 Xinxing Xie1†Jinhui Gan1†
Xinxing Xie1†Jinhui Gan1† Zezhang Liu2,3†Yulian Zhou4Kun Yuan2,3
Zezhang Liu2,3†Yulian Zhou4Kun Yuan2,3 Zhigang Chen2,3Shiping Chen2Rui Zhou2,3Lipei Liu2,5Xiaoyan Huang2Yan Zhang2,3Qian Liu2,3Wenqian Zhang2,3*
Zhigang Chen2,3Shiping Chen2Rui Zhou2,3Lipei Liu2,5Xiaoyan Huang2Yan Zhang2,3Qian Liu2,3Wenqian Zhang2,3* Jungao Huang1*
Jungao Huang1* Junkun Chen1*
Junkun Chen1*- 1Ganzhou Maternal and Child Health Hospital, Ganzhou, Jiangxi, China
- 2BGI Genomics, Shenzhen, China
- 3Clin Lab, BGI Genomics, Wuhan, China
- 4Dayu Maternal and Child Health Hospital, Ganzhou, Jiangxi, China
- 5Clin Lab, BGI Genomics, Tianjin, China
α-globin gene triplication carriers were not anemic in general, while some studies found that α-globin gene triplication coinherited with heterozygous β-thalassemia may cause adverse clinical symptoms, which yet lacks sufficient evidence in large populations. In this study, we investigated the prevalence and distribution of α-globin gene triplication as well as the phenotypic characteristics of α-globin gene triplication coinherited with heterozygous β-thalassemia in Ganzhou city, southern China. During 2021-2022, a total of 73,967 random individuals who received routine health examinations before marriage were genotyped for globin gene mutations by high-throughput sequencing. Among them, 1,443 were α-globin gene triplication carriers, with a carrier rate of 1.95%. The most prevalent mutation was αααanti3.7/αα (43.10%), followed by αααanti4.2/αα (38.12%). 42 individuals had coinherited α-globin gene triplication and heterozygous β-thalassemia. However, they did not differ from the individuals with heterozygous β-thalassemia and normal α-globin (αα/αα) in terms of mean corpuscular volume (MCV) and mean corpuscular hemoglobin (MCH) levels. In addition, heterogenous clinical phenotypes were found in two individuals with the same genotype. Our study established a database of Ganzhou α-globin gene triplication and provided practical advice for the clinical diagnosis of α-globin gene triplication.
1 Introduction
Thalassemia is an autosomal recessive genetic disease with one or more mutations in globin chains. It is highly prevalent in malaria endemic areas as an adaptive response to malaria (Vlok et al., 2021). Multiple studies from Guangdong, Guangxi, Fujian, Yunnan and Guizhou provinces showed that thalassemia is highly prevalent in China with an overall carrier rate of 10.09%, and Guangxi province is the most prevalent region with a carrier rate of 19.04% (Lai et al., 2017). According to the mutated globin genes, thalassemia is mainly divided into two types: α-thalassemia and β-thalassemia (Muncie and Campbell, 2009). The imbalance between the α- and β-globin chains can result in a variety of clinical phenotypes, varying from completely healthy with normal red blood cell indices to severe anemia requiring lifelong blood transfusion. The latter poses a huge financial burden to the affected families and is one of the major public health and social problem that deserves increased efforts in investigation and prevention (Gharaibeh et al., 2018; Pepe et al., 2022).
α-globin gene triplication is the result of unequal crossing over between misaligned homologous segments in the α-globin gene cluster on chromosome 16 during meiosis, leading to an increased accumulation of α-globin (Weatherall and Clegg, 2001). In the carriers of α-globin gene triplication with a normal β-globin gene, the α-globin accumulation does not result in any clinical symptoms or significant hematological changes. However, co-inheritance of α-globin gene triplication and heterozygous β-thalassemia can increase the synthetic imbalance between α-globin and β-globin, and alter the hematological features of the carriers (Mehta et al., 2015). As a result, the co-inheritance may convert the clinical phenotype from β-thalassemia heterozygotes to β-thalassemia intermedia or β-thalassemia major (Farashi et al., 2015; Majid et al., 2015). In contrast, another study reported that there was no significant difference in the hematological indices between those co-inherited carriers and the β-thalassemia carriers with normal α-globin gene or healthy individuals (Hamid et al., 2021). Therefore, the interaction between α-globin gene triplication and heterozygous β-thalassemia is yet not clear.
Previous studies indicated that the frequencies of α-globin gene triplication in different populations varied from 0.4% (out of 125 individuals) in Sardinians, 1.2% (out of 3,500 individuals) in Netherlands to 5% (out of 50 individuals) in Greek (Goossens et al., 1980; Giordano, Bakker-Verwij, and Harteveld, 2009). In an Iran population of 4005 β-thalassemia carriers, the frequency of α-globin gene triplication was 1.67% (Hamid et al., 2021). In China, Wu et al. (2016) reported the frequency of α-globin gene triplication in 1,169 individuals from southern region was 1.2% and stressed the importance of α-globin gene triplication information in genetic counseling. More recently, the frequencies of α-globin gene triplication were found to be 0.77% (out of 7,644 individuals) in Guizhou province and 0.84% (out of 23,900 individuals) in southern Guangxi (X. Luo et al., 2021; Long and Liu, 2021). The low frequency of α-globin gene triplication in these studies suggested that a large sample size is necessary to determine the prevalence of α-globin gene triplication and its interaction with heterozygous β-thalassemia.
