of the human heart, which is critical for studying structural defects of CHDs (Figure 2). Though expensive large animal models such as pig and non-human Primates provide a more promising and more efficient mammalian in vivo model system to identify key genes and mechanisms critical for heart development and function that served as prototypes for mammalian studies due to the high degree of conservation of genetic pathways and reduced genetic complexity. In particular, non-human Primates because of their evolutionary proximity to humans, the similarity of the two species’ cardiovascular systems can be considered a better candidate to model heart disease. Many CHDs are caused by mutations in early cardiac TFs such as GATA4, TBX5, and NKX2-5, which steer broad gene expression programs leading to changes in cell identity. Understanding how these transcriptional changes from various models’ systems can alter the transcriptional and epigenomic landscape requires multi-omic approaches, such as single-cell RNA-seq, single nucleus, and total and spatial transcriptomes which are described in subsequent sessions.
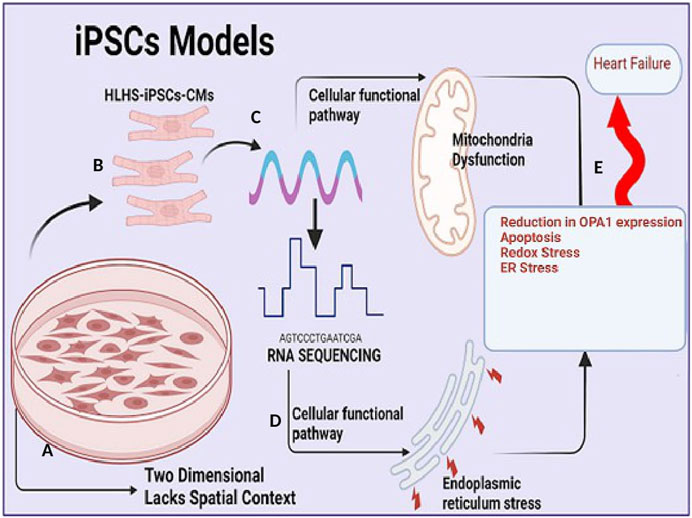
FIGURE 2. Induced pluripotent stem cell (iPSc) model in understanding congenital heart disease (A). Induced pluripotent stem cell (iPSc) model allows for large scalability of cardiomyocytes and mimics human biological mechanism but they are two dimensional and lacks spatial context (B). Cardiomyocytes (CM) cells from HLHS patients are generated and expanded from induced pluripotent stem cells (iPSc) via standardized protocol utilizing broad-spectrum pharmacological inhibitors (C). Extraction of RNA, total/bulk RNA or single cell sequencing of cardiomyocytes. (D). RNA sequencing analysis capable of identifying functional, biological pathway and signaling transduction involved in regulating mitochondria function and endoplasmic stress. (E). Reduction in Optic atrophy 1 (OPA1) expression (a mitochondria fusion protein) results in increased apoptosis (an important mechanism of cell death in heart failure (HF), redox stress and endoplasmic reticulum stress which predisposes to adult heart failure.
4 Transcriptomes in congenital heart diseases
4.1 Transfer RNA fragments: a predictive biomarker for CHD
The rapid development of high-throughput sequencing technologies has revealed that most regulatory RNAs function without involvement in protein translation (Li and Liu, 2019). Beyond the central dogma of molecular biology lies the noncoding transcriptome, in which, unlike messenger RNA (mRNA), transcribed RNA is not translated into protein. Such noncoding RNAs (ncRNAs) comprise a sizable fraction of transcriptome by mass and play crucial roles in human biology and disease (Pallazo and Lee, 2015). Coding RNAs/mRNA encodes protein to act as various components including enzymes, cell structures, and signal transductors. Transfer RNA (tRNA) are 76–90 nucleotides, that serve as the physical link between transcriptome and proteome (the mRNA and the amino acid sequence of proteins respectively) decoding information present in mRNA sequences into specific proteins (Peng et al., 2022). In cardiovascular diseases, a wide range of RNA modifications including m6A (N6-adenosine methylation), m5C (5-methylcytidin), Nm (2′-O-ribose-methylation), (pseudouridine), m7G (N7-methylguanosine), and m1A (N1-adenosine methylation) have been found in tRNA, rRNA, mRNA which are involved in metabolic syndrome, heart failure, coronary heart disease, and hypertension (Wu et al., 2019). Transfer RNAs (tRNA) are involved in gene expression regulation, protein synthesis, inhibiting translation, and signal transduction ((Xie et al., 2020). tRNA was first thought to be stable, however, recent developments in unbiased high-throughput sequencing facilitated the discovery of a new class of tRNA: tRNA halves (31–40 nucleotides) and tRNA-derived fragments (tRFs, 14–30 nucleotides) (Cole et al., 2009; Lee et al., 2009; Liao et al., 2010). While our current understanding of the functional role of transfer RNA fragment (tRF) in CHD is still in its infancy, the pioneering study of Lu et al., 2023 has shown that distinct tRNA fragments might serve as a biomarker for accurate diagnosis of CHD during pregnancy. Here, 18 tRFs/tiRNAs were selected as predictive biomarkers of CHDs. By further analysis, tRF-58:74-Gly-GCC-1 and tiRNA-1:35-Leu-CAG-1-M2 were validated as promising biomarkers of CHD (Lu et al., 2023). The onset and progress of CHD, mainly in early pregnancy, is a complex and relatively long-term process involving many genetic and epigenetic alterations. The recent study of Li and others is the only study that has explored tFRNA in CHD, future studies with large sample sizes would add another dimension to this concept and open up a new perspective to understanding the relationship between the change of tRFs/tiRNAs expression level and the time of pregnancy, the causal relationship between tRFs/tiRNAs expression level and the occurrence of CHD. Thus, additional evidence of the role of transfer RNA fragments in the pathogenesis of CHD is warranted.
4.2 Small nucleolar RNAs in congenital heart disease
Small nucleolar RNAs (snoRNAs) represent a class of regulatory RNAs responsible for telomerase activity and posttranscriptional maturation of ribosomal RNAs (rRNAs) (Caveille et al., 1996; Ganot et al., 1997). RNAs that are highly similar in structure and function to snoRNAs are the so-called small cajal body associated RNAs (scaRNAs), which assemble in cajal bodies to modify spliceosomal small nuclear (sno)RNAs (Jady et al., 2003; Cao et al., 2018; Bergstrand et al., 2022). Failure of sno/scaRNAs has been implicated in pathologies such as congenital heart anomalies, neuromuscular disorders, and various malignancies. More precisely, scaRNA has been shown to have a role in splicing, and defects in splicing may contribute to severe congenital heart anomalies (Patil et al., 2015). Current information on the functional role of sno/sna RNA in CHDs is thus scarce and only a short list of reports has provided evidence of the involvement of sno/sna in CHDs. The first attempts by Obrien et al., show that in TOF samples, the targeted nucleotides of differentially expressed snoRNAs were concentrated in the 28S, 18S ribosomal RNAs, and 2 spliceosomal RNAs (U2 and U6) (Obrien et al., 2012). They further observed splicing variants in 51% of genes in the myocardium from children with TOF, which are critical for cardiac development (Obrien et al., 2012). Using RNA-seq, Patil et al., identified 12 scaRNAs downregulated in the right ventricle of infants with TOF. However, these 12 scaRNAs affected targeted only U2 and U6 snRNAs (Patil et al., 2015). Consistent with this, the snoRNAs that were downregulated in TOF samples in the work of Ciao and others targeted the spliceosomal RNAs of the U2 spliceosome (Cao et al., 2018). Small Nucleolar RNA have been identified in CHDs caused by septation defects with a high incidence reported in VSD. Small Nucleolar RNA Host Gene 6 (SNHG6) is highly expressed in fetal cardiac tissues of VSD patients (Jiang et al., 2019). While experimentally testing differentially expressed genes they demonstrate that SNHG6 gain-of-function simultaneously blocked cardiomyocyte proliferation while enhancing apoptosis. Although the mechanism by which SNHG6 blocks cardiomyocyte proliferation is not known with certitude, SNHG6 contributes to ventricular septal defect formation via negative regulation of miR-101 and activation of Wnt/β-catenin pathway, suggesting a plausible mechanistic link between SNHG6 upregulation, impaired miR-100 expression, Wnt/β-catenin activation and the formation of VSD. Overall, these observations suggested a link between levels of snoRNA that target spliceosomal RNAs, spliceosomal function, and heart development and translation of developmentally important gene that possibly contributes to the cardiac defect.
