 Ekaterina M. Dvorianinova1*
Ekaterina M. Dvorianinova1* Elena N. Pushkova1
Elena N. Pushkova1 Nadezhda L. Bolsheva1
Nadezhda L. Bolsheva1 Elena V. Borkhert1
Elena V. Borkhert1 Tatiana A. Rozhmina2
Tatiana A. Rozhmina2 Daiana A. Zhernova1,3
Daiana A. Zhernova1,3 Roman O. Novakovskiy1
Roman O. Novakovskiy1 Anastasia A. Turba1
Anastasia A. Turba1 Elizaveta A. Sigova1,4
Elizaveta A. Sigova1,4 Nataliya V. Melnikova1
Nataliya V. Melnikova1 Alexey A. Dmitriev1*
Alexey A. Dmitriev1*- 1Engelhardt Institute of Molecular Biology, Russian Academy of Sciences, Moscow, Russia
- 2Federal Research Center for Bast Fiber Crops, Torzhok, Russia
- 3Faculty of Biology, Lomonosov Moscow State University, Moscow, Russia
- 4Moscow Institute of Physics and Technology, Moscow, Russia
Sequencing whole plant genomes provides a solid foundation for applied and basic studies. Genome sequences of agricultural plants attract special attention, as they reveal information on the regulation of beneficial plant traits. Flax is a valuable crop cultivated for oil and fiber. Genome sequences of its representatives are rich sources of genetic information for the improvement of cultivated forms of the plant. In our work, we sequenced the first genome of flax with the dehiscence of capsules—Linum usitatissimum convar. сrepitans (Boenn.) Dumort—on the Oxford Nanopore Technologies (ONT) and Illumina platforms. We obtained 23 Gb of raw ONT data and 89 M of 150 + 150 paired-end Illumina reads and tested different tools for genome assembly and polishing. The genome assembly produced according to the Canu—Racon ×2—medaka—POLCA scheme had optimal contiguity and completeness: assembly length—412.6 Mb, N50—5.2 Mb, L50—28, and complete BUSCO—94.6% (64.0% duplicated, eudicots_odb10). The obtained high-quality genome assembly of L. usitatissimum convar. crepitans provides opportunities for further studies of evolution, domestication, and genome regulation in the section Linum.
1 Introduction
Plant genomes demonstrate high variability in size and content (Li et al., 2022; Sun et al., 2022). Genome sequences enable studying the beneficial features of agricultural plants and modifying and improving the desired traits (Nützmann et al., 2016; Sedeek et al., 2019; Wang et al., 2020; Choudhury et al., 2022; Singh et al., 2022; Tello-Ruiz et al., 2022). Therefore, plant genomics and pan-genomics open vast opportunities for breeding and agriculture. Knowledge of genome structure can unveil the mechanisms of regulation of key agricultural traits and highlight possible large-scale differences between the representatives of a taxon. In addition, plant genomes can spur studies on the evolution, domestication, and adaptation processes and the emergence of metabolic diversity driven by whole genome duplication (Song et al., 2021; El Karkouri et al., 2022; Petereit et al., 2022; Zhou and Liu, 2022; Bartlett et al., 2023).
Linum usitatissimum L. is a dual-purpose agricultural plant providing two main raw products of multipurpose use—seed and fiber (Nag et al., 2015). Flax seed is a source of biologically active compounds beneficial for human health (Kezimana et al., 2018; Sirotkin, 2023). Flax seed in animal feed also causes positive effects on immunity and growth (Salem et al., 2023). Flax oil of a certain fatty acid composition is actively used in the coating industry (Wang and Padua, 2005; Dmitriev et al., 2020a). In addition to the use in textile production (Van der Werf and Turunen, 2008), flax fiber serves as a component of composite materials (More, 2022). Flax biomass can be used as a source of bioenergy (Batog et al., 2023). Thus, L. usitatissimum is an important agricultural crop, and data on its diversity at the genome level can be implicated in breeding and understanding the evolution in the section Linum.
Genome sequences of flax representatives are useful sources of information for both basic and applied studies. Currently, seven L. usitatissimum assemblies are available in the databases (NCBI and Zenodo) (You et al., 2018; Dmitriev et al., 2020b; Zhang et al., 2020; Sa et al., 2021; Dvorianinova et al., 2022; Zhao et al., 2023). In the species of the section Linum, apart from L. usitatissimum, a genome of Linum bienne Mill. (considered to be a wild ancestor of cultivated flax) is available in the NCBI database (Zhang et al., 2020). Our study aimed at assembling a high-quality genome of L. usitatissimum convar. crepitans (Boenn.) Dumort. Convar. crepitans is a group of flax varieties with spontaneously opening capsules. It has been cultivated for fiber in Europe, but now it is not in use since seed shattering significantly complicates harvesting. However, it can be found in germplasm collections. The main feature of the convar. crepitans is the dehiscence of its capsules, but in other ways, it is quite similar to L. usitatissimum convar. usitatissimum (Muir and Westcott, 2003). The genetic resource of the convar. crepitans is limited (Diederichsen, 2019). Nevertheless, the investigation of this convar. can significantly broaden the data on genetic diversity and domestication of L. usitatissimum. The genome assembly of the convar. crepitans can be incorporated in pan-genomic studies of flax, including the construction of pan-genome, mining key agricultural traits, and establishing the evolution of flax forms.