Jiangxi province, located in southern China, is highly prevalent with thalassemia (Zhao et al., 2018; Qiu et al., 2020). Ganzhou has been identified as the most prevalent city in southern Jiangxi with a carrier rate of 9.49%, whereas middle and northern Jiangxi has carrier rates of 3.90% and 2.63% respectively (Lin et al., 2014). Recently, a large-scale study found that the carrier rate of thalassemia in Ganzhou City is as high as 14.54% (Yang et al., 2023). However, the prevalence and distribution patterns of α-globin gene triplication, as well as its interaction with heterozygous β-thalassemia in Ganzhou city are still unknown. In this study, we performed a large-scale retrospective analysis of α-globin gene triplication in Ganzhou city based on 73,967 subjects who were genotyped using next-generation sequencing (NGS). The prevalence and distribution patterns of α-globin gene triplication were determined. In addition, we explored the interaction of α-globin gene triplication and heterozygous β-thalassemia according to the hematological parameters. This study will aid in better understanding α-globin gene triplication and providing more information for the clinical diagnosis of α-globin gene triplication.
2 Materials and methods
2.1 Population samples
A total of 73,967 subjects (37,001 males, 36,968 females, age ranged from 18 to 50 years old) receiving routine health examinations before marriage in Ganzhou city of Jiangxi province in China were genotyped for globin gene mutations including α-globin gene triplication from August 2021 to September 2022. These subjects came from 18 counties of Ganzhou, including Xingguo, Ningdu, Shicheng, Huichang, Yudu, Ganxian, Nankang, Longnan, Zhanggong, Shangyou, Chongyi, Dayu, Xinfeng, Anyuan, Xunwu, Dingnan, Quannan, and Ruijin.
2.2 Sample collection and blood analysis
After obtaining the informed consent of the subjects, peripheral blood was collected in EDTA tubes. All the blood samples were stored at −20°C before the experiment. The hematological parameters of all the blood samples were automatically measured using an automated XS-1000i Hematology Analyzer System (Lincolnshire, IL, United States). Mean Corpuscular Volume (MCV), Mean Corpuscular Hemoglobin (MCH) and Hemoglobin (Hb) concentration were determined and statistically analyzed using ANOVA. The relative proportions of HbA, HbA2 and HbF were measured by CAPILLARYS 3 OCTA (SEBIA, France) at Ganzhou Maternal and Child Health Hospital.
2.3 DNA extraction
We utilized the Q Kingfisher Flex system (Thermo Scientific, Rockford, IL) to extract genomic DNA from whole blood. Subsequently, the GenMag Nucleic Acid Isolation kit (Magnetic bead method) (GenMagBio, Beijing, China) was used for DNA isolation. To quantify the concentrations of DNA samples, we employed a NanoDrop-8000 spectrophotometer (Thermo Scientific, Waltham, MA, United States). The samples were subjected to restriction based on the following criteria: a DNA concentration exceeding 20 ng/mL and an A260/A280 ratio range of 1.8–2.0.
2.4 Thalassemia genotyping
We utilized a combined approach of Gap-PCR and NGS for thalassemia detection (Zhang et al., 2019). Briefly, seven types of deletion mutations (α deletions: -SEA, --THAI, -α3.7, -α4.2; β deletions: SEA-HPFH, Chinese Gγ+(Aγδβ)0, Taiwanese deletion) were characterized by the Gap-PCR. Other mutations were detected using NGS. Firstly, the target sequences of HBA1, HBA2, and HBB were amplified and enriched through multiplex PCR. Secondly, sequencing libraries were subsequently prepared following the MGISEQ-2000 sequencing library preparation protocol (MGI, Shenzhen, China). Finally, sequencing was carried out using the paired-end tag (PE100) on an MGISEQ-2000 chip (MGI, Shenzhen, China). The protocol for bioinformatic analysis of identifying hemoglobin gene mutations was previously described (Xi et al., 2023). Furthermore, for individuals indicated as carriers of α-globin gene triplication in the analysis results, we confirmed this through PCR following the internationally recognized literature (Wang et al., 2003).
2.5 Multiplex ligation-dependent probe amplification
Multiplex ligation-dependent probe amplification (MLPA) was conducted with the commercial kit SALSA MLPA P140 HBA probemix (MRC-Holland, the Netherlands) by Beijing Yinda Xin Technology Co., Ltd.
2.6 Definition of anemia degree
According to WHO recommendations (WHO, 2011), non-anemia, mild anemia, moderate anemia and severe anemia were diagnosed using hemoglobin levels. All the subjects in this study are non-pregnant women (15 years or older) and men (15 years or older) living at altitudes lower than 1,000 m above sea level. Consequently, anemia cut-offs were 12 g/dL or higher (non-pregnant women) and 13 g/dL or higher (men) for non-anemia, 11–11.9 g/dL (non-pregnant women) and 11–12.9 g/dL (men) for mild anemia, 8–10.9 g/dL for moderate anemia, lower than 8 g/dL for severe anemia.
2.7 Statistical analysis
All the statistical analyses were performed using the SPSS 19 software (SPSS Inc., Chicago, IL, United States). Continuous variables were expressed as means ± SD, while categorical variables were expressed as numbers or percentages. The Chi-square test and one-way ANOVA were employed for statistical difference analysis. p-value <0.05 was considered as significant (*p-value <0.05; **p-value <0.01; ***p-value <0.001).