4.3 Long noncoding RNA in congenital heart disease
Long-noncoding RNA is a heterogeneous group of non-coding transcripts more than 200 nt long that are involved in many biological processes. This class of ncRNA makes up the largest portion of the mammalian non-coding transcriptome (Mercer et al., 2009). Recent studies suggest that circulating plasma lncRNAs have significant potential as a novel diagnostic biomarker in predicting CHDs. Gu et al. (2016) identified five differentially expressed lncRNAs- ENST00000436681, ENST00000422826, AA584040, AA709223, and BX478947 across fetal CHD samples and controls suggesting that circulating plasma lncRNA may serve as novel biomarkers for CHD diagnosis (Gu et al., 2016). As aptly demonstrated (Figure 3), another line of evidence supporting the role of lncRNAs as promising biomarkers for CHDs is the detection of the high expression level of HOTAIR in right atrial biopsies of patients with ASD and VSD (Jiang et al., 2019), the validation of two candidate lncRNAs, ENST00000513542 and RP11-473L15.2 significantly associated to VSD and ASD (Song et al., 2013), and more recently the identification of a MALAT1 polymorphism associated to VSD and ASD (Li et al., 2018). Besides the role of lncRNAs in cardiac septal defect, it is hypothesized that high HA117 expression is associated with adverse outcomes in TOF patients (Wang et al., 2018). However, the mechanism of action of HA117 in TOF remains unclear. Additional studies are required to fully elucidate the functional relevance of lncRNAs in this context. Interestingly a novel lncRNA, SAP30-2:1 with an unknown function was significantly downregulated in damaged cardiac tissues from patients with CHD. Further, the knockdown of SAP30-2:1 decreased the expression of the HAND2 gene suggesting that SAP30-2:1 may be involved in heart development by targeting HAND2 and may thus represent a novel therapeutic target for CHD (Ma et al., 2021). More work is required to understand the function mechanism of lncRNA SAP30-2:1 in TOF.
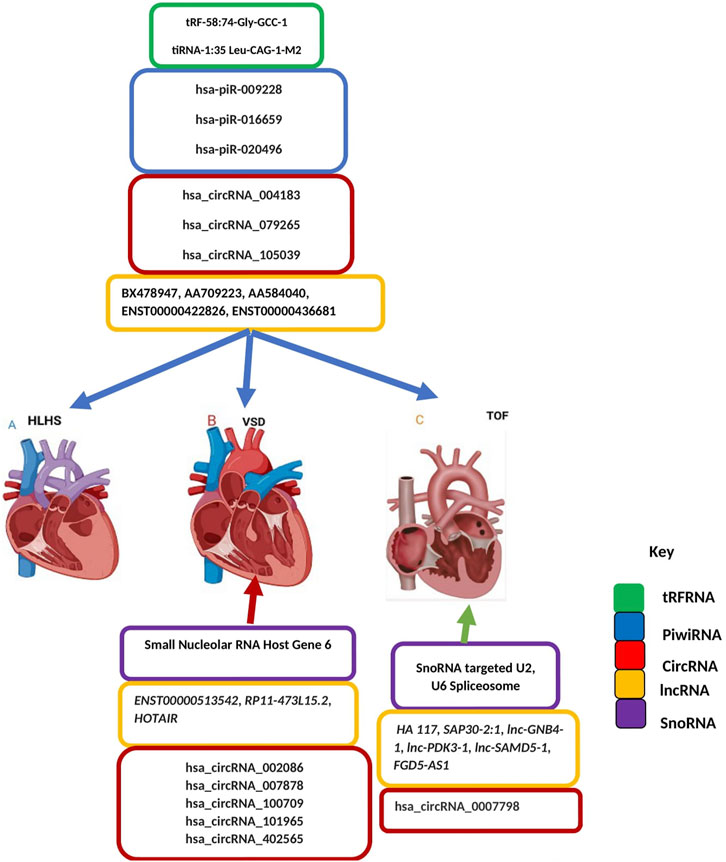
FIGURE 3. Non-coding Transcriptomes in Congenital Heart Disease. Colored boxes indicate class of non-coding RNA across each type of CHD as represented on the key (A) Left-sided obstructive lesions CHD (B) Septation defects CHD (C) Cyanotic heart diseases CHD. Blue arrow describes transfer RNA, Piwi RNA, Circular RNA, long noncoding RNA and small nucleolar RNA associated with all types of CHD including left-sided obstructive lesions CHD, septation defects CHD and cyanotic heart diseases CHD. Red an-ow indicates circular RNA, long noncoding RNA and small nucleolar RNA associated with septation defect CHD. Green arrow indicates circular RNA, long noncoding RNA and small nucleolar RNA associated with Cyanotic heart diseases CHD.
4.4 Micro RNA in congenital heart disease
miRNAs are small ncRNAs of 21- to 23-nucleotide that mediate post-transcriptional gene silencing by controlling the translation of mRNA into proteins through binding to complementary sites predominantly in the 3’ untranslated regions (UTR) of pre-messenger RNA (mRNA, protein-coding) (He and Hannon, 2004; Mendell, 2005; Treiber et al., 2019). A concise summary of micro-RNA in congenital heart disease has been reviewed elsewhere (Duenas et al., 2019). The majority of microRNA have been implicated in left-sided obstruction defects especially hypoplastic left heart syndrome (Figure 4), yet a reference micro-RNA diagnostic biomarker has not been confidently used in the clinical setting due to a limited understanding of the functional mechanism. In HLHS, three microRNA species (miR-99a, miR-100, and miR-145a) were downregulated immediately after the stage 1 operation of the Norwood procedure while values returned to control levels after stage 3 operation, indicating a strong influence of altered blood flow conditions on microRNA expression. Further, the downregulation of micro RNAs in the RV of HLHS patients, directly regulate the expression of FOG2 that modulates the expression of GATA4, GATA5, and GATA6, key players in cardiac development (Sucharov et al., 2015) The most relevant microRNA in CHDs is miR-1 (van Rooij and Olson, 2007; Wei et al., 2014). miR-1 simultaneously targets the cardiac transcription factor HAND2 to negatively control cardiogenesis and also targets histone deacetylase (HDAC) 4 to inhibit downstream MEF2 and several other regulators of cardiac growth during development leading to complex heart defects (van Rooij and Olson, 2007; Bruneau, 2008; Wei et al., 2014). In addition to experimental evidence, microRNAs are attractive clinical biomarkers as they remain stable in blood, urine, and other biological fluids and evade RNA-degrading enzymes. By identifying four significantly upregulated microRNAs (miR-19, miR-22, miR-29c, and miR-375) in mothers carrying fetuses with CHD. Yu et al. (Yu et al., 2011; Zhu et al., 2013., demonstrated, that microRNAs in maternal serum are candidate biomarkers for prenatal detection of fetal CHD in early pregnancy. Both studies also demonstrated that miR-19b and miR-29c were significantly upregulated in VSDs and all four microRNAs upregulated in TOF. Several studies have shown there is a genetic association between microRNA and the occurrence of TOF (Bittel et al., 2011; Obrien et al., 2012; Zhang et al., 2013; Liang et al., 2014; Huang et al., 2015; Low et al., 2015; Abu Halima et al., 2017; Lai et al., 2017; Grunert et al., 2019), however, the functional mechanism by which these micro-RNAs are upregulated in TOF remains to be elucidated.
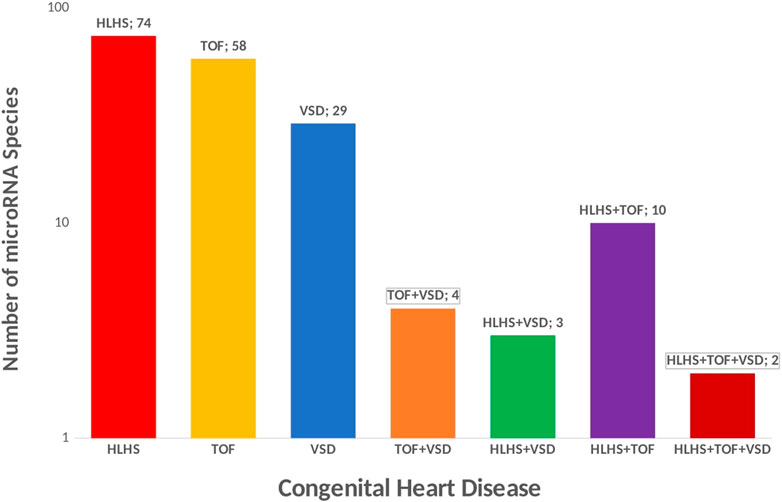
FIGURE 4. Micro RNA in Congenital Heart Disease. Micro-RNA species in CHDs could be potential biomarker for treatment. 74 unique microRNA species have been reported in HLHS, 58 unique microRNA species have been reported in TOF, 29 unique microRNA species have been reported in VSD, 4 micro-RNA species are commonly shared across CHD associated with septation defect and cyanotic heart disease (TOF and VSD), 3 micro-RNA species are commonly associated with HLHS and VSD, 2 micro-RNA species are commonly associated with HLHS, TOF and VSD.