2 Material and methods
2.1 Plant material
The seeds of L. usitatissimum convar. crepitans K-1531 were provided by the Institute for Flax (Torzhok, Russia). The seeds were sterilized in a 1% NaClO solution for 5 min and then germinated on Petri dishes. High-quality seedlings were transplanted into the soil and grown for 3–4 weeks. After that, the tops of the plant branches were covered with a dark cloth to prevent exposure to light for 1 week. This step was important to minimize the level of metabolites in flax leaves before DNA extraction. The leaves were collected, frozen in liquid nitrogen, and stored at −70°C until DNA isolation.
2.2 DNA extraction
Nucleus isolation and DNA extraction were performed according to the previously developed protocol (Dvorianinova et al., 2022). Additionally, part of the DNA was purified using the Circulomics Short Read Eliminator kit (SRE kit, Circulomics, United States). DNA concentration and quality were assessed using a Qubit fluorometer (Thermo Fisher Scientific, United States) and a NanoDrop spectrophotometer (Thermo Fisher Scientific), as well as by electrophoresis in 0.3% agarose gel.
2.3 Nanopore and Illumina sequencing
Three libraries were prepared for Nanopore sequencing according to the manufacturer’s protocol for SQK-LSK109 and SQK-LSK114 kits (Oxford Nanopore Technologies (ONT), United Kingdom). The first one was prepared from the SRE-purified DNA using the SQK-LSK109 kit (ONT) and sequenced on the FLO-MIN-106D R9.4.1 flow cell (ONT). The second one was prepared from the SRE-purified DNA using the SQK-LSK114 kit (ONT) and sequenced on the FLO-MIN-114 R10.4.1 flow cell (400 bps mode, ONT). The third one was prepared from the non-treated DNA using the SQK-LSK109 kit (ONT) and sequenced on the FLO-MIN-106D R9.4.1 flow cell (ONT).
An Illumina DNA library was prepared using the NEBNext Ultra II DNA Library Prep Kit for Illumina (New England BioLabs, United Kingdom) according to the manufacturer’s protocol. Sequencing was performed on a NovaSeq 6000 (Illumina, United States) instrument with a read length of 150 + 150 bp.
2.4 Data analysis
Raw FAST5 sequences were converted into FASTQ format by Guppy 6.4.6 using the super accuracy flip-flop algorithm (dna_r9.4.1_450bps_sup.cfg, dna_r10.4.1_e8.2_400bps_sup.cfg) and quality filtering (--min_qscore 8). The adapter sequences were removed with Porechop (https://github.com/rrwick/Porechop).
Draft genomes were assembled using Canu 2.2 (Koren et al., 2017) (set parameter: genome size = 400 m), Flye 2.9 (Kolmogorov et al., 2019) (set parameters: “--genome size 400 m,” “--nano-raw”), GoldRush (Wong et al., 2023) (set parameter: G = 4e6), and NECAT (Chen et al., 2021). Assembly quality was evaluated by the QUAST parameters (QUAST 5.0.2) (Gurevich et al., 2013) and the presence of universal single-copy orthologs (BUSCO 4.1.2, eudicots_odb10) (Simão et al., 2015). For the reference-based QUAST assessment, we used the first version of the genome and annotation of the L. usitatissimum variety CDC Bethune (https://ftp.ncbi.nlm.nih.gov/genomes/genbank/plant/Linum_usitatissimum/all_assembly_versions/GCA_000224295.1_LinUsi_v1.1/, GCA_000224295.1_LinUsi_v1.1_genomic.fna.gz, GCA_000224295.1_LinUsi_v1.1_genomic.gff.gz, and GCA_000224295.1).
To improve the quality of the draft assembly of the convar. crepitans genome, polishing was performed using ONT reads: Racon 1.5.0 (Vaser et al., 2017) (polishing with both R9.4.1 and R10.4.1 reads) and medaka 1.8.0 (https://github.com/nanoporetech/medaka) (-m r1041_e82_400bps_fast_g615; polishing with R10.4.1 reads). Illumina reads were trimmed (trailing:30) and filtered (minlen:50) using Trimmomatic 0.38 (Bolger et al., 2014) and then used for final polishing by POLCA from MaSuRCA 4.0.1 (Zimin et al., 2013).
To align Illumina reads to the final genome assembly of the convar. crepitans, BWA-MEM (Li, 2013) was used. To calculate the coverage percentage of the final genome assembly with Illumina reads, SAMtools depth (Li et al., 2009) (set parameters: -q0 -Q0) was run on the generated bam file, and the number of covered positions was calculated with the “wc -l” bash command.
Repeat content of L. usitatissimum genome assemblies was calculated with LTR_retriever 2.9.0 (Ou and Jiang, 2018), which includes the BuildDatabase (default parameters), RepeatModeler (“-engine ncbi”), and RepeatMasker (“consensi.fa.classified” file as input) modules.
3 Results
To assemble a high-quality genome of L. usitatissimum convar. crepitans, we performed whole-genome sequencing on the ONT and Illumina platforms. Three DNA libraries were prepared for ONT sequencing. In two DNA pools, short fragments were eliminated using the SRE kit (Circulomics, United States). For these DNA pools enriched with long fragments, we received 7.2 Gb (R9.4.1 flow cell) and 6.2 Gb (R10.4.1 flow cell) of raw ONT data with an N50 of 22.9 and 21.8 kb, respectively. For the library from the non-treated DNA, we received 9.6 Gb (R9.4.1 flow cell) of raw ONT data with an N50 of 17.3 kb. After basecalling and adapter trimming, a total of 15.2 Gb of ONT data with an N50 of 21.8 kb remained. Then, we assembled draft genomes using Canu and Flye, which performed best in our previous study (Dvorianinova et al., 2022), as well as GoldRush and NECAT, which were not tested by us earlier.