3 Results
3.1 The characteristic of α-globin gene triplication in Ganzhou
From 2021 to 2022, 1,443 of 73,967 subjects were diagnosed as α-globin gene triplication carriers with a carrier rate of 1.95%. There is no significant difference (p-value = 0.17) in the carrier rate between males (n = 701) and females (n = 742, Figure 1A). As shown in Figure 1, 12 different genotypes of α-globin gene triplications were detected. Among them, the most prevalent genotype was αααanti3.7/αα, which had a carrier rate of 0.84% (622/73,967) and accounted for 43.10% (622/1,443) of the α-globin gene triplication carriers, followed by αααanti4.2/αα and αααanti4.2/-α3.7, which had carrier rates of 0.74% (550/73,967) and 0.26% (193/73,967), respectively, and accounted for 38.12% (550/1,443) and 13.37% (193/1,443) of the α-globin gene triplication carriers (Figures 1B, C). The remaining nine genotypes of αααanti3.7/--SEA, αααanti4.2/--SEA, αααanti3.7/-α3.7, αααanti3.7/-α4.2, αααanti4.2/αWSα, αααanti3.7/αααanti4.2, αααanti4.2/HKαα/--SEA, αααanti4.2/αCSα, and αααanti3.7/Hb Phnom Penh accounted for only 5.41% (78/1,443) of all the α-globin gene triplication carriers (Figures 1B, C). In addition, only one carrier of αααanti4.2/αCSα and one carrier of αααanti3.7/Hb Phnom were found.
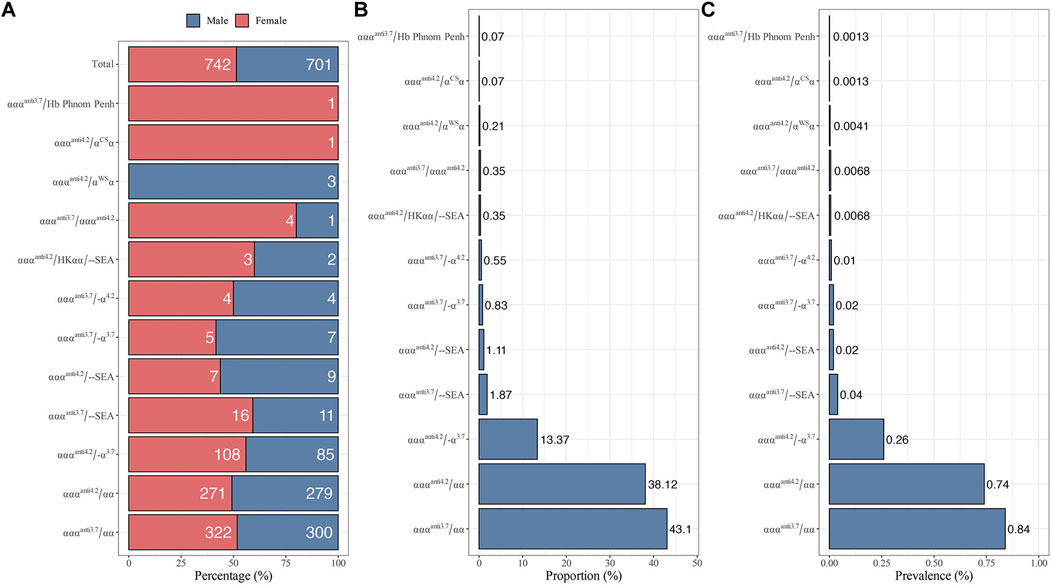
FIGURE 1. Distribution of different genotypes with α-globin gene triplication in 73,967 subjects. (A) Gender distribution. (B) The proportion of different genotypes among 1,443 α-globin gene triplication carriers. (C) The prevalence of different genotypes.
3.2 Geographical distribution
The carrier rates of 18 counties in Ganzhou ranged from 1.32% to 2.51% (Figure 2 and Supplementary Table S1). Shicheng county in the northeast Ganzhou had the highest carrier rate at 2.51% (63/2,514), followed by Yudu at 2.33% (241/10,351), Ruijin at 2.31% (135/5,840), Dayu at 2.23% (36/1,614) and Huichang at 2.15% (113/5256). Ganxian in the west of Ganzhou had the lowest carrier rate at 1.32% (67/5,078), which was significantly lower than Ruijin (p = 0.001), Yudu (p = 0.001), Shicheng (p = 0.005), Huichang (p = 0.012) and Dayu (p = 0.026), but not significantly different from Ganxian and Quannan (p = 0.633), Longnan (p = 0.480), Xunwu (p = 0.304), Chongyi (p = 0.222) and Xingguo (p = 0.168). Unexpectedly, in contrast to the overall distribution pattern of thalassemia observed in China, which showed high prevalence in the south and low prevalence in the north, the distribution pattern of α-globin gene triplication in Ganzhou appeared to be random.
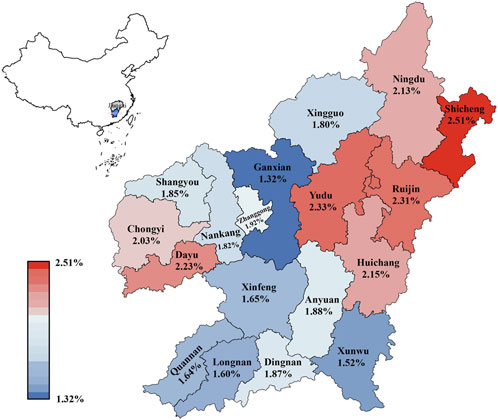
FIGURE 2. Distribution of the prevalence of α-globin gene triplication in different regions of Ganzhou city.