4.5 Circular RNA in congenital heart disease
Similar to miRNAs and long noncoding RNAs (lncRNAs), circRNAs are becoming a new research hotspot in the field of RNA and could be used as a predictive tool to understand the biological mechanism of several CHDs. CircRNAs are single-stranded and created by back splicing of 3′-5′ linear coding or non-coding RNAs, forming covalently closed loops (Carrara et al., 2018). Seminal studies in VSD and TOF patients identified CircRNAs as biomarkers supporting their use as predictive tools. Liu and others identified 20 differentially expressed circRNAs in VSD and recommended 5 circular RNAs differentiated expressed in VSD myocardial tissues compared with controls. hsa_circRNA_002086 was highly expressed and hsa_circRNA_007878, hsa_circRNA_100,709, hsa_circRNA_101,965, hsa_circRNA_402,565 were underexpressed in VSD samples (Liu et al., 2018). Yu et al. provided a comprehensive understanding of the circular RNA network involved in the biology of TOF. In their work, 276 differentially expressed circRNAs; 214 upregulated and 62 downregulated circRNAs were identified in TOF samples. Additional assay validated hsa_circRNA_0007798 as a significant biomarker and therapeutic target for TOF (Yu et al., 2021). Seven differentially expressed circRNAs were identified in CHD patients. Among these 7 circRNA, hsa_circRNA_004183, hsa_circRNA_079265, and hsa_circRNA_105,039 were under-expressed in plasma from children with CHD (Wu et al., 2019). These circRNAs may be crucial in the development of CHD and may serve as novel non-invasive biomarkers for the diagnosis of CHD in children. Interestingly, individual circRNAs have a unique characteristic of inhibiting microRNAs (miRs), by binding/sponging microRNAs, thereby regulating the corresponding miR target genes (Hansen et al., 2013; Morten et al., 2022). In a recent study, hsa_circ_105,039 acted as a sponge for miR-17. miRNAs possess positive effects on the progression of CHDs, therefore by acting as a sponge for miR-17, hsa_circ_105,039 may be a promising biomarker for prognosis and therapeutic target for CHD (Yu t al., 2021). Most recently, hsa_circ_0003416 was found to be significantly downregulated in children with pulmonary arterial hypertension associated with congenital heart disease (PAH-CHD), through a poorly understood mechanism (Huang et al., 2022). Collectively, these studies provide some fundamental data that suggest the important role of circRNAs in heart development and their potential as promising predictive tools and therapeutic targets for CHD. Additional studies are needed to elucidate the functional and biological mechanism of circRNAs in the development of CHDs.
4.6 PIWI-interacting RNAs in congenital heart disease
PIWI-interacting RNAs (piRNAs) are small RNAs, 24–31 nucleotides in length with big functions, they associate with PIWI proteins to form the Piwi-piRNA pathway which mediates epigenetic programming and posttranscriptional regulation (Iwasaki et al., 2015). To date, only a limited number of reports have provided evidence of the differential expression of piRNAs in CHDs (Jia et al., 2021). Here, they identified a biomarker panel of three pregnancy-associated exosomal piRNAs (hsa-piR-009228, hsa-piR-016659, and hsa-piR-020496) which distinguished fetuses with congenital malformations from normal fetuses. However, whether they can serve as bona fide biomarkers for all classes of CHDs remains to be elucidated. Other types of ncRNAs whose biological functions are poorly defined include promoter-associated small RNAs (PASRs) (Kapranov et al., 2007), TSS-associated RNAs (TSSa-RNAs) (Seila et al., 2008), promoter upstream transcripts (PROMPTs) (Preker et al., 2008) and transcription initiation RNAs (tiRNAs) (Taft et al., 2009). No study has provided evidence of a genetic association between them and the occurrence of CHD.
5 Transcriptome sequencing technology
5.1 Insights from total RNA sequencing
The discovery of genes and variants with a potential causal link to CHD combined with analyses of gene expression during development and functional studies in model systems provide crucial evidence for assigning causality in the clinical setting (Richards et al., 2015; Strande et al., 2017). With the popularity of sequencing technologies, RNA sequencing of tissue samples (bulk RNA-seq) has generated a huge amount of data about transcriptomic alterations in cardiovascular diseases (Wang et al., 2009; Ramachandran et al., 2022). Pediatric tissue samples are rare and exceedingly difficult to obtain (Brisson et al., 2012) this has hampered or slowed down the tissue molecular biology of CHDs, limiting a thorough understanding of the biological mechanism of CHDs. Samples assayed recently were explanted cardiac tissue from heart transplant patients and donor samples (Hill et al., 2022). Two major complicating factors when analyzing human heart tissue (compared to other human tissue types) are that it contains a large proportion of fibrous tissue and has a low cell density, so disrupting the cells and extracting their total RNA is challenging. Thus, rigorous precautions must be taken to avoid degradation of RNA during its extraction, and (hence) impairment of both RNA quality and yields. Detailed RNA-seq studies of healthy and failing human myocardium have revealed remarkable similarities between upregulated genes in the failing heart and fetal myocardium (Akat et al., 2014). Therefore, considering the complication of procuring pediatric tissue to study congenital heart disease, the inability to create a perfect CHD model, and the fact that heart failure is a major long-term complication of CHD. Researchers could gain more insight into the biological mechanism of CHD by creating heart failure surgical models. Pathway analysis approach has been employed to study RNA seq data, investigate enrichment for biologically relevant pathways and functions and pinpoint differentially expressed genes in CHD. Our cross-study comparison based on data from several CHD cohorts describes some definitive transcripts, prominent differentially expressed genes, and enrichment pathways in CHD by applying bulk RNA sequencing technology (Table 1).
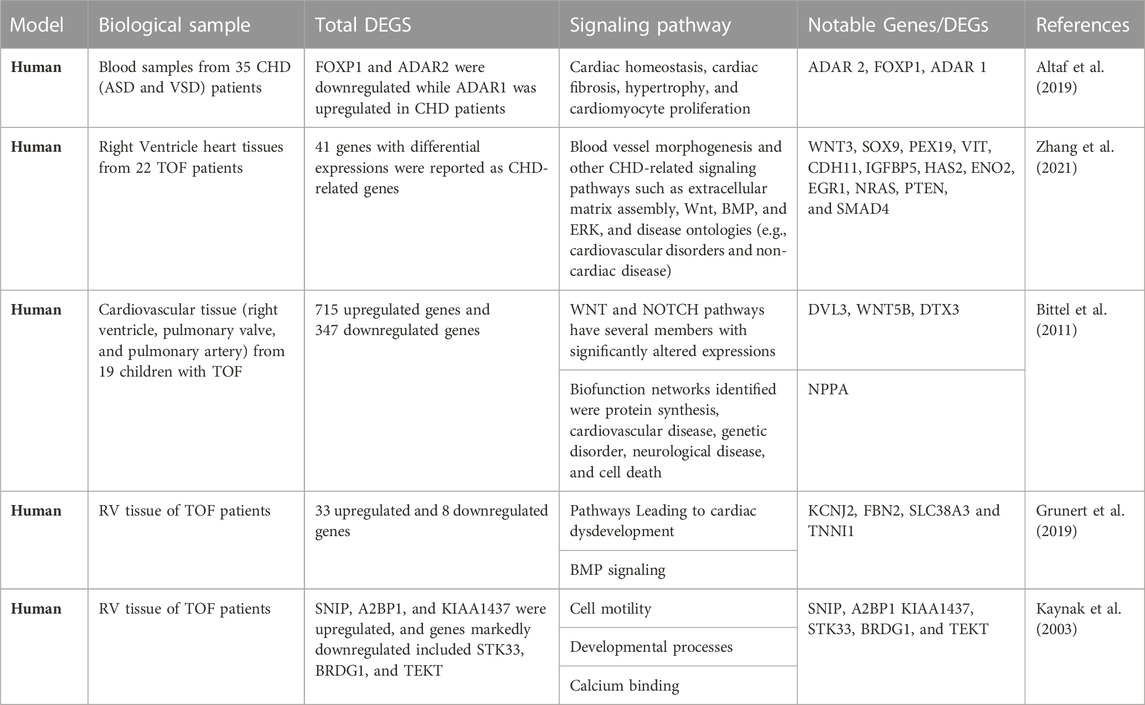
TABLE 1. Notable Genes and Functional Pathway associated with congenital heart disease in Human Patients.