The expected size of the L. usitatissimum genome was 400–450 Mb (You et al., 2018; Sa et al., 2021; Dvorianinova et al., 2022). Given the same size for the convar. crepitans, only Canu produced an assembly of a reasonable length—416.3 Mb (Figure 1). The assembly had an N50 of 5.2 Mb and the BUSCO completeness of 94.2% (eudicots_odb10). The assembly by NECAT had the same percentage of complete BUSCO and the highest N50 (7.2 Mb). However, the assembly length (374.5 Mb) was smaller than the expected one and might indicate the absence of important non-coding elements, e.g., repeats. Flye produced an assembly of an even smaller length—323.9 Mb. GoldRush demonstrated the worst performance among the tested software. The assembly was only 298.9 Mb long, had an N50 in the kb-range, and the BUSCO completeness of 66.6%.
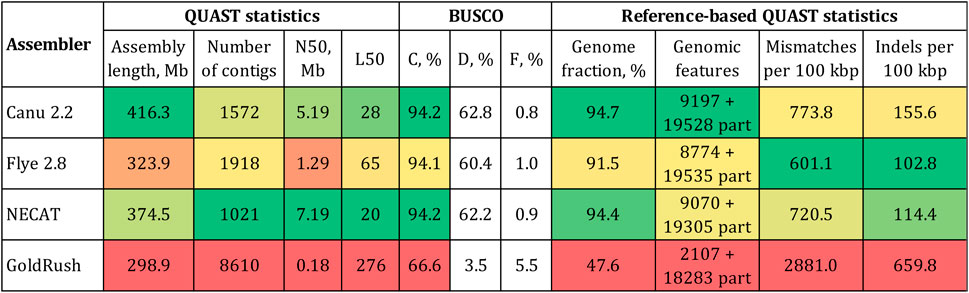
FIGURE 1. QUAST and BUSCO statistics for the L. usitatissimum convar. crepitans genome assemblies obtained with different tools. The green (best)–yellow–red (worst) color scale represents the quality of the values. BUSCO (eudicots_odb10): C—complete, D—duplicated, and F—fragmented. Genomic features: complete + partial; the detected feature from a reference genome is considered partial if it is covered by at least 100 bp.
The received draft assemblies were also assessed by the reference-based QUAST statistics (Figure 1). As a reference, we used the first version of the L. usitatissimum CDC Bethune genome (GCA_000224295.1) because it was assembled from accurate Illumina reads and annotated. Using a reference genome based on Illumina data is beneficial, as it contains errors different from those in a genome assembled from ONT data. In addition, the availability of annotation of the reference genome enabled us to calculate important QUAST statistics, e.g., the number of reference genomic features. The assembly by Canu had the highest fraction of the reference genome covered and the highest number of complete reference genomic features. The assembly by Flye had the lowest relative number of mismatches/indels. However, the accuracy of the obtained genome sequences can be improved by the polishing procedure. Therefore, we chose the assembly by Canu as optimal due to its length and the received parameters of contiguity and completeness.
Next, we improved the accuracy of the Canu-assembled sequences by polishing. To select polishers, we relied on the results of our previous studies. Two rounds of genome polishing with Racon and one round of polishing with medaka was the best combination for ONT reads (Dmitriev et al., 2020b; Krasnov et al., 2020; Melnikova et al., 2021; Dvorianinova et al., 2022). Therefore, we used this scheme for the genome of the convar. crepitans (Figure 2). Two iterations of Racon significantly decreased the relative number of mismatches and especially indels (by ∼2 times). The percentage of complete BUSCO and the number of complete reference genomic features increased. Polishing using medaka further improved the reference-based QUAST statistics. However, it slightly reduced the percentage of complete BUSCO (by 0.1%) and strongly reduced the percentage of duplicated BUSCO (by 1.9%). After all iterations of polishing with ONT reads (Racon ×2—medaka), the assembly length decreased by ∼4 Mb, compared to that of the draft assembly.
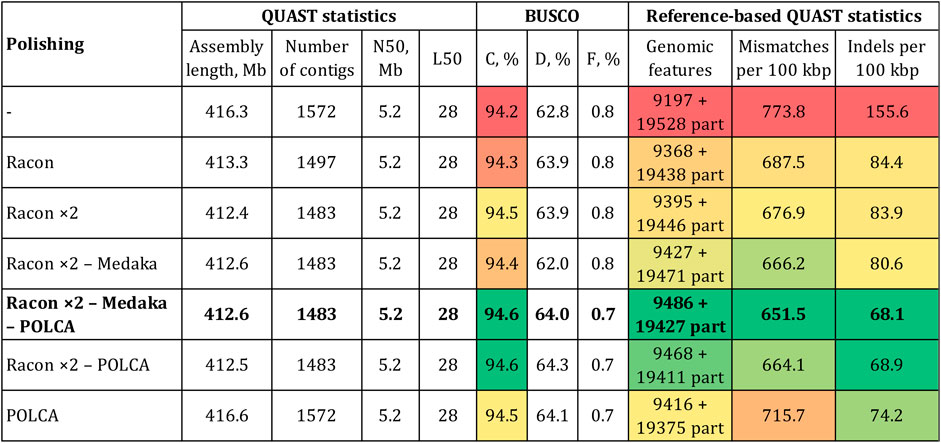
FIGURE 2. QUAST and BUSCO statistics for the Canu-assembled L. usitatissimum convar. crepitans genome polished with different tools. The green (best)–yellow–red (worst) color scale represents the quality of the values. BUSCO (eudicots_odb10): C—complete, D—duplicated, and F—fragmented. Genomic features: complete + partial; the detected feature from a reference genome is considered partial if it is covered by at least 100 bp.