3.3 Hematological parameters in the population
In order to understand the genotype-phenotype associations in α-globin gene triplication carriers, the carriers were divided into six groups (C to H) according to the genotypes of α and β globin genes (Table 1). In addition, two control groups (A and B) were randomly selected from the normal α globin (αα/αα) subjects with normal β globin (βN/βN, n = 80) or heterozygous β-thalassemia (βmut/βN, n = 80). Since all the individuals of this study were in childbearing age, the average age of each group was between 22.67 and 28.00 (Table 1). Compared with the group A with genotype αα/αα and βN/βN, the hematological parameters of group C with genotype αααanti3.7/αα and βN/βN, group D with genotype αααanti4.2/αα and βN/βN and group E with genotype αααanti3.7/-α3.7 and βN/βN did not differ significantly, which were within the normal range (Table 1). This result suggested that α-globin gene triplication did not affect hematological parameters of the subjects with normal β globin gene at the population level. On the contrary, the MCV and MCH of group B with genotype αα/αα and βmut/βN (MCV 66.09 ± 7.86 fL, MCH 21.15 ± 2.95 pg), group F with genotype αααanti3.7 or anti4.2/αα (MCV 68.57 ± 10.22 fL, MCH 21.46 ± 3.73 pg) and βmut/βN and group G with genotype αααanti4.2/-α3.7 and βmut/βN (MCV 63.47 ± 3.35 fL, MCH 20.27 ± 1.02 pg) were significantly lower than those of group A (MCV 89.51 ± 5.89 fL, MCH 29.73 ± 2.21 pg, Table 1). Moreover, the MCV and MCH of group H (αααanti3.7/--SEA and βmut/βN) with only one subject were also lower than those of group A (Table 1). However, the hematological parameters of subjects in group F and G showed no significant difference in comparison with that of group B. In addition, Hb level did not vary significantly among the eight groups. These results indicated that at the population level, the α-globin gene triplication did not have large effect on the carriers in terms of hematological parameters including MCV, MCH, and Hb levels.
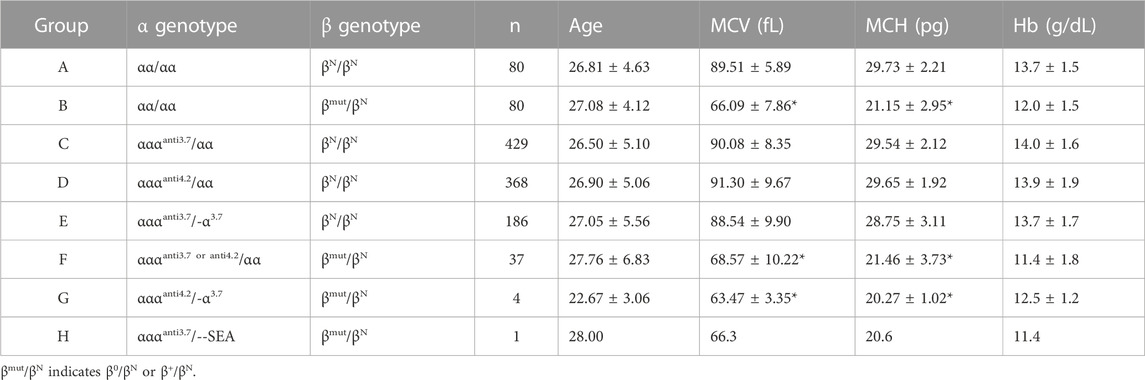
TABLE 1. Hematological parameters of samples with α gene triplication.
3.4 Clinical features in subjects
To gain a further understanding of the effect of α-globin gene triplication, eight subjects with α-globin gene triplication and heterozygous β-thalassemia were successfully follow-up visited. None of them had blood transfusion history. MLPA results validated that they had triplicated α-globin genes, which was consistent with the NGS results (Supplementary Table S2 and Supplementary Figure S1). Hematological analysis revealed that all of eight subjects had Hb A
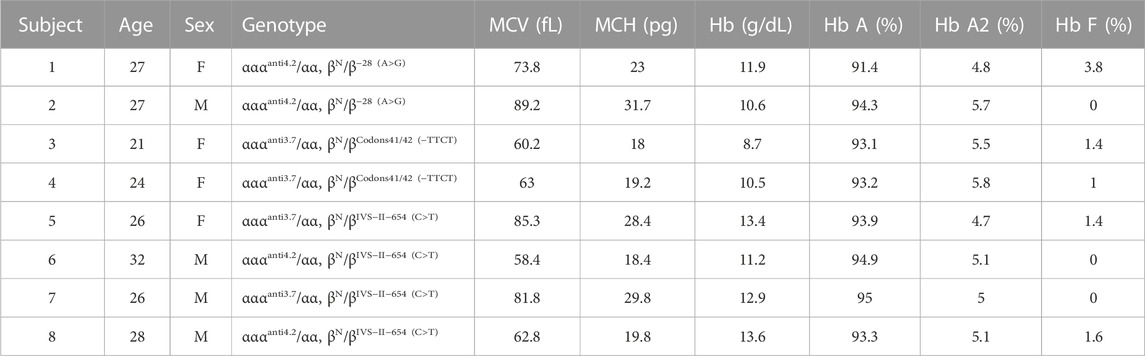
TABLE 2. Hematological and molecular data from carriers with α-globin gene triplication and heterozygous β-thalassemia.
4 Discussion
Since the discovery of thalassemia, pathophysiology characteristics and molecular basis of thalassemia has been studied extensively (Fibach and Rachmilewitz, 2017; Mettananda and Higgs, 2018). It is generally recognized that the imbalance between the α- and β-globin chain can result in hemoglobin disorders, such as hemoglobin H disease characterized by hemolysis with varied severities, and Bart’s Hydrops fetalis characterized by death in utero. However, the α-globin gene triplication was not identified until the 1980s (Higgs et al., 1980). Because α-globin gene triplication carriers are usually asymptomatic, the effects of α-globin gene triplication on thalassemia were easily ignored in epidemiological surveys (Zhuang et al., 2020; Peng et al., 2021). However, co-inheritance of α-globin gene triplication and heterozygous β-thalassemia might result in β-thalassemia intermedia (Quek and Thein, 2007). Therefore, it is necessary to investigate the prevalence of α-globin gene triplication and the phenotypic effects resulting from the co-inheritance of α-globin gene triplication and heterozygous β-thalassemia.
