 Yeping Wang
Yeping Wang Fang Sheng1
Fang Sheng1 Zhaonan Yu
Zhaonan Yu Haoyi Wang
Haoyi Wang
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Genet., 20 October 2023
Sec. Human and Medical Genomics
Volume 14 - 2023 | https://doi.org/10.3389/fgene.2023.1267241
Background: Research on fetal loss related to germline mutations in single genes remains limited. Disruption of CEP55 has recently been established in association with perinatal deaths characterized by hydranencephaly, renal dysplasia, oligohydramnios, and characteristic dysmorphisms. We herein present a Chinese family with recurrent fetal losses due to compound heterozygous nonsense CEP55 variants.
Case presentations: The Chinese couple had a history of five pregnancies, with four of them proceeding abnormally. Two stillbirths (II:3 and II:4) sequentially occurred in the third and fourth pregnancy. Prenatal ultrasound scans revealed phenotypic similarities between fetuses II:3 and II:4, including oligohydramnios, bilateral renal dysplasia and hydrocephalus/hydranencephaly. Clubfoot and syndactyly were also present in both stillborn babies. Fetus II:3 presented with endocardial cushion defects while fetus II:4 did not. With the product of conception in the fourth pregnancy, whole exome sequencing (WES) on fetus II:4 identified compound heterozygous nonsense CEP55 variants comprised of c.190C>T(p.Arg64*) and c.208A>T(p.Lys70*). Both variants were expected to result in lack of the TSG101 and ALIX binding domain. Sanger sequencing confirmed the presence and cosegregation of both variants.
Conclusion: This is the fifth reported family wherein biallelic CEP55 variants lead to multiple perinatal deaths. Our findings, taken together with previously described phenotypically similar cases and even those with a milder and viable phenotype, broaden the genotypic and phenotypic spectrum of CEP55-associated lethal fetal syndrome, highlighting the vital biomolecular function of CEP55.
Fetal loss, one of the most severe adverse outcomes of pregnancy, can lead to long-lasting grief, guilt, anxiety, self-blame, post-traumatic stress disorder, and marriage breakdown (Robinson, 2014; Smith et al., 2022). The annual prevalence of fetal loss at ≥20 weeks (stillbirth) is estimated at 13.2 and 5.74 per 1,000 births in China and the United States, respectively (Zhu et al., 2021; Gregory et al., 2022). A subgroup of fetal losses have been attributed to Mendelian diseases caused by pathogenic single-nucleotide variants or small insertions/deletions, though the relevant data are limited (Shamseldin et al., 2018; Sahlin et al., 2019; Stanley et al., 2020).
The Centrosomal Protein 55 kDa (CEP55) gene located at chromosome 10q23 encodes a coiled-coil centrosomal protein that promotes the abscission process, the second stage of cytokinesis, through the recruitment of Endosomal Sorting Complex Required for Transport (ESCRT) machinery to the midbody (Fabbro et al., 2005; Lee et al., 2008). This protein harbors 464 amino acids with three central coiled-coil domains, and phosphorylation of the three C-terminal serine residues (