5.2 Insights from single-cell transcriptomics
The advent of single-nuclear RNA sequencing and single-cell RNA sequencing (scRNA-seq) technology has enabled a detailed characterization of the many cells that populate the human mature heart (Cao et al., 2019; Litvinukova et al., 2020). Both technologies are outstanding approaches to exploring transcriptome dynamics at the resolution of a single cell (Shapiro et al., 2013) and identifying transcriptional heterogeneity between different cell lineages and distinct cell states. The healthy hearts in human and mouse reveals that heart tissue is typically composed of a variety of cell types, including but not limited to four subsets of ventricular cardiomyocytes and five subsets of atrial cardiomyocytes endothelial cell, fibroblast, macrophages, and smooth muscle cell (Litvinukova et al., 2020; Tabula Muris Consortium, 2020; Wang et al., 2021). Genes of the Notch signaling pathway are highly enriched in endocardial cells at gestational week 7, when compaction of the myocardium occurs, whereas genes related to the BMP pathway are expressed in endocardial and fibroblast-like cells from gestational week 5 to week 25, reflecting periods of endocardial-to-mesenchymal transition (Cui et al., 2019). Single-cell RNA sequencing provides a powerful tool to study DEG profiles in the cell subpopulations of interest at the single-cell level. This could enhance the understanding of the underlying mechanisms of CHD at both the cellular and molecular levels and highlight potential targets for the treatment of CHD. However, only a few studies have yet investigated the development of various categories of CHDs using scRNA-seq analysis. Among them, scRNA-seq studies of heart development and gene expression signatures in cells derived from CHD and normal control tissues have mostly been based on animal models, and less so on human cardiac tissue. scRNA-seq studies of human fetal hearts, although more limited in scope than studies on adult hearts, have identified transcriptional signatures of cardiac muscle cells (TNNI3 and TNNT2), fibroblast-like cells (COL1A1, COL1A2, and POSTN), endothelial cells (PECAM1 and KDR) and valvular cells (SOX9) (Cui et al., 2019). With scRNA-seq analyses of healthy and diseased hearts, Gladka and others found that CKAP4 could modulate the activation of fibroblasts, showing positive correlations with known myofibroblast markers (Gladka et al., 2018). Yang et al. suggested that selective expression of NEXN, an F-actin-binding protein, could lead to ASD by inhibiting GATA4 (Yang et al., 2014). Duong et al. showed that Nr2f1a is expressed in differentiated atrial cardiomyocytes and that it mediates the size of the atrial and atrial-atrioventricular canal by regulating the differentiation of atrial cardiomyocytes (Duong et al., 2018). Single-nucleus RNA sequencing analysis on 157,273 nuclei from control heart tissues and heart tissues from patients with hypoplastic left heart syndrome (HLHS), tetralogy of Fallot, and dilated and hypertrophic cardiomyopathies a recent study found CHD-specific cell states in cardiomyocytes, which showed evidence of insulin resistance and increased expression of genes associated with FOXO signaling and CRIM1. Cardiac fibroblasts in HLHS were enriched in a low-Hippo and high-YAP cell state characteristic of activated cardiac fibroblasts (Hill et al., 2022). scRNA-seq analysis offers a promising paradigm for the identification of functionally relevant pathways, validated markers, and therapeutic targets. A spectrum of changes associated with various cell subpopulations in ASD was captured in various clusters in the development of ASD. (Wang et al., 2021). Cells in cardiomyocyte clusters showed significantly higher expression of FABP4, CD36, TNNT3, and AQP1, which are markers of cardiomyocytes. Markers of endothelial cells, including SELE, ACKR1, PLVAP, DNASE1L3, and CCL14, were highly expressed in Cluster 5 also known as the endothelial cell cluster. Clusters 4 and 6 were considered smooth muscle cell clusters due to the high expression of markers RGS5, GJA4, TAGLN, ACTA2, MYL9, and SOD3. Markers of fibroblasts, including DCN, COL1A2, LUM, COL1A1, FBLN1, and TCF21, were highly expressed in Clusters 2, 3, and 7. Hence, Clusters 2, 3, and 7 were defined as fibroblast clusters. Cluster 8 was considered a macrophage cluster due to the high expression of markers AIF1, CD163, and CD68. ASD showed a decreased proportion of cardiomyocytes and an increased proportion of fibroblasts. There was more cellular crosstalk among cardiomyocytes, fibroblasts, and macrophages, especially between fibroblasts and macrophages. For all cell types, the majority of the DEGs were downregulated in ASD samples. For cardiomyocytes, there were 199 DEGs (42 upregulated and 157 downregulated) between ASD and normal samples (Wang et al., 2021).
5.3 Insights from spatial transcriptomics sequencing
Despite the many advantages of total/bulk RNA sequencing and scRNA-seq, there are several pitfalls. In standard bulk RNA-seq, whole tissue biopsies are homogenized, and only average representations of expression profiles within the entire sample are obtained. With single-cell transcriptomic sequencing, the coverage of gene expression quantification in scRNA-seq data (usually covering approximately up to 10,000 genes) is also substantially compromised in comparison with bulk RNA-seq data (usually covering 100,000 genes) (Hou et al., 2020) Further, scRNA-seq only isolate individual cells in droplets and does not preserve the tissue structure that is a fundamental component of every biological organism. As a result, information on spatial patterns of gene expression is lost and signals from subpopulations of cells with deviant profiles, such as those with low-level marker gene expression, are obscured.
To overcome these deficiencies, spatial transcriptomics (ST) technology (Ståhl et al., 2016) offers a promising paradigm enabling spatial analysis of important marker genes expressed at low levels within whole tissue sections. Spatial transcriptomic profiling provides the genomic information of single cells as they are intricately spatially organized within their native tissue environment (Li and Peng, 2022). Expanding our knowledge on the expression of transcripts meanwhile preserving the morphology of tissues will facilitate the understanding of cell type heterogeneity, cell-cell interaction, and cell fate dynamics in normal and abnormal biological contexts. As an illustration, hPSC-based CHD models which are more likely to capture Human-specific biology are two-dimensional and thus lack the spatial context of the Human heart, which is critical for studying structural defects of CHDs. Spatial transcriptomics technologies hold the promise of overcoming this limitation as transcriptomics expressions are captured while preserving spatial anatomy (Mantri et al., 2021). To the best of our knowledge, spatial mapping of transcriptional differences across ventricle and atrial tissue pediatric heart tissue in pediatric congenital heart disease has not been previously described. However, by combining single-cell and spatial transcriptomics, Stahl and others studied the development of the chicken heart from the early to late four-chambered heart stage and identified anatomically restricted expression patterns, including the expression of genes implicated in congenital heart disease. They also discovered a persistent enrichment of the small, secreted peptide, thymosin beta-4, throughout coronary vascular development and uncovered an intricate interplay between cellular differentiation and morphogenesis (Mantri et al., 2021). While SPTseq is gaining popularity in translational cardiovascular research and facilitating transcriptomics assays in the heart while preserving spatial anatomy, current spatial transcriptomics approaches lack single-cell resolution. A better approach to unraveling cellular interaction in spatially resolved gene expression entails combining both single-cell and spatial transcriptomic sequencing with algorithms for data integration. As explicitly described here (Mantri et al., 2021; Shi et al., 2022; Longo et al., 2021), spatial transcriptomics data can be integrated with the scRNA-seq data using Seurat-v3 anchor-based integration (Hie et al., 2019; Butler et al., 2018). These techniques along with the use of humans, animals, and the hPSC-based CHD model can help in understanding the development of CHD and pinpointing specific therapeutic targets.
6 Conclusion and future directions
Despite the progress of next-generation sequencing, there are still gaps in the literature regarding transcriptomic profiling in congenital heart disease and identifying consistent therapeutic markers. Relying on diagnostic biomarkers is a promising approach for the early detection of CHDs in infant blood. For example, transcriptome changes in the blood (cells or plasma) may help to better diagnose or determine the prognosis of patients. NcRNA signatures provide valuable molecular insight into patient phenotypes and could add to traditional markers and established clinical variables. LncRNA biomarkers are also being investigated as novel predictive tools to monitor therapeutic effectiveness and stratify patients. However, it cannot be excluded that there are still some CHDs that cannot be detected throughout pregnancy due to a lack of serum biomarkers for more comprehensive screening and diagnosis of CHD (McCracken et al., 2018). Understanding the functional mechanism of novel non-coding transcriptomes (described in Figure 3) could still lend a clue or open up avenues for deciphering diagnostic makers, especially for CHDs like HLHS that present with poor prognoses even after multiple surgeries. Numerous short-circulating miRNAs have been implicated in CHDs. miR-329, and miR-222 species commonly shared across all types of CHD are promising biomarkers to inform tailored treatment selection and monitor ongoing efficacy and thus deserve further investigation. Spatial transcriptomics technologies hold the promise of overcoming the limitations of single-cell transcriptomics as transcriptomics expressions are captured while preserving spatial anatomy (Mantri et al., 2021), as such its represent a powerful tool for researcher seeking to deploy embryo model for studying congenital heart disease. Additionally, generating 3D models in tissue organoids and generating a post-natal model of CHD are important topics for future research.
Author contributions
NO: Conceptualization, Data curation, Formal Analysis, Investigation, Methodology, Project administration, Resources, Software, Validation, Visualization, Writing–original draft, Writing–review and editing. CH: Funding acquisition, Investigation, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing–review and editing. TN: Conceptualization, Data curation, Formal Analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing–review and editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. The authors acknowledge the generous support from the Todd and Karen Wanek Family Program for Hypoplastic Left Heart Syndrome, at the Mayo Clinic Foundation, Rochester, MN.
Acknowledgments
The authors acknowledge the generous support from the Todd and Karen Wanek Family Program for Hypoplastic Left Heart Syndrome, at Mayo Clinic Foundation, Rochester, MN.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
CHD, congenital heart disease; ER, Endoplasmic reticulum; OPA, optic atrophy type 1 (OPA1; bHLH, Basic helix loop helix; IAA, interrupted aortic arch (IAA); TOF, Tetralogy of fallot; HLHS, Hypoplastic left heart syndrome; VSD, Ventricular septal defect; PTA, persistent truncus arteriosus; DORV, Double outlet right ventricle; CRISPR, clustered regularly interspaced short palindromic repeat; NIH, National Institute of Health; PBF, pulmonary blood flow; hPSC, Human pluripotent stem cells; FFPE, Formalin fixed paraffin embedded; DEG, Differentially expressed genes; RIN, RNA integrity number; cDNA, complementary DNA; mRNA, messenger RNA; ST, Spatial transcripome; WNT, Wingless/Integrated’ signaling; LncRNA, Long coding RNA; Sno/Sna RNA, Small nucleolar RNA; ScaRNA, Small Cajal body-specific RNA; CircRNA, Circular RNA; MiRNA, Micro RNA; piRNA, Piwi interacting RNA; RV, Right Ventricle; LV, Left ventricle.