4.1 Prevalence of triplicated α-globin gene
The prevalence of α-globin gene triplication has been described in different populations. In a cohort of Sri Lanka of 620 β-thalassemia carriers, the carrier rate of α-globin gene triplication was 2.0% (Fisher et al., 2003). Another study studied 106 sickle cell patients living in the Democratic Republic of Congo, and found 11.3% patients were α-globin gene triplication carriers coinherited with α3.7 (Mikobi et al., 2018). In the Iranian cohort, the α-globin gene triplication carrier rate was much lower, at 0.9% in patients with sickle cell anemia and 1.67% in patients with β-thalassemia (Hamid et al., 2021). In this study, we reported the prevalence of α-globin gene triplication in Ganzhou city for the first time in a large and randomly recruited cohort of 73967 subjects. In our study, the carrier rate of α-globin gene triplication was 1.95% (1,443/73,967) (Figure 1C), which was close to the carrier rate of 2.3% of heterozygous β-thalassemia in Ganzhou city (Lin et al., 2014). Compared to previous studied, Ganzhou had a higher carrier rate of α-globin gene triplication than the Iranian population (1.2%) (Farashi et al., 2015), Dutch population (1%) (Giordano, Bakker-Verwij, and Harteveld, 2009), Guangdong province (1.2%) (Xie et al., 2015), Indians (1.1%) (Nadkarni et al., 2008) and Guizhou province (0.77%) (X. Luo et al., 2021). Unpublished data showed that the frequency of β-thalassemia carriers is 3.7% (2768/73967), of which 1.51% (42/2768) are α-globin gene triplication carriers. This frequency was slightly lower than that in Iran (1.67%) (Hamid et al., 2021). These differences were consistent with the findings that the carrier rates of the α-globin gene triplication varies among different populations (Goossens et al., 1980). Therefore, our result more accurately reflects the real prevalence of α-globin gene triplication in Ganzhou city through NGS, and 42 subjects with co-inheritance of α-globin gene triplication and heterozygous β-thalassemia were vital genetic resources for studying the influence of α-globin gene triplication on β-thalassemia.
4.2 Geographical distribution of triplicated α-globin gene
In China, the prevalence of thalassemia was higher in the north than in the south, which had been proved in several previous studies (Lin et al., 2014; Lai et al., 2017). In Guizhou province, the carrier rate of α-globin gene triplication was significantly higher in Qiannan (2.23%) than in other regions of Qiandongnan (1.34%), Qianbei (0.62%) and Qianxi (0.43%) (X. Luo et al., 2021), which was in accordance with the geographical distribution of thalassemia in China. Although published data showed that the carrier rate of thalassemia in the 18 counties of Ganzhou city followed this pattern (Yang et al., 2023), the distribution of α-globin gene triplication in these counties seemed to be random (Figure 1). In the south of Ganzhou, low prevalence was found in Quannan (1.64%), Xunwu (1.52%) and Xinfeng (1.65%), while higher prevalence was found in Dingnan (2.01%). In the north of Ganzhou, higher prevalence was found in Ningdu (2.01%) and Shicheng (2.39%), while lower prevalence was found in Xingguo (1.73%). These findings suggested that the distribution pattern of α-globin gene triplication was distinct from that of thalassemia and appeared to be random across the counties in Ganzhou city.
4.3 Interaction of triplicated α-globin gene and heterozygous β-thalassemia
Our study identified 42 carriers of both heterozygous β-thalassemia and α-globin gene triplication, providing a valuable resource to assess the interaction between heterozygous β-thalassemia and α-globin gene triplication in the Chinese population. The imbalance between α- and β-globin chains is the key to cause thalassemia. Several researchers have proposed that due to the increased α-globin accumulation, the co-inheritance of α-globin gene triplication and heterozygous β-thalassemia could result in poor clinical symptoms, such as β-thalassemia intermedia (Ma et al., 2001; Mehta et al., 2015; Ropero et al., 2022). In consistence with these studies, despite our clinical follow-up results indicated that eight carriers with both heterozygous β-thalassemia and α-globin gene triplication had no history of blood transfusion, 6 carriers exhibited anemic symptoms (subjects 1, 2, 3, 4, 6, and 7), including three moderate anemic carriers (subjects 2, 3, and 4). Moreover, the hemoglobin results revealed heterogenous anemia degrees among the subjects with identical genotype. This observation applies to subjects 1 and 2 with genotype αααanti4.2/αα and βN/β−28 (A>G), as well as subjects 6 and 8 with genotype αααanti3.7/αα and βN/βIVS−II−654 (C>T). It is intriguing to note that Camaschella et al. (1997) also observed phenotypic differences among the individuals with identical genotypes (Camaschella et al., 1997). Therefore, it is recommended to provide increased clinical attentions to the anemic individuals who had both α-globin gene triplication and heterozygous β-thalassemia.
Contrarily, a previous study conducted in an Iranian population suggested that there is no necessity to allocate additional attention to α-globin gene triplication in the carriers of heterozygous β-thalassemia (Hamid et al., 2021). Moreover, another study reported that 10 carriers of heterozygous β-thalassemia with α-globin gene triplication were asymptomatic (Mehta et al., 2015). In accordance with these findings, our findings indicated that α-globin gene triplication did not result in significant change on the mean values of hematological parameters (including MCV, MCH, and Hb levels) in either heterozygous β-thalassemia population or normal population, as demonstrated in Table 1.