To improve the assembly accuracy to the maximum extent, we additionally polished the convar. crepitans genome (Canu—Racon ×2—medaka) with the generated Illumina data (89 M of 150 + 150 paired-end reads). According to our previous studies, POLCA was the most effective tool for Illumina reads (Dmitriev et al., 2020b; Krasnov et al., 2020; Melnikova et al., 2021). As a result of this procedure, the BUSCO completeness increased to 94.6% (by 0.2%), and the percentage of duplicated BUSCO increased to 64.0% (by 2.0%) (Figure 2). Thus, it eliminated the negative effect of medaka polishing, which caused the reduction in the parameter. Polishing with Illumina data also significantly increased assembly accuracy, according to the reference-based QUAST statistics.
In addition to polishing the draft Canu-assembled genome with both ONT and Illumina data, we tested whether it was possible to reach the same or better results using only Illumina reads or omitting the step of polishing by medaka. Thus, we polished the Canu and Canu—Racon ×2 assemblies with POLCA. The assembly polished using Racon (two iterations), medaka, and POLCA was more complete and accurate than the assemblies polished by Racon and POLCA or only POLCA (Figure 2). It had more complete reference genomic features and a lower relative number of mismatches and indels. However, the Canu–Racon ×2—medaka—POLCA assembly had a slightly lower percentage of duplicated BUSCO than the other two polished assemblies (Canu—Racon ×2—POLCA and Canu—POLCA), by 0.3% and 0.1%, respectively. Compared to genome assemblies polished using both ONT and Illumina reads, the assembly polished only using Illumina reads (Canu—POLCA) had significantly worse statistics of accuracy. Therefore, polishing with ONT reads could not be replaced with polishing only with short accurate reads.
Thus, the Canu—Racon ×2—medaka—POLCA scheme produced the most contiguous and complete assembly: length—412.6 Mb, N50—5.2 Mb, L50—28, and complete BUSCO—94.6%. Mapping Illumina reads to the convar. crepitans genome revealed that more than 398.5 million nucleotide positions were covered (96.6% of the sequence). According to SAMtools flagstat, 98.6% of the passed Illumina reads were mapped to the assembled genome. This indicated that the obtained genome assembly is of reasonable length and high completeness.
To compare the assembly of L. usitatissimum convar. crepitans with the available L. usitatissimum and L. bienne assemblies, the genomes were downloaded from the NCBI and Zenodo databases: 3896 (GCA_030674075.1), Atlant (GCA_014858635.1), Neiya No. 9 (https://zenodo.org/record/7811972), YY5 v.2 (https://zenodo.org/record/4872894), CDC Bethune v.1 and v.2 (GCA_000224295.1, GCA_000224295.2), Heiya 14 (GCA_010665265.1), Longya 10 (GCA_010665275.2), and L. bienne 15003 (GCA_010665285.1). For the downloaded assemblies, QUAST statistics were taken from the NCBI and Zenodo assembly descriptions (for the contig level) or calculated. To calculate BUSCO statistics, the eudicots_odb10 dataset was used. Among the analyzed genomes, the assembly of the convar. crepitans had one of the highest N50 and was the second most complete genome (after the assembly of Neiya No. 9), according to BUSCO statistics (Table 1).
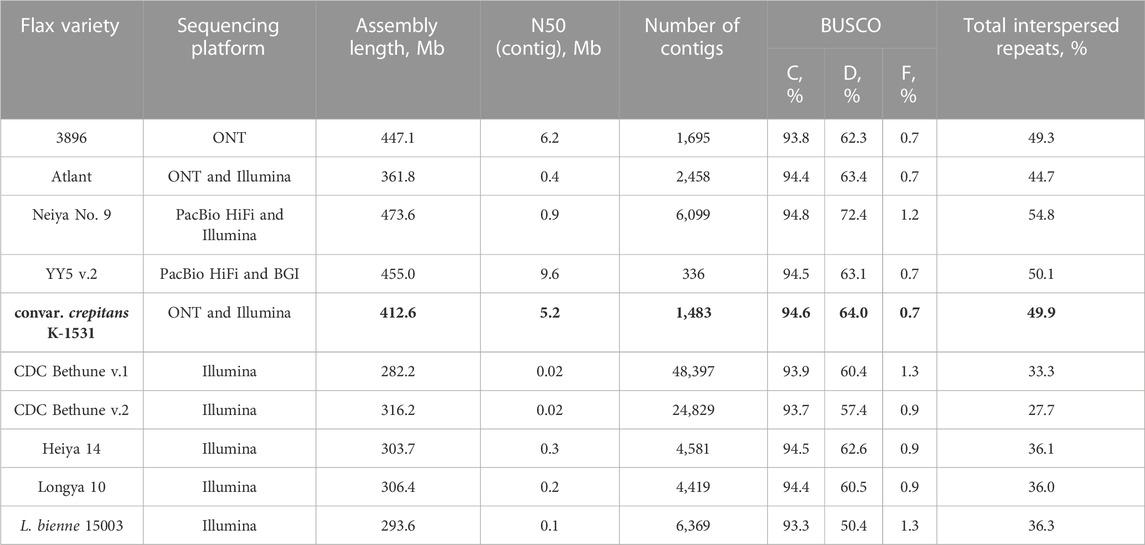
TABLE 1. QUAST and BUSCO (C—complete, D—duplicated, and F—fragmented; eudicots_odb10) statistics and repeat content for the obtained L. usitatissimum convar. crepitans assembly (marked bold) and L. usitatissimum and L. bienne genome assemblies available in databases.