References
Abu-Halima, M., Meese, E., Keller, A., Abdul-Khaliq, H., and Rädle-Hurst, T. (2017). Analysis of circulating microRNAs in patients with repaired Tetralogy of Fallot with and without heart failure. J. Transl. Med. 15, 156. doi:10.1186/s12967-017-1255-z
Akat, K. M., Moore-McGriff, D., Morozov, P., Brown, M., Gogakos, T., Correa Da Rosa, J., et al. (2014). Comparative RNA-sequencing analysis of myocardial and circulating small RNAs in human heart failure and their utility as biomarkers. Proc. Natl. Acad. Sci. U. S. A. 111, 11151–11156. doi:10.1073/pnas.1401724111
Altaf, F., Vesely, C., Sheikh, A. M., Munir, R., Shah, S. T. A., and Tariq, A. (2019). Modulation of ADAR mRNA expression in patients with congenital heart defects. PloS one 14 (4), e0200968. doi:10.1371/journal.pone.0200968
Ang, Y. S., Rivas, R. N., Ribeiro, A. J. S., Srivas, R., Rivera, J., Stone, N. R., et al. (2016). Disease model of GATA4 mutation reveals transcription factor cooperativity in human cardiogenesis. Cell. 167 (7), 1734–1749. doi:10.1016/j.cell.2016.11.033
Bakker, M. K., Bergman, J. E. H., Krikov, S., Amar, E., Cocchi, G., Cragan, J., et al. (2019). Prenatal diagnosis and prevalence of critical congenital heart defects: an international retrospective cohort study. BMJ Open 9, e028139. doi:10.1136/bmjopen-2018-028139
Barrett-Connor, E., Wingard, D., Wong, N., and Goldberg, R. (2018). “Heart disease and diabetes,” in Diabetes in America. Editor C. C. Cowie (USA: National Institute of Diabetes and Digestive and Kidney Diseases US).
Benson, D. W., Sharkey, A., Fatkin, D., Lang, P., Basson, C. T., McDonough, B., et al. (1998). Reduced penetrance, variable expressivity, and genetic heterogeneity of familial atrial septal defects. Circulation 97 (20), 2043–2048. doi:10.1161/01.cir.97.20.2043
Bergstrand, S., O’Brien, E. M., Coucoravas, C., Hrossova, D., Peirasmaki, D., Schmidli, S., et al. (2022). Small Cajal body-associated RNA 2 (scaRNA2) regulates DNA repair pathway choice by inhibiting DNA-PK. Nat. Commun. 13, 1015. doi:10.1038/s41467-022-28646-5
Bittel, D. C., Butler, M. G., Kibiryeva, N., Marshall, J. A., Chen, J., Lofland, G. K., et al. (2011). Gene expression in cardiac tissues from infants with idiopathic conotruncal defects. BMC Med. genomics 4, 1. doi:10.1186/1755-8794-4-1
Blakeslee, W. W., Demos-Davies, K. M., Lemon, D. D., Lutter, K. M., Cavasin, M. A., Payne, S., et al. (2017). Histone deacetylase adaptation in single ventricle heart disease and a young animal model of right ventricular hypertrophy. Pediatr. Res. 82, 642–649. doi:10.1038/pr.2017.126
Brisson, A. R., Matsui, D., Rieder, M. J., and Fraser, D. D. (2012). Translational research in pediatrics: tissue sampling and biobanking. Pediatrics 129 (1), 153–162. doi:10.1542/peds.2011-0134
Bruneau, B. G. (2008). The developmental genetics of congenital heart disease. Nature 451, 943–948. doi:10.1038/nature06801
Butler, A., Hoffman, P., Smibert, P., Papalexi, E., and Satija, R. (2018). Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nature biotechnology 36 (5), 411–420. doi:10.1038/nbt.4096
Cao, J., Spielmann, M., Qiu, X., Huang, X., Ibrahim, D. M., Hill, A. J., et al. (2019). The single-cell transcriptional landscape of mammalian organogenesis. Nature 566 (7745), 496–502. doi:10.1038/s41586-019-0969-x
Cao, T., Rajasingh, S., Samanta, S., Dawn, B., Bittel, D. C., and Rajasingh, J. (2018). Biology and clinical relevance of noncoding sno/scaRNAs. Trends Cardiovasc. Med. 28 (2), 81–90. doi:10.1016/j.tcm.2017.08.002
Carrara, M., Fuschi, P., Ivan, C., and Martelli, F. (2018). Circular RNAs: methodological challenges and perspectives in cardiovascular diseases. J. Cell. Mol. Med. 22, 5176–5187. doi:10.1111/jcmm.13789
Cavaillé, J., Nicoloso, M., and Bachellerie, J. P. (1996). Targeted ribose methylation of RNA in vivo directed by tailored antisense RNA guides. Nature 383 (6602), 732–735. doi:10.1038/383732a0
Cole, C., Sobala, A., Lu, C., Thatcher, S. R., Bowman, A., Brown, J. W., et al. (2009). Filtering of deep sequencing data reveals the existence of abundant Dicer-dependent small RNAs derived from tRNAs. RNA 15, 2147–2160. doi:10.1261/rna.1738409
Crick, F. (1970). Central dogma of molecular biology. Nature 227 (5258), 561–563. doi:10.1038/227561a0
Cui, Y., Zheng, Y., Liu, X., Yan, L., Fan, X., Yong, J., et al. (2019). Single-cell transcriptome analysis maps the developmental track of the human heart. Cell. Rep. 26 (7), 1934–1950. doi:10.1016/j.celrep.2019.01.079
Diller, G. P., Kempny, A., Alonso-Gonzalez, R., Swan, L., Uebing, A., Li, W., et al. (2015). Survival prospects and circumstances of death in contemporary adult congenital heart disease patients under follow-up at a large tertiary centre. Circulation 132 (22), 2118–2125. doi:10.1161/CIRCULATIONAHA.115.017202
Dueñas, A., Expósito, A., Aranega, A., and Franco, D. (2019). The role of non-coding RNA in congenital heart diseases. J. Cardiovasc. Dev. Dis. 6 (2), 15. doi:10.3390/jcdd6020015
Duong, T. B., Ravisankar, P., Song, Y. C., Gafranek, J. T., Rydeen, A. B., Dohn, T. E., et al. (2018). Nr2f1a balances atrial chamber and atrioventricular canal size via BMP signaling-independent and -dependent mechanisms. Dev. Biol. 434 (1), 7–14. doi:10.1016/j.ydbio.2017.11.010
Fahed, A. C., Gelb, B. D., Seidman, J. G., and Seidman, C. E. (2013). Genetics of congenital heart disease: the glass half empty. Circ. Res. 112, 707–720. doi:10.1161/CIRCRESAHA.112.300853
Friedberg, M. K., and Reddy, S. (2019). Right ventricular failure in congenital heart disease. Curr. Opin. Pediatr. 31, 604–610. doi:10.1097/MOP.0000000000000804
Ganot, P., Bortolin, M.-L., and Kiss, T. (1997). Site-specific pseudouridine formation in preribosomal RNA is guided by small nucleolar RNAs. Cell. 89 (5), 799–809. doi:10.1016/S0092-8674(00)80263-9
GBD 2017 Congenital Heart Disease Collaborators (2020). Global, regional, and national burden of congenital heart disease, 1990-2017: a systematic analysis for the Global Burden of Disease Study 2017. Child Adolesc. health 4 (3), 185–200. doi:10.1016/S2352-4642(19)30402-X
George, R. M., and Firulli, A. B. (2019). Hand factors in cardiac development. Anat. Rec. Hob. N. J. 2007) 302 (1), 101–107. doi:10.1002/ar.23910
Gladka, M. M., Molenaar, B., de Ruiter, H., van der Elst, S., Tsui, H., Versteeg, D., et al. (2018). Single-cell sequencing of the healthy and diseased heart reveals cytoskeleton-associated protein 4 as a new modulator of fibroblasts activation. Circulation 138 (2), 166–180. doi:10.1161/CIRCULATIONAHA.117.030742
Grunert, M., Appelt, S., Dunkel, I., Berger, F., and Sperling, S. R. (2019). Altered microRNA and target gene expression related to Tetralogy of Fallot. Sci. Rep. 9 (1), 19063. doi:10.1038/s41598-019-55570-4
Grunert, M., Dorn, C., Cui, H., Dunkel, I., Schulz, K., Schoenhals, S., et al. (2016). Comparative DNA methylation and gene expression analysis identifies novel genes for structural congenital heart diseases. Cardiovasc. Res. 112 (1), 464–477. doi:10.1093/cvr/cvw195
GTEx Consortium (2013). The genotype-tissue expression (GTEx) project. Nat. Genet. 45, 580–585. doi:10.1038/ng.2653
Gu, M., Zheng, A., Tu, W., Zhao, J., Li, L., Li, M., et al. (2016). Circulating LncRNAs as novel, non-invasive biomarkers for prenatal detection of fetal congenital heart defects. Cell. Physiol. biochem. 38, 1459–1471. doi:10.1159/000443088
Hansen, T. B., Jensen, T. I., Clausen, B. H., Bramsen, J. B., Finsen, B., Damgaard, C. K., et al. (2013). Natural RNA circles function as efficient microRNA sponges. Nature 495 (7441), 384–388. doi:10.1038/nature11993