We speculate that such discrepant findings observed in different studies may be attributed to the subjects analyzed. Some studies only focused on the symptomatic patients who sought medical attentions at hospitals (Ma et al., 2001; Theodoridou et al., 2020; Ropero et al., 2022), whereas our study, along with the Iranian cohort, analyzed both asymptomatic and symptomatic individuals (Hamid et al., 2021). We recommend that α-globin gene triplication should not be considered as a high-risk factor during prenatal diagnosis decisions or genetic counseling, and that the presence of α-globin gene triplication in healthy individuals should not receive extensive clinical attention. Certainly, in heterozygous β-thalassemia carriers who exhibit corresponding symptoms, clinical attention should be directed towards the potential involvement of α-globin gene triplication.
In this study, we have discovered that the α-globin gene triplication has a limited impact on the anemia phenotype. Although we thoroughly assessed all the globin gene mutations in triplication carriers, some information has not been covered, such as HBA12 which has garnered substantial attention in the research community (S.Q. Luo et al., 2020; Borgio et al., 2014). Additionally, a previous study has suggested a link between α globin gene copy number and chronic kidney disease, as well as end-stage kidney disease (Ruhl et al., 2022). Therefore, we recommend that future research endeavors continue to investigate the influence of other co-inherited variants with the triplication on the phenotype. Furthermore, it is advisable to consider monitoring the triplication’s effects on kidney function in subsequent clinical studies.
4.4 Conclusion
In conclusion, this study conducted an extensive survey to evaluate the occurrence and distribution of α-globin gene triplication in Ganzhou city. The results revealed that α-globin gene triplication is randomly distributed among various counties, with an overall carrier rate of 1.95%. The study also emphasized the diverse clinical manifestations of β-thalassemia with α-globin gene triplication, underscoring the importance of cautious assessment in clinical practice. These findings enrich the existing genetic data on α-globin gene triplication and offer valuable insights for diagnosing atypical cases in the future.
Data availability statement
In accordance with ethical considerations, the data presented in the current study are available from the corresponding authors upon request.
Ethics statement
This study has been reviewed and approved by the Ethics Review Committees of Maternal and Child health hospital of Ganzhou city (No. 81 of 2022) and BGI (BGI-IRB 23045). Consent forms were signed by all the subjects. The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author contributions
XX: Formal Analysis, Writing–original draft, Writing–review and editing. JG: Formal Analysis, Writing–original draft, Writing–review and editing. ZL: Conceptualization, Formal Analysis, Validation, Writing–original draft, Writing–review and editing. YuZ: Data curation, Investigation, Writing–original draft. KY: Data curation, Investigation, Conceptualization, Writing–original draft. ZC: Formal Analysis, Validation, Conceptualization, Writing–original draft. SC: Methodology, Validation, Conceptualization, Writing–original draft. RZ: Conceptualization, Methodology, Writing–original draft. LL: Methodology, Validation, Conceptualization, Writing–original draft. XH: Methodology, Validation, Conceptualization, Writing–original draft. YaZ: Methodology, Validation, Conceptualization, Writing–original draft. QL: Methodology, Validation, Conceptualization, Writing–original draft. WZ: Conceptualization, Supervision, Writing–review and editing. JH: Conceptualization, Supervision, Writing–review and editing. JC: Conceptualization, Supervision, Writing–review and editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This study is supported by Jiangxi Provincial Natural Science Foundation (20232BAB216098).
Acknowledgments
We thank the patients for their willingness to participate in this study.
Conflict of interest
Authors ZL, KY, ZC, SC, RZ, LL, XH, YaZ, QL, and WZ were employed by the company BGI Genomics.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2023.1267892/full#supplementary-material
References
Borgio, J. F., AbdulAzeez, S., Al-Nafie, A. N., Naserullah, Z. A., Al-Jarrash, S., Al-Madan, M. S., et al. (2014). A novel HBA2 gene conversion in cis or trans: "α12 allele" in a Saudi population. Blood Cells Mol. Dis. 53 (4), 199–203. doi:10.1016/j.bcmd.2014.07.001
Camaschella, C., Kattamis, A. C., Petroni, D., Roetto, A., Sivera, P., Sbaiz, L., et al. (1997). Different hematological phenotypes caused by the interaction of triplicated alpha-globin genes and heterozygous beta-thalassemia. Am. J. Hematol. 55 (2), 83–88. doi:10.1002/(sici)1096-8652(199706)55:2<83:aid-ajh6>3.0.co;2-z
Farashi, S., Bayat, N., Faramarzi Garous, N., Ashki, M., Montajabi Niat, M., Vakili, S., et al. (2015). Interaction of an α-globin gene triplication with β-globin gene mutations in Iranian patients with β-thalassemia intermedia. Hemoglobin 39 (3), 201–206. doi:10.3109/03630269.2015.1027914