The obtained convar. crepitans assembly contained 49.9% of total interspersed repeats (Table 1) per ∼413 Mb (assembly length). Meanwhile, L. usitatissimum genome assemblies from long reads had 44.7%–54.8% of repetitive sequences per 362–474 Mb. Flax genome assemblies from short reads comprised only 27.7%–36.3% of total interspersed repeats and had a smaller size (294–316 Mb).
4 Discussion
Plant genomes became the foundation of studies on the regulation of genetic features and their involvement in metabolic pathways, species evolution, and adaptation. Currently, genome sequencing is routine but relevant for agricultural plants. The genomes of crops are indispensable for modern breeding based on molecular procedures and targeted improvement of valuable plant features (Dmitriev et al., 2022). Furthermore, the availability of several diverse genome sequences for a species is key to the discovery of novel useful agricultural traits. For L. usitatissimum, seven genome sequences of different varieties were received earlier (You et al., 2018; Dmitriev et al., 2020b; Zhang et al., 2020; Sa et al., 2021; Dvorianinova et al., 2022; Zhao et al., 2023). In this work, we sequenced the genome of L. usitatissimum convar. crepitans which is no longer cultivated due to the dehiscence of capsules. However, such unused genomic material can still be the source of valuable agricultural features.
To obtain the genome of the convar. crepitans, we performed DNA sequencing on the Oxford Nanopore Technologies and Illumina platforms. We assembled the received data using a range of software and calculated quality statistics. Different assemblers were tested in our previous work on their efficacy in constructing the genome of L. usitatissimum line 3896 (Dvorianinova et al., 2022). Most of the tested software (miniasm, NextDenovo, Raven, Shasta, SMARTdenovo, and wtdbg2) demonstrated poor QUAST and BUSCO statistics or assembled a genome of a significantly smaller size than the expected one. In our work on sequencing the genome of the Atlant cultivar, two tested tools (Shasta and wtdbg2) also showed poor performance (Dmitriev et al., 2020b). Therefore, we decided not to include the aforementioned assemblers in our current analysis and focused on the recently released tools and those that showed the best results.
Thus, to obtain draft assemblies, we used Canu, Flye, NECAT, and GoldRush. Canu, the most CPU time-consuming tool, still demonstrated the best performance in terms of assembly completeness and contiguity, including assembly size. NECAT produced the assembly with the highest N50 and the fewest number of contigs but of a size smaller than the expected one (400–450 Mb) and ∼42 Mb smaller than that for the assembly by Canu. Both assemblies had the same BUSCO completeness. Flye assembled a genome with QUAST and BUSCO statistics that was significantly worse but comparable to those of the assemblies by Canu and NECAT. At the same time, the assembly by Flye had the smallest relative number of mismatches/indels. Possibly, this could be due to the included polishing module (Kolmogorov et al., 2019). However, despite the achieved accuracy, the whole genome sequence still missed 20%–30% of the expected genome size. GoldRush was unable to produce a genome with reasonable statistics. Thus, we considered the assembly by Canu optimal.
To improve the accuracy of the obtained genome assembly, one can apply a polishing procedure. The Canu-assembled genome was polished using ONT reads by the Racon (two iterations, both R.9.4.1 and R10.4.1 reads) and medaka (R10.4.1 reads) polishers. Each of the two rounds of Racon increased BUSCO completeness and the number of complete reference genomic features in the assembly. The procedure also decreased the relative number of mismatches and indels by 12.5% and 46.1%, respectively. Sequencing data from R9.4.1 flow cells are more inaccurate than those from R10.4.1 flow cells (Sereika et al., 2022). Thus, in our previous study, polishing with ONT data only from R9.4.1 flow cells had a less dramatic effect (Dvorianinova et al., 2022). Polishing with medaka showed the same trend in statistic values as polishing with Racon. Final polishing with Illumina reads by POLCA also improved QUAST and BUSCO parameters. However, skipping polishing with ONT reads and polishing only with Illumina reads was not as beneficial as using both ONT and Illumina data. BUSCO completeness was almost the same for assemblies obtained according to Canu—POLCA and Canu—Racon ×2—medaka—POLCA. However, more mismatches/indels remained in the assembly polished only with Illumina reads. Thus, the final optimal assembly was obtained using the Canu—Racon ×2—medaka—POLCA scheme (Figure 3). The assembly had a size of 412.6 Mb, consisted of 1,483 contigs, had an N50 of 5.2 Mb, and a BUSCO completeness of 94.6%.
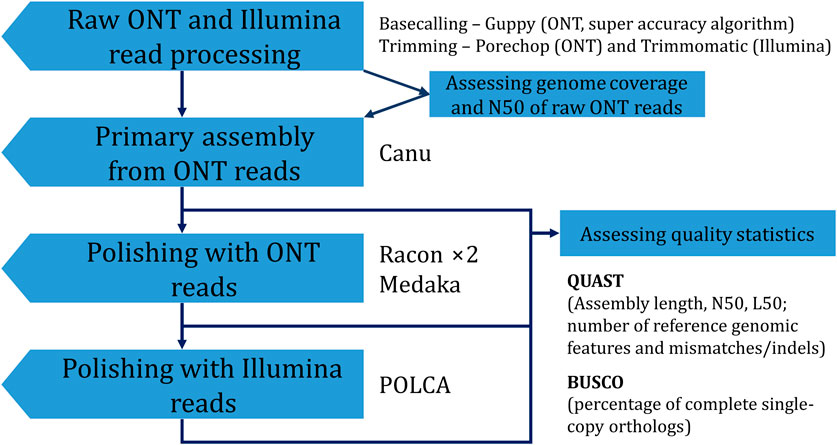
FIGURE 3. The scheme used to obtain the final genome assembly of L. usitatissimum convar. crepitans.