He, L., and Hannon, G. J. (2004). MicroRNAs: small RNAs with a big role in gene regulation. Nat. Rev. Genet. 5, 522–531. doi:10.1038/nrg1379
Heikinheimo, M., Scandrett, J. M., and Wilson, D. B. (1994). Localization of transcription factor GATA-4 to regions of the mouse embryo involved in cardiac development. Dev. Biol. 164, 361–373. doi:10.1006/dbio.1994.1206
Hie, B, Bryson, B., and Berger, B. (2019). Efficient integration of heterogeneous 122 single-cell transcriptomes using Scanorama. Nature biotechnology 37 (6), 685–691. doi:10.1038/s41587-019-0113-3
Hill, M. C., Kadow, Z. A., Long, H., Morikawa, Y., Martin, T. J., Birks, E. J., et al. (2022). Integrated multi-omic characterization of congenital heart disease. Nature 608 (7921), 181–191. doi:10.1038/s41586-022-04989-3
Hoffman, J. L., and Kaplan, S. (2002). The incidence of congenital heart disease. J. Am. Coll. Cardiol. 39 (12), 1890–1900. doi:10.1016/s0735-1097(02)01886-7
Hou, W, Ji, Z., Ji, H., and Hicks, S. C. (2020). A systematic evaluation of single-cell RNA-sequencing imputation methods. Genome biology 21 (1), 218. doi:10.1186/s13059-020-02132-x
Hsu, D. T., and Pearson, G. D. (2009). Heart failure in children: part II: diagnosis, treatment, and future directions. Circ. Heart. Fail. 2, 490–498. doi:10.1161/CIRCHEARTFAILURE.109.856229
Huang, J. B., Mei, J., Jiang, L. Y., Jiang, Z. L., Liu, H., Zhang, J. W., et al. (2015). MiR-196a2 rs11614913 T>C polymorphism is associated with an increased risk of tetralogy of Fallot in a Chinese population. Acta Cardiol. Sin. 31, 18–23. doi:10.6515/acs20140310b
Huang, Y., Su, D., Ye, B., Huang, Y., Qin, S., Chen, C., et al. (2022). Expression and clinical significance of circular RNA hsa_circ_0003416 in pediatric pulmonary arterial hypertension associated with congenital heart disease. J. Clin. laboratory analysis 36 (4), e24273. doi:10.1002/jcla.24273
Iascone, M., Ciccone, R., Galletti, L., Marchetti, D., Seddio, F., Lincesso, A. R., et al. (2012). Identification of de novo mutations and rare variants in hypoplastic left heart syndrome. Clin. Genet. 81, 542–554. doi:10.1111/j.1399-0004.2011.01674.x
Iwasaki, Y. W., Siomi, M. C., and Siomi, H. (2015). PIWI-interacting RNA: its biogenesis and functions. Annu. Rev. Biochem. 84, 405–433. doi:10.1146/annurev-biochem-060614-034258
Jády, B. E., Darzacq, X., Tucker, K. E., Matera, A. G., Bertrand, E., and Kiss, T. (2003). Modification of Sm small nuclear RNAs occurs in the nucleoplasmic Cajal body following import from the cytoplasm. EMBO J. 22 (8), 1878–1888. doi:10.1093/emboj/cdg187
Jarlstad Olesen, M. T., and Kristensen, L. S. (2021). Circular RNAs as microRNA sponges: evidence and controversies. Essays Biochem. 65 (4), 685–696. doi:10.1042/EBC20200060
Jia, S., Zhang, Q., Wang, Y., Wang, Y., Liu, D., He, Y., et al. (2021). PIWI-interacting RNA sequencing profiles in maternal plasma-derived exosomes reveal novel non-invasive prenatal biomarkers for the early diagnosis of nonsyndromic cleft lip and palate. EBioMedicine 65, 103253. doi:10.1016/j.ebiom.2021.103253
Jiang, Y., Zhuang, J., Lin, Y., Wang, X., Chen, J., and Han, F. (2019). Long noncoding RNA SNHG6 contributes to ventricular septal defect formation via negative regulation of miR-101 and activation of Wnt/β-catenin pathway. Die Pharm. 74 (1), 23–28. doi:10.1691/ph.2019.8736
Kapranov, P., Cheng, J., Dike, S., Nix, D. A., Duttagupta, R., Willingham, A. T., et al. (2007). RNA maps reveal new RNA classes and a possible function for pervasive transcription. Sci. (New York, N.Y.) 316 (5830), 1484–1488. doi:10.1126/science.1138341
Kaynak, B., von Heydebreck, A., Mebus, S., Seelow, D., Hennig, S., Vogel, J., et al. (2003). Genome-wide array analysis of normal and malformed human hearts. Circulation 107 (19), 2467–2474. doi:10.1161/01.CIR.0000066694.21510.E2
Komuro, I., and Izumo, S. (1993). Csx: a murine homeobox-containing gene specifically expressed in the developing heart. Proc. Natl. Acad. Sci. 90, 8145–8149. doi:10.1073/pnas.90.17.8145
Koon, A. C., and Chan, H. Y. E. (2017). Drosophila melanogaster as a model organism to study RNA toxicity of repeat expansion-associated neurodegenerative and neuromuscular diseases. Front. Cell. Neurosci. 11, 70. doi:10.3389/fncel.2017.00070
Koutsourakis, M., Langeveld, A., Patient, R., Beddington, R., and Grosveld, F. (1999). The transcription factor GATA6 is essential for early extraembryonic development. Development 126, 723–732. doi:10.1242/dev.126.9.723
Krishna, M. R., and Kumar, R. K. (2020). Diagnosis and management of critical congenital heart diseases in the newborn. Indian J. Pediatr. 87, 365–371. doi:10.1007/s12098-019-03163-4
Lai, C. T. M., Ng, E. K. O., Chow, P. C., Kwong, A., and Cheung, Y. F. (2017). Circulating MicroRNA in patients with repaired tetralogy of Fallot. Eur. J. Clin. Investig. 47, 574–582. doi:10.1111/eci.12778
Lee, Y. S., Shibata, Y., Malhotra, A., and Dutta, A. (2009). A novel class of small RNAs: tRNA-derived RNA fragments (tRFs). Genes. Dev. 23, 2639–2649. doi:10.1101/gad.1837609
Leirgul, E., Fomina, T., Brodwall, K., Greve, G., Holmstrøm, H., Vollset, S. E., et al. (2014). Birth prevalence of congenital heart defects in Norway 1994–2009-A nationwide study. Am. Heart J. 168, 956–964. doi:10.1016/j.ahj.2014.07.030
Li, B., Qing, T., Zhu, J., Wen, Z., Yu, Y., Fukumura, R., et al. (2017). A comprehensive mouse transcriptomic BodyMap across 17 tissues by RNA-seq. Sci. Rep. 7, 4200. doi:10.1038/s41598-017-04520-z
Li, D., Ji, L., Liu, L., Liu, Y., Hou, H., Yu, K., et al. (2014). Characterization of circulating microRNA expression in patients with a ventricular septal defect. PLoS ONE 9, e106318. doi:10.1371/journal.pone.0106318
Li, J., and Liu, C. (2019). Coding or noncoding, the converging concepts of RNAs. Front. Genet. 10, 496. doi:10.3389/fgene.2019.00496
Li, Q., Zhu, W., Zhang, B., Wu, Y., Yan, S., Yuan, Y., et al. (2018). The MALAT1 gene polymorphism and its relationship with the onset of congenital heart disease in Chinese. Biosci. Rep. 38, BSR20171381. doi:10.1042/BSR20171381
Li, Q. Y., Newbury-Ecob, R. A., Terrett, J. A., Wilson, D. I., Curtis, A. R. J., Yi, C. H., et al. (1997). Holt-Oram syndrome is caused by mutations in TBX5, a member of the Brachyury (T) gene family. Nat. Genet. 15, 21–29. doi:10.1038/ng0197-21
Li, Z., and Peng, G. (2022). Spatial transcriptomics: new dimension of understanding biological complexity. Biophys. Rep. 8 (3), 119–135. doi:10.52601/bpr.2021.210037
Liang, D., Xu, X., Deng, F., Feng, J., Zhang, H., Liu, Y., et al. (2014). miRNA-940 reduction contributes to human Tetralogy of Fallot development. J. Cell. Mol. Med. 18, 1830–1839. doi:10.1111/jcmm.12309
Liao, J. Y., Ma, L. M., Guo, Y. H., Zhang, Y. C., Zhou, H., Shao, P., et al. (2010). Deep sequencing of human nuclear and cytoplasmic small RNAs reveals an unexpectedly complex subcellular distribution of miRNAs and tRNA 3′ trailers. PLoS ONE 5, e10563. doi:10.1371/journal.pone.0010563
Litviňuková, M., Talavera-López, C., Maatz, H., Reichart, D., Worth, C. L., Lindberg, E. L., et al. (2020). Cells of the adult human heart. Nature 588 (7838), 466–472. doi:10.1038/s41586-020-2797-4
Liu, H., Hu, Y., Zhuang, B., Yin, J., Chen, X. H., Wang, J., et al. (2018). Differential expression of CircRNAs in embryonic heart tissue associated with ventricular septal defect. Int. J. Med. Sci. 15 (7), 703–712. doi:10.7150/ijms.21660