Fibach, E., and Rachmilewitz, E. A. (2017). Pathophysiology and treatment of patients with beta-thalassemia - an update. F1000Res 6, 2156. doi:10.12688/f1000research.12688.1
Fisher, C. A., Premawardhena, A., de Silva, S., Perera, G., Rajapaksa, S., Olivieri, N. A., et al. (2003). The molecular basis for the thalassaemias in Sri Lanka. Br. J. Haematol. 121 (4), 662–671. doi:10.1046/j.1365-2141.2003.04346.x
Gharaibeh, H., Barqawi, M. A., Al-Awamreh, K., and Al Bashtawy, M. (2018). Clinical burdens of β-thalassemia major in affected children. J. Pediatr. Hematol. Oncol. 40 (3), 182–187. doi:10.1097/mph.0000000000001104
Giordano, P. C., Bakker-Verwij, M., and Harteveld, C. L. (2009). Frequency of alpha-globin gene triplications and their interaction with beta-thalassemia mutations. Hemoglobin 33 (2), 124–131. doi:10.1080/03630260902827684
Goossens, M., Dozy, A. M., Embury, S. H., Zachariades, Z., Hadjiminas, M. G., Stamatoyannopoulos, G., et al. (1980). Triplicated alpha-globin loci in humans. Proc. Natl. Acad. Sci. U. S. A. 77 (1), 518–521. doi:10.1073/pnas.77.1.518
Hamid, M., Keikhaei, B., Galehdari, H., Saberi, A., Sedaghat, A., Shariati, G., et al. (2021). Alpha-globin gene triplication and its effect in beta-thalassemia carrier, sickle cell trait, and healthy individual. EJHaem 2 (3), 366–374. doi:10.1002/jha2.262
Higgs, D. R., Old, J. M., Pressley, L., Clegg, J. B., and Weatherall, D. J. (1980). A novel alpha-globin gene arrangement in man. Nature 284 (5757), 632–635. doi:10.1038/284632a0
Lai, K., Huang, G., Su, L., and He, Y. (2017). The prevalence of thalassemia in mainland China: evidence from epidemiological surveys. Sci. Rep. 7 (1), 920. doi:10.1038/s41598-017-00967-2
Lin, M., Zhong, T. Y., Chen, Y. G., Wang, J. Z., Wu, J. R., Lin, F., et al. (2014). Molecular epidemiological characterization and health burden of thalassemia in Jiangxi Province, P. R. China. PLoS One 9 (7), e101505. doi:10.1371/journal.pone.0101505
Long, J., and Liu, E. (2021). The carriage rates of αααanti3.7, αααanti4.2, and HKαα in the population of Guangxi, China measured using a rapid detection qPCR system to determine CNV in the α-globin gene cluster. Gene 768, 145296. doi:10.1016/j.gene.2020.145296
Luo, S. Q., Chen, X. Y., Tang, N., Huang, J., Zhong, Q. Y., Cai, R., et al. (2020). Pedigree analysis of nonhomologous sequence recombination of HBA1 and HBA2 genes. Hemoglobin 44 (5), 329–333. doi:10.1080/03630269.2020.1807355
Luo, X., Zhang, X. M., Wu, L. S., Chen, J., and Chen, Y. (2021). Prevalence and clinical phenotype of the triplicated α-globin genes and its ethnic and geographical distribution in Guizhou of China. BMC Med. Genomics 14 (1), 97. doi:10.1186/s12920-021-00944-9
Ma, S. K., Au, W. Y., Chan, A. Y., and Chan, L. C. (2001). Clinical phenotype of triplicated alpha-globin genes and heterozygosity for beta0-thalassemia in Chinese subjects. Int. J. Mol. Med. 8 (2), 171–175.doi:10.3892/ijmm.8.2.171
Majid, N., Moghaddam Ebrahim, M., Akbar, D., Shaban, A., Shadi, T., and Masoud, P. (2015). Case report: a patient with coinheritance of alpha-globin gene triplication and ivsi-5 mutation of beta-globin gene. J. Zanjan Univ. Med. Sci. Health Serv. 8 (12), 1. doi:10.17795/zjrms975
Mehta, P. R., Upadhye, D. S., Sawant, P. M., Gorivale, M. S., Nadkarni, A. H., Shanmukhaiah, C., et al. (2015). Diverse phenotypes and transfusion requirements due to interaction of β-thalassemias with triplicated α-globin genes. Ann. Hematol. 94 (12), 1953–1958. doi:10.1007/s00277-015-2479-8
Mettananda, S., and Higgs, D. R. (2018). Molecular basis and genetic modifiers of thalassemia. Hematol. Oncol. Clin. North Am. 32 (2), 177–191. doi:10.1016/j.hoc.2017.11.003
Mikobi, T. M., Lukusa, P. T., Aloni, M. N., Lumaka, A., Akilimali, P. Z., Devriendt, K., et al. (2018). Association between sickle cell anemia and alpha thalassemia reveals a high prevalence of the α3.7 triplication in congolese patients than in worldwide series. J. Clin. Lab. Anal. 32 (1), e22186. doi:10.1002/jcla.22186
Muncie, H. L., and Campbell, J. (2009). Alpha and beta thalassemia. Am. Fam. Physician 80 (4), 339–344.
Nadkarni, A., Phanasgaonkar, S., Colah, R., Mohanty, D., and Ghosh, K. (2008). Prevalence and molecular characterization of alpha-thalassemia syndromes among Indians. Genet. Test. 12 (2), 177–180. doi:10.1089/gte.2007.0080
Peng, Q., Zhang, Z., Li, S., Cheng, C., Li, W., Rao, C., et al. (2021). Molecular epidemiological and hematological profile of thalassemia in the dongguan region of Guangdong province, southern China. J. Clin. Lab. Anal. 35 (2), e23596. doi:10.1002/jcla.23596
Pepe, A., Pistoia, L., Gamberini, M. R., Cuccia, L., Lisi, R., Cecinati, V., et al. (2022). National networking in rare diseases and reduction of cardiac burden in thalassemia major. Eur. Heart J. 43 (26), 2482–2492. doi:10.1093/eurheartj/ehab851
Qiu, Y., Mao, L., Chen, S.-P., Li, H., Wang, H., Guan, L., et al. (2020). Large-scale screening of thalassemia in Ji’an, P.R. China. Journal of Women's Health and Development.