BUSCO completeness of the obtained assembly was higher than that of the available assemblies for L. usitatissimum. Its length and repeat content were expectedly greater than these parameters of the assemblies obtained from short reads (varieties CDC Bethune, Longya 10, Heiya 14; L. bienne 15003). However, the repeat content in the genome of the convar. crepitans was similar to that of the assemblies from long reads (varieties 3896, Atlant, Neiya No. 9, and YY5 v.2). Thus, the non-coding sequences in the assembly are likely complete. The percentage of duplicated BUSCO in the obtained assembly was also high (above 60%) and comparable to that of L. usitatissimum assemblies. This fact correlates with the idea of L. usitatissimum origin. The species might have originated from the crossing of two Linum species. Then, the genome of the progeny probably underwent diploidization. Thus, the resulting ploidy of most genomic features is four (Bolsheva et al., 2017).
The assembled genome of the convar. crepitans has a quality comparable to that of the line 3896—the NCBI reference genome for L. usitatissimum. The line 3896 genome was assembled and polished using ONT reads (Dvorianinova et al., 2022). Meanwhile, the genome of the convar. crepitans was assembled from ONT data and additionally polished with both ONT and Illumina reads. Thus, the assembly of the convar. crepitans has more complete BUSCO likely due to the improvement with accurate Illumina data. However, its contig N50 is lower than that of the assembly of line 3896 or variety YY5. In general, the obtained genome of the convar. crepitans has a quality close to that of most flax assemblies from long reads and outperformed the assemblies from short reads. Nevertheless, its level can still be upgraded to the chromosome one, e.g., using Hi-C data.
In this work, we sequenced the first genome of L. usitatissimum convar. crepitans. The volume and quality of the obtained data were sufficient to produce a high-quality assembly with QUAST and BUSCO statistics that were superior or close to those of the available L. usitatissimum genomes. Its quality level can be additionally upgraded to the scaffold and chromosome level. Our data allow investigating the diversity and evolution of the section Linum as well as mining key traits for breeding.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found as follows: https://www.ncbi.nlm.nih.gov/, PRJNA1006423.
Author contributions
ED: writing–original draft, writing–review and editing, and investigation. EP: writing–review and editing and investigation. NB: writing–review and editing and investigation. EB: writing–review and editing and investigation. TR: writing–review and editing and investigation. DZ: writing–review and editing and investigation. RN: writing–review and editing and investigation. AT: writing–review and editing and investigation. ES: writing–review and editing and investigation. NM: conceptualization, writing–original draft, writing–review and editing and investigation. AD: conceptualization, writing–original draft, writing–review and editing, and investigation.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. The work was financially supported by the Ministry of Science and Higher Education of the Russian Federation, grant number 075-15-2021-1064.
Acknowledgments
The authors would like to thank the Center for Precision Genome Editing and Genetic Technologies for Biomedicine, EIMB RAS for providing the computing power and techniques for the data analysis. This work was performed using the equipment of EIMB RAS “Genome” center (http://www.eimb.ru/ru1/ckp/ccu_genome_ce.php).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Bartlett, M. E., Moyers, B. T., Man, J., Subramaniam, B., and Makunga, N. P. (2023). The power and perils of de novo domestication using genome editing. Annu. Rev. Plant Biol. 74 (1), 727–750. doi:10.1146/annurev-arplant-053122-030653
Batog, J., Wawro, A., Gieparda, W., Bujnowicz, K., Foksowicz-Flaczyk, J., Rojewski, S., et al. (2023). Effective use of flax biomass in biorefining processes. Appl. Sci. 13 (13), 7359. doi:10.3390/app13137359
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30 (15), 2114–2120. doi:10.1093/bioinformatics/btu170
Bolsheva, N. L., Melnikova, N. V., Kirov, I. V., Speranskaya, A. S., Krinitsina, A. A., Dmitriev, A. A., et al. (2017). Evolution of blue-flowered species of genus Linum based on high-throughput sequencing of ribosomal RNA genes. BMC Evol. Biol. 17 (2), 253. doi:10.1186/s12862-017-1105-x
Chen, Y., Nie, F., Xie, S.-Q., Zheng, Y.-F., Dai, Q., Bray, T., et al. (2021). Efficient assembly of nanopore reads via highly accurate and intact error correction. Nat. Commun. 12 (1), 60. doi:10.1038/s41467-020-20236-7
Choudhury, A., Deb, S., Kharbyngar, B., Rajpal, V. R., and Rao, S. R. (2022). Dissecting the plant genome: through new generation molecular markers. Genet. Resour. Crop Evol. 69 (8), 2661–2698. doi:10.1007/s10722-022-01441-3
Diederichsen, A. (2019). “A taxonomic view on genetic resources in the genus Linum L. For flax breeding,” in Genetics and genomics of Linum. Editor C. A. Cullis (Cham: Springer International Publishing), 1–15.
Dmitriev, A., Pushkova, E., and Melnikova, N. (2022). Plant genome sequencing: modern technologies and novel opportunities for breeding. Mol. Biol. 56 (4), 495–507. doi:10.1134/s0026893322040045
Dmitriev, A. A., Kezimana, P., Rozhmina, T. A., Zhuchenko, A. A., Povkhova, L. V., Pushkova, E. N., et al. (2020a). Genetic diversity of SAD and FAD genes responsible for the fatty acid composition in flax cultivars and lines. BMC Plant Biol. 20 (1), 301. doi:10.1186/s12870-020-02499-w
Dmitriev, A. A., Pushkova, E. N., Novakovskiy, R. O., Beniaminov, A. D., Rozhmina, T. A., Zhuchenko, A. A., et al. (2020b). Genome sequencing of fiber flax cultivar Atlant using Oxford Nanopore and Illumina platforms. Front. Genet. 11, 590282. doi:10.3389/fgene.2020.590282
Dvorianinova, E. M., Bolsheva, N. L., Pushkova, E. N., Rozhmina, T. A., Zhuchenko, A. A., Novakovskiy, R. O., et al. (2022). Isolating Linum usitatissimum L. nuclear DNA enabled assembling high-quality genome. Int. J. Mol. Sci. 23 (21), 13244. doi:10.3390/ijms232113244
El Karkouri, K., Ghigo, E., Raoult, D., and Fournier, P.-E. (2022). Genomic evolution and adaptation of arthropod-associated Rickettsia. Sci. Rep. 12 (1), 3807. doi:10.1038/s41598-022-07725-z
Gurevich, A., Saveliev, V., Vyahhi, N., and Tesler, G. (2013). QUAST: quality assessment tool for genome assemblies. Bioinformatics 29 (8), 1072–1075. doi:10.1093/bioinformatics/btt086
Kezimana, P., Dmitriev, A. A., Kudryavtseva, A. V., Romanova, E. V., and Melnikova, N. V. (2018). Secoisolariciresinol diglucoside of flaxseed and its metabolites: biosynthesis and potential for nutraceuticals. Front. Genet. 9, 641. doi:10.3389/fgene.2018.00641
Kolmogorov, M., Yuan, J., Lin, Y., and Pevzner, P. A. (2019). Assembly of long, error-prone reads using repeat graphs. Nat. Biotechnol. 37 (5), 540–546. doi:10.1038/s41587-019-0072-8
Koren, S., Walenz, B. P., Berlin, K., Miller, J. R., Bergman, N. H., and Phillippy, A. M. (2017). Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 27 (5), 722–736. doi:10.1101/gr.215087.116
Krasnov, G. S., Pushkova, E. N., Novakovskiy, R. O., Kudryavtseva, L. P., Rozhmina, T. A., Dvorianinova, E. M., et al. (2020). High-quality genome assembly of Fusarium oxysporum f. sp. lini. Front. Genet. 11, 959. doi:10.3389/fgene.2020.00959
Li, H. (2013). Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. Available at: https://arxiv.org/abs/1303.3997.
Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., et al. (2009). The sequence alignment/map format and SAMtools. bioinformatics 25 (16), 2078–2079. doi:10.1093/bioinformatics/btp352
Li, W., Liu, J., Zhang, H., Liu, Z., Wang, Y., Xing, L., et al. (2022). Plant pan-genomics: recent advances, new challenges, and roads ahead. J. Genet. Genomics 49 (9), 833–846. doi:10.1016/j.jgg.2022.06.004
Melnikova, N. V., Pushkova, E. N., Dvorianinova, E. M., Beniaminov, A. D., Novakovskiy, R. O., Povkhova, L. V., et al. (2021). Genome assembly and sex-determining region of male and female Populus × sibirica. Front. Plant Sci. 12, 625416. doi:10.3389/fpls.2021.625416
More, A. P. (2022). Flax fiber–based polymer composites: a review. Adv. Compos. Hybrid Mater. 5 (1), 1–20. doi:10.1007/s42114-021-00246-9
Nag, S., Mitra, J., and Karmakar, P. (2015). An overview on flax (Linum usitatissimum L.) and its genetic diversity. Int. J. Agric. Environ. Biotechnol. 8 (4), 805–817. doi:10.5958/2230-732x.2015.00089.3
Nützmann, H.-W., Huang, A., and Osbourn, A. (2016). Plant metabolic clusters – from genetics to genomics. New Phytol. 211 (3), 771–789. doi:10.1111/nph.13981
Ou, S., and Jiang, N. (2018). LTR_retriever: a highly accurate and sensitive program for identification of long terminal repeat retrotransposons. Plant physiol. 176 (2), 1410–1422. doi:10.1104/pp.17.01310
Petereit, J., Bayer, P. E., Thomas, W. J. W., Tay Fernandez, C. G., Amas, J., Zhang, Y., et al. (2022). Pangenomics and crop genome adaptation in a changing climate. Plants 11 (15), 1949. doi:10.3390/plants11151949
Sa, R., Yi, L., Siqin, B., An, M., Bao, H., Song, X., et al. (2021). Chromosome-level genome assembly and annotation of the fiber flax (Linum usitatissimum) genome. Front. Genet. 12, 735690. doi:10.3389/fgene.2021.735690
Salem, M. O. A., Taştan, Y., Bilen, S., Terzi, E., and Sönmez, A. Y. (2023). Dietary flaxseed (Linum usitatissimum) oil supplementation affects growth, oxidative stress, immune response, and diseases resistance in rainbow trout (Oncorhynchus mykiss). Fish Shellfish Immunol. 138, 108798. doi:10.1016/j.fsi.2023.108798
Sedeek, K. E. M., Mahas, A., and Mahfouz, M. (2019). Plant genome engineering for targeted improvement of crop traits. Front. Plant Sci. 10, 114. doi:10.3389/fpls.2019.00114
Sereika, M., Kirkegaard, R. H., Karst, S. M., Michaelsen, T. Y., Sørensen, E. A., Wollenberg, R. D., et al. (2022). Oxford Nanopore R10.4 long-read sequencing enables the generation of near-finished bacterial genomes from pure cultures and metagenomes without short-read or reference polishing. Nat. Methods 19 (7), 823–826. doi:10.1038/s41592-022-01539-7
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V., and Zdobnov, E. M. (2015). BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31 (19), 3210–3212. doi:10.1093/bioinformatics/btv351
Singh, K. S., van der Hooft, J. J., van Wees, S. C., and Medema, M. H. (2022). Integrative omics approaches for biosynthetic pathway discovery in plants. Nat. Product. Rep. 39 (9), 1876–1896. doi:10.1039/d2np00032f
Sirotkin, A. V. (2023). Influence of flaxseed (Linum usitatissimum) on female reproduction. Planta Medica 89, 608–615. doi:10.1055/a-2013-2966
Song, J.-M., Zhang, Y., Zhou, Z.-W., Lu, S., Ma, W., Lu, C., et al. (2021). Oil plant genomes: current state of the science. J. Exp. Bot. 73 (9), 2859–2874. doi:10.1093/jxb/erab472
Sun, Y., Shang, L., Zhu, Q.-H., Fan, L., and Guo, L. (2022). Twenty years of plant genome sequencing: achievements and challenges. Trends Plant Sci. 27, 391–401. doi:10.1016/j.tplants.2021.10.006
Tello-Ruiz, M. K., Jaiswal, P., and Ware, D. (2022). “Gramene: a resource for comparative analysis of plants genomes and pathways,” in Plant bioinformatics: methods and protocols (Berlin, Germany: Springer), 101–131.
Van der Werf, H. M., and Turunen, L. (2008). The environmental impacts of the production of hemp and flax textile yarn. Industrial Crops Prod. 27 (1), 1–10. doi:10.1016/j.indcrop.2007.05.003
Vaser, R., Sović, I., Nagarajan, N., and Šikić, M. (2017). Fast and accurate de novo genome assembly from long uncorrected reads. Genome Res. 27 (5), 737–746. doi:10.1101/gr.214270.116
Wang, H., Cimen, E., Singh, N., and Buckler, E. (2020). Deep learning for plant genomics and crop improvement. Curr. Opin. Plant Biol. 54, 34–41. doi:10.1016/j.pbi.2019.12.010
Wang, Q., and Padua, G. W. (2005). Properties of zein films coated with drying oils. J. Agric. Food Chem. 53 (9), 3444–3448. doi:10.1021/jf047994n
Wong, J., Coombe, L., Nikolić, V., Zhang, E., Nip, K. M., Sidhu, P., et al. (2023). Linear time complexity de novo long read genome assembly with GoldRush. Nat. Commun. 14 (1), 2906. doi:10.1038/s41467-023-38716-x
You, F. M., Xiao, J., Li, P., Yao, Z., Jia, G., He, L., et al. (2018). Chromosome-scale pseudomolecules refined by optical, physical and genetic maps in flax. Plant J. 95 (2), 371–384. doi:10.1111/tpj.13944
Zhang, J., Qi, Y., Wang, L., Wang, L., Yan, X., Dang, Z., et al. (2020). Genomic comparison and population diversity analysis provide insights into the domestication and improvement of flax. Iscience 23 (4), 100967. doi:10.1016/j.isci.2020.100967
Zhao, X., Yi, L., Zuo, Y., Gao, F., Cheng, Y., Zhang, H., et al. (2023). High-quality genome assembly and genome-wide association study of male sterility provide resources for flax improvement. Plants 12 (15), 2773. doi:10.3390/plants12152773
Zhou, X., and Liu, Z. (2022). Unlocking plant metabolic diversity: a (pan)-genomic view. Plant Commun. 3, 100300. doi:10.1016/j.xplc.2022.100300
Keywords: flax, Linum usitatissimum convar. crepitans, section Linum, nanopore sequencing, de novo genome assembly
Citation: Dvorianinova EM, Pushkova EN, Bolsheva NL, Borkhert EV, Rozhmina TA, Zhernova DA, Novakovskiy RO, Turba AA, Sigova EA, Melnikova NV and Dmitriev AA (2023) Genome of Linum usitatissimum convar. crepitans expands the view on the section Linum. Front. Genet. 14:1269837. doi: 10.3389/fgene.2023.1269837
Received: 30 July 2023; Accepted: 24 October 2023;
Published: 22 November 2023.
Edited by:
Fakhriddin Kushanov, Academy of Sciences Republic of Uzbekistan (UzAS), UzbekistanReviewed by:
Ritwika Das, Indian Agricultural Statistics Research Institute, Indian Council of Agricultural Research, IndiaEman Tawfik, Helwan University, Egypt
Xuming Li, Hugo Biotechnologies Co., Ltd., China
Wang Xiao, Chinese Academy of Sciences (CAS), China
Copyright © 2023 Dvorianinova, Pushkova, Bolsheva, Borkhert, Rozhmina, Zhernova, Novakovskiy, Turba, Sigova, Melnikova and Dmitriev. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ekaterina M. Dvorianinova, ZHZvcmlhbmlub3ZhLmVtQHBoeXN0ZWNoLmVkdQ==; Alexey A. Dmitriev, YWxleF8yNDVAbWFpbC5ydQ==