Liu, X., Yagi, H., Saeed, S., Bais, A. S., Gabriel, G. C., Chen, Z., et al. (2017). The complex genetics of hypoplastic left heart syndrome. Nat. Genet. 49, 1152–1159. doi:10.1038/ng.3870
Liu, Y., Chen, S., Zühlke, L., Black, G. C., and Choy, M. K. (2019). Global birth prevalence of congenital heart defects 1970–2017: updated systematic review and meta-analysis of 260 studies. Int. J. Epidemiol. 48, 455–463. doi:10.1093/ije/dyz009
Longo, S. K., Guo, M. G., Ji, A. L., and Khavari, P. A. (2021). Integrating single-cell and spatial transcriptomics to elucidate intercellular tissue dynamics. Nature reviews. Genetics, 22 (10), 627–644. doi:10.1038/s41576-021-00370-8
Low, K. J., Buxton, C. C., and Newbury-Ecob, R. A. (2015). Tetralogy of Fallot, microcephaly, short stature, and brachymesophalangy is associated with hemizygous loss of noncoding MIR17HG and coding GPC5. Clin. Dysmorphol. 24, 113–114. doi:10.1097/MCD.0000000000000069
Lu, E., Wu, L., Chen, B., Xu, S., Fu, Z., Wu, Y., et al. (2023). Maternal serum tRNA-derived fragments (tRFs) as potential candidates for diagnosis of fetal congenital heart disease. J. Cardiovasc. Dev. Dis. 10 (2), 78. doi:10.3390/jcdd10020078
Ma, J., Chen, S., Hao, L., Sheng, W., Chen, W., Ma, X., et al. (2021). Long non-coding RNA SAP30-2:1 is downregulated in congenital heart disease and regulates cell proliferation by targeting HAND2. Front. Med. 15 (1), 91–100. doi:10.1007/s11684-020-0778-5
Mantri, M., Scuderi, G. J., Abedini-Nassab, R., Wang, M. F. Z., McKellar, D., Shi, H., et al. (2021). Spatiotemporal single-cell RNA sequencing of developing chicken hearts identifies interplay between cellular differentiation and morphogenesis. Nat. Commun. 12 (1), 1771. doi:10.1038/s41467-021-21892-z
McCracken, C., Spector, L. G., Menk, J. S., Knight, J. H., Vinocur, J. M., Thomas, A. S., et al. (2018). Mortality following pediatric congenital heart surgery: an analysis of the causes of death derived from the national death index. J. Am. Heart Assoc. 7 (22), e010624. doi:10.1161/JAHA.118.010624
Mendell, J. T. (2005). MicroRNAs: critical regulators of development, cellular physiology, and malignancy. Cell. Cycle 4, 1179–1184. doi:10.4161/cc.4.9.2032
Mercer, T. R., Dinger, M. E., and Mattick, J. S. (2009). Long non-coding RNAs: insights into functions. Nat. Rev. Genet. 10, 155–159. doi:10.1038/nrg2521
Morrisey, E. E., Ip, H. S., Tang, Z., Lu, M. M., and Parmacek, M. S. (1997). GATA-5: a transcriptional activator expressed in a novel temporally and spatially-restricted pattern during embryonic development. Dev. Biol. 183, 21–36. doi:10.1006/dbio.1996.8485
Morton, S. U., Quiat, D., Seidman, J. G., and Seidman, C. E. (2022). Genomic frontiers in congenital heart disease. Nat. Rev. Cardiol. 19 (1), 26–42. doi:10.1038/s41569-021-00587-4
Ntiloudi, D., Giannakoulas, G., Parcharidou, D., Panagiotidis, T., Gatzoulis, M. A., and Karvounis, H. (2016). Adult congenital heart disease: a paradigm of epidemiological change. Int. J. Cardiol. 218, 269–274. doi:10.1016/j.ijcard.2016.05.046
O'Brien, J. E., Kibiryeva, N., Zhou, X. G., Marshall, J. A., Lofland, G. K., Artman, M., et al. (2012). Noncoding RNA expression in myocardium from infants with tetralogy of Fallot. Cardiovasc. Genet. 5 (3), 279–286. doi:10.1161/CIRCGENETICS.111.961474
Palazzo, A. F., and Lee, E. S. (2015). Non-coding RNA: what is functional and what is junk? Front. Genet. 6, 2. doi:10.3389/fgene.2015.00002
Patil, P., Kibiryeva, N., Uechi, T., Marshall, J., O'Brien, J. E., Artman, M., et al. (2015). scaRNAs regulate splicing and vertebrate heart development. Biochimica biophysica acta 1852 (8), 1619–1629. doi:10.1016/j.bbadis.2015.04.016
Peng, R., Santos, H. J., and Nozaki, T. (2022). Transfer RNA-derived small RNAs in the pathogenesis of parasitic Protozoa. Genes. 13 (2), 286. doi:10.3390/genes13020286
Preker, P., Nielsen, J., Kammler, S., Lykke-Andersen, S., Christensen, M. S., Mapendano, C. K., et al. (2008). RNA exosome depletion reveals transcription upstream of active human promoters. Science 322, 1851–1854. doi:10.1126/science.1164096
Ramachandran, V., Bhagavatheeswaran, S., Shanmugam, S., Vasudevan, M., Ragunathan, M., Cherian, K. M., et al. (2022). Deep sequencing unveils altered cardiac miRNome in congenital heart disease. Mol. Genet. genomics MGG 297 (4), 1123–1139. doi:10.1007/s00438-022-01908-z
Rao, A., Barkley, D., França, G. S., and Yanai, I. (2021). Exploring tissue architecture using spatial transcriptomics. Nature 596 (7871), 211–220. doi:10.1038/s41586-021-03634-9
Rao, K. S., Kameswaran, V., and Bruneau, B. G. (2022). Modeling congenital heart disease: lessons from mice, hPSC-based models, and organoids. Genes. & Dev. 36 (11-12), 652–663. doi:10.1101/gad.349678.122
Reddy, S., Zhao, M., Hu, D. Q., Fajardo, G., Hu, S., Ghosh, Z., et al. (2012). Dynamic microRNA expression during the transition from right ventricular hypertrophy to failure. Physiol. Genomics 44, 562–575. doi:10.1152/physiolgenomics.00163.2011
Reddy, S., Zhao, M., Hu, D. Q., Fajardo, G., Katznelson, E., Punn, R., et al. (2013). Physiologic and molecular characterization of a murine model of right ventricular volume overload. Am. J. Physiol. Heart Circ. Physiol. 304, H1314–H1327. doi:10.1152/ajpheart.00776.2012
Reller, M. D., Strickland, M. J., Riehle-Colarusso, T., Mahle, W. T., and Correa, A. (2008). Prevalence of congenital heart defects in metropolitan Atlanta, 1998-2005. J. Pediatr. 153, 807–813. doi:10.1016/j.jpeds.2008.05.059
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genet. Med. official J. Am. Coll. Med. Genet. 17 (5), 405–424. doi:10.1038/gim.2015.30
Schroeder, A. M., Allahyari, M., Vogler, G., Missinato, M. A., Nielsen, T., Yu, M. S., et al. (2019). Model system identification of novel congenital heart disease gene candidates: focus on RPL13. Hum. Mol. Genet. 28, 3954–3969. doi:10.1093/hmg/ddz213
Seila, A. C., Calabrese, J. M., Levine, S. S., Yeo, G. W., Rahl, P. B., Flynn, R. A., et al. (2008). Divergent transcription from active promoters. Science 322, 1849–1851. doi:10.1126/science.1162253
Shapiro, E., Biezuner, T., and Linnarsson, S. (2013). Single-cell sequencing-based technologies will revolutionize whole-organism science. Nat. Rev. Genet. 14 (9), 618–630. doi:10.1038/nrg3542
Shi, X., Zhang, L., Li, Y., Xue, J., Liang, F., and Ni, H. W. (2022). Integrative Analysis of Bulk and Single-Cell RNA Sequencing Data Reveals Cell Types Involved in Heart Failure. Frontiers in bioengineering and biotechnology, 9, 779225. doi:10.3389/fbioe.2021.779225
Song, G., Shen, Y., Zhu, J., Liu, H., Liu, M., Shen, Y. Q., et al. (2013). Integrated analysis of dysregulated lncRNA expression in fetal cardiac tissues with ventricular septal defect. PLoS ONE 8, e77492. doi:10.1371/journal.pone.0077492
Souidi, A., and Jagla, K. (2021). Drosophila heart as a model for cardiac development and diseases. Cells 10 (11), 3078. doi:10.3390/cells10113078
Srivastava, D. (2006). Making or breaking the heart: from lineage determination to morphogenesis. Cell. 126, 1037–1048. doi:10.1016/j.cell.2006.09.003
Ståhl, P. L, Salmén, F., Vickovic, S., Lundmark, A., Navarro, J. F, Magnusson, J., et al. (2016). Visualization and analysis of gene expression in tissue sections by spatial transcriptomics. Science 353, 78–82. doi:10.1126/science.aaf2403
Stankiewicz, P., Sen, P., Bhatt, S. S., Storer, M., Xia, Z., Bejjani, B. A., et al. (2009). Genomic and genic deletions of the FOX gene cluster on 16q24.1 and inactivating mutations of FOXF1 cause alveolar capillary dysplasia and other malformations. Am. J. Hum. Genet. 84, 780–791. doi:10.1016/j.ajhg.2009.05.005
Strande, N. T., Riggs, E. R., Buchanan, A. H., Ceyhan-Birsoy, O., DiStefano, M., Dwight, S. S., et al. (2017). Evaluating the clinical validity of gene-disease associations: an evidence-based framework developed by the clinical genome resource. Am. J. Hum. Genet. 100 (6), 895–906. doi:10.1016/j.ajhg.2017.04.015
Strausberg, R. L., Feingold, E. A., Klausner, R. D., and Collins, F. S. (1999). The mammalian gene collection. Sci. (New York, N.Y.) 286 (5439), 455–457. doi:10.1126/science.286.5439.455
Sucharov, C. C., Sucharov, J., Karimpour-Fard, A., Nunley, K., Stauffer, B. L., and Miyamoto, S. D. (2015). Micro-RNA expression in hypoplastic left heart syndrome. J. Card. Fail. 21, 83–88. doi:10.1016/j.cardfail.2014.09.013
Tabula Muris Consortium (2020). A single-cell transcriptomic atlas characterizes ageing tissues in the mouse. Nature 583 (7817), 590–595. doi:10.1038/s41586-020-2496-1
Taft, R. J., Glazov, E. A., Cloonan, N., Simons, C., Stephen, S., Faulkner, G. J., et al. (2009). Tiny RNAs associated with transcription start sites in animals. Nat. Genet. 41, 572–578. doi:10.1038/ng.312
The GTEx Consortium Science (2015). Human genomics. The Genotype-Tissue Expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science 348, 648–660. doi:10.1126/science.1262110
Theis, J. L., Hrstka, S. C., Evans, J. M., O'Byrne, M. M., de Andrade, M., O'Leary, P. W., et al. (2015a). Compound heterozygous NOTCH1 mutations underlie impaired cardiogenesis in a patient with hypoplastic left heart syndrome. Hum. Genet. 134, 1003–1011. doi:10.1007/s00439-015-1582-1
Theis, J. L., Zimmermann, M. T., Evans, J. M., Eckloff, B. W., Wieben, E. D., Qureshi, M. Y., et al. (2015b). Recessive MYH6 mutations in hypoplastic left heart with reduced ejection fraction. Circ. Cardiovasc. Genet. 8, 564–571. doi:10.1161/CIRCGENETICS.115.001070
Thomas, P. S. (1980). Hybridization of denatured RNA and small DNA fragments transferred to nitrocellulose. Proc. Natl. Acad. Sci. U. S. A. 77, 5201–5205. doi:10.1073/pnas.77.9.5201
Tian, J., Fratz, S., Hou, Y., Lu, Q., Görlach, A., Hess, J., et al. (2011). Delineating the angiogenic gene expression profile before pulmonary vascular remodeling in a lamb model of congenital heart disease. Physiological genomics, 43 (1), 87–98. doi:10.1152/physiolgenomics.00135.2010
Treiber, T., Treiber, N., and Meister, G. (2019). Regulation of microRNA biogenesis and its crosstalk with other cellular pathways. Nat. Rev. Mol. Cell. Biol. 20, 5–20. doi:10.1038/s41580-018-0059-1
Tripathi, D., and Reddy, S. (2022). iPSC model of congenital heart disease predicts disease outcome. Cell. stem Cell. 29 (5), 659–660. doi:10.1016/j.stem.2022.04.010
Urashima, T., Zhao, M., Wagner, R., Fajardo, G., Farahani, S., Quertermous, T., et al. (2008). Molecular and physiological characterization of RV remodeling in a murine model of pulmonary stenosis. Am. J. Physiol. Heart Circ. Physiol. 295, H1351-H1368. doi:10.1152/ajpheart.91526.2007
van Rooij, E., and Olson, E. N. (2007). MicroRNAs: powerful new regulators of heart disease and provocative therapeutic targets. J. Clin. Investig. 117, 2369–2376. doi:10.1172/JCI33099
Vissers, L. E. L. M., Kalvakuri, S., de Boer, E., Geuer, S., Oud, M., van Outersterp, I., et al. (2020). De novo variants in CNOT1, a central component of the CCR4-NOT complex involved in gene expression and RNA and protein stability, cause neurodevelopmental delay. Am. J. Hum. Genet. 107 (1), 164–172. doi:10.1016/j.ajhg.2020.05.017
Wang, B., Shi, G., Zhu, Z., Chen, H., and Fu, Q. (2018). Sexual difference of small RNA expression in Tetralogy of Fallot. Sci. Rep. 8, 12847. doi:10.1038/s41598-018-31243-6
Wang, Z., Gerstein, M., and Snyder, M. (2009). RNA-Seq: a revolutionary tool for transcriptomics. Nat. Rev. Genet. 10 (1), 57–63. doi:10.1038/nrg2484
Wang, Z., Wang, H., Zhang, Y., Yu, F., Yu, L., and Zhang, C. (2021). Single-cell RNA sequencing analysis to characterize cells and gene expression landscapes in atrial septal defect. J. Cell. Mol. Med. 25 (20), 9660–9673. doi:10.1111/jcmm.16914
Wei, Y., Peng, S., Wu, M., Sachidanandam, R., Tu, Z., Zhang, S., et al. (2014). Multifaceted roles of miR-1s in repressing the fetal gene program in the heart. Cell. Res. 24, 278–292. doi:10.1038/cr.2014.12
Williams, K., Carson, J., and Lo, C. (2019). Genetics of congenital heart disease. Biomolecules 9 (12), 879. doi:10.3390/biom9120879
Wu, J., Li, J., Liu, H., Yin, J., Zhang, M., Yu, Z., et al. (2019). Circulating plasma circular RNAs as novel diagnostic biomarkers for congenital heart disease in children. J. Clin. laboratory analysis 33 (9), e22998. doi:10.1002/jcla.22998
Xie, Y., Yao, L., Yu, X., Ruan, Y., Li, Z., and Guo, J. (2020). Action mechanisms and research methods of tRNA-derived small RNAs. Sig Transduct. Target Ther. 5, 109. doi:10.1038/s41392-020-00217-4
Yang, F., Zhou, L., Wang, Q., You, X., Li, Y., Zhao, Y., et al. (2014). NEXN inhibits GATA4 and leads to atrial septal defects in mice and humans. Cardiovasc. Res. 103 (2), 228–237. doi:10.1093/cvr/cvu134
Yu, B., Li, M., Han, S. P., Yu, Z., and Zhu, J. (2021a). Circular RNA hsa_circ_105039 promotes cardiomyocyte differentiation by sponging miR-17 to regulate cyclinD2 expression. Mol. Med. Rep. 24 (6), 861. doi:10.3892/mmr.2021.12501
Yu, H., Wang, X., and Cao, H. (2021b). Construction and investigation of a circRNA-associated ceRNA regulatory network in Tetralogy of Fallot. BMC Cardiovasc. Disord. 21 (1), 437. doi:10.1186/s12872-021-02217-w
Yu, Z., Han, S., Hu, P., Zhu, C., Wang, X., Qian, L., et al. (2011). Potential role of maternal serum microRNAs as a biomarker for fetal congenital heart defects. Med. Hypotheses. 76, 424–426. doi:10.1016/j.mehy.2010.11.010
Zaidi, S., and Brueckner, M. (2017). Genetics and genomics of congenital heart disease. Circ. Res. 120, 923–940. doi:10.1161/CIRCRESAHA.116.309140
Zhang, J., Chang, J. J., Xu, F., Ma, X. J., Wu, Y., Li, W. C., et al. (2013). MicroRNA deregulation in right ventricular outflow tract myocardium in nonsyndromic tetralogy of Fallot. Can. J. Cardiol. 29, 1695–1703. doi:10.1016/j.cjca.2013.07.002
Zhang, X., Gao, Y., Zhang, X., Zhang, X., Xiang, Y., Fu, Q., et al. (2021). FGD5-AS1 is a hub lncRNA ceRNA in hearts with tetralogy of Fallot which regulates congenital heart disease genes transcriptionally and epigenetically. Front. Cell. Dev. Biol. 9, 630634. doi:10.3389/fcell.2021.630634
Zhu, H. (2016). Forkhead box transcription factors in embryonic heart development and congenital heart disease. Life Sci. 144, 194–201. doi:10.1016/j.lfs.2015.12.001
Keywords: pediatric congenital heart disease, RNA sequencing, transcriptomes, animal model, iPSC models, gene expression, noncoding transcriptomes
Citation: Odogwu NM, Hagen C and Nelson TJ (2023) Transcriptome studies of congenital heart diseases: identifying current gaps and therapeutic frontiers. Front. Genet. 14:1278747. doi: 10.3389/fgene.2023.1278747
Received: 17 August 2023; Accepted: 16 November 2023;
Published: 13 December 2023.
Edited by:
Andrew Landstrom, Duke University, United StatesReviewed by:
Abdelaziz Beqqali, University of Edinburgh, United KingdomBhagyalaxmi Mohapatra, Banaras Hindu University, India
Copyright © 2023 Odogwu, Hagen and Nelson. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nkechi Martina Odogwu, T2RvZ3d1Lk5rZWNoaUBtYXlvLmVkdQ==; Timothy J. Nelson, TmVsc29uLlRpbW90aHlAbWF5by5lZHU=