Quek, L., and Thein, S. L. (2007). Molecular therapies in beta-thalassaemia. Br. J. Haematol. 136 (3), 353–365. doi:10.1111/j.1365-2141.2006.06408.x
Ropero, P., González Fernández, F. A., Nieto, J. M., Torres-Jiménez, W. M., and Benavente, C. (2022). β-Thalassemia intermedia: interaction of α-globin gene triplication with β-thalassemia heterozygous in Spain. Front. Med. (Lausanne) 9, 866396. doi:10.3389/fmed.2022.866396
Ruhl, A. P., Jeffries, N., Yang, Y., Naik, R. P., Patki, A., Pecker, L. H., et al. (2022). Alpha globin gene copy number is associated with prevalent chronic kidney disease and incident end-stage kidney disease among black Americans. J. Am. Soc. Nephrol. 33 (1), 213–224. doi:10.1681/asn.2021050653
Theodoridou, S., Balassopoulou, A., Boutou, E., Delaki, E. E., Yfanti, E., Vyzantiadis, T. A., et al. (2020). Coinheritance of triplicated alpha-globin gene and beta-thalassemia mutations in adulthood: ten years of referrals in northern Greece. J. Pediatr. Hematol. Oncol. 42 (8), e762–e764. doi:10.1097/mph.0000000000001730
Vlok, M., Buckley, H. R., Miszkiewicz, J. J., Walker, M. M., Domett, K., Willis, A., et al. (2021). Forager and farmer evolutionary adaptations to malaria evidenced by 7000 years of thalassemia in Southeast Asia. Sci. Rep. 11 (1), 5677. doi:10.1038/s41598-021-83978-4
Wang, W., Ma, E. S., Chan, A. Y., Prior, J., Erber, W. N., Chan, L. C., et al. (2003). Single-tube multiplex-PCR screen for anti-3.7 and anti-4.2 alpha-globin gene triplications. Clin. Chem. 49 (10), 1679–1682. doi:10.1373/49.10.1679
Weatherall, D. J., and Clegg, J. B. (2001). “The α thalassaemias and their interactions with structural haemoglobin variants,” in The thalassaemia syndromes, 484–525.
WHO (2011). Haemoglobin concentrations for the diagnosis of anaemia and assessment of severity. World Health Organization.
Wu, M. Y., Zhou, J. Y., Li, J., and Li, D. Z. (2016). The frequency of α-globin gene triplication in a southern Chinese population. Indian J. Hematol. Blood Transfus. 32 (1), 320–322. doi:10.1007/s12288-015-0588-0
Xi, H., Liu, Q., Xie, D. H., Zhou, X., Tang, W. L., Tang, G., et al. (2023). Epidemiological survey of hemoglobinopathies based on next-generation sequencing platform in hunan province, China. Biomed. Environ. Sci. 36 (2), 127–134. doi:10.3967/bes2023.016
Xie, X. M., Wu, M. Y., and Li, D. Z. (2015). Evidence of selection for the α-globin gene deletions and triplications in a southern Chinese population. Hemoglobin 39 (6), 442–444. doi:10.3109/03630269.2015.1072551
Yang, T., Luo, X., Liu, Y., Lin, M., Zhao, Q., Zhang, W., et al. (2023). Next-generation sequencing analysis of the molecular spectrum of thalassemia in Southern Jiangxi, China. Hum. Genomics 17 (1), 77. doi:10.1186/s40246-023-00520-5
Zhang, H., Li, C., Li, J., Hou, S., Chen, D., Yan, H., et al. (2019). Next-generation sequencing improves molecular epidemiological characterization of thalassemia in Chenzhou Region, P.R. China. J. Clin. Lab. Anal. 33 (4), e22845. doi:10.1002/jcla.22845
Zhao, P., Wu, H., and Weng, R. (2018). Molecular analysis of hemoglobinopathies in a large ethnic Hakka population in southern China. Med. Baltim. 97 (45), e13034. doi:10.1097/md.0000000000013034
Keywords: thalassemia, α-globin gene triplication, prevalence, geographic distribution, Ganzhou, molecular diagnosis
Citation: Xie X, Gan J, Liu Z, Zhou Y, Yuan K, Chen Z, Chen S, Zhou R, Liu L, Huang X, Zhang Y, Liu Q, Zhang W, Huang J and Chen J (2023) Prevalence and genetic analysis of triplicated α-globin gene in Ganzhou region using high-throughput sequencing. Front. Genet. 14:1267892. doi: 10.3389/fgene.2023.1267892
Received: 27 July 2023; Accepted: 26 September 2023;
Published: 19 October 2023.
Edited by:
Giovanni Malerba, University of Verona, ItalyReviewed by:
Hans Ackerman, National Institute of Allergy and Infectious Diseases, United StatesXianke Zeng, National Institute of Health, in collaboration with reviewer HA
J. Francis Borgio, Imam Abdulrahman Bin Faisal University, Saudi Arabia
Copyright © 2023 Xie, Gan, Liu, Zhou, Yuan, Chen, Chen, Zhou, Liu, Huang, Zhang, Liu, Zhang, Huang and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wenqian Zhang, emhhbmd3ZW5xaWFuNTZAZ21haWwuY29t; Jungao Huang, anVuZ2FvaHVhbmdAc2luYS5jb20=; Junkun Chen, MTkwODE5MzU1M0BxcS5jb20=
†These authors have contributed equally to this work and share first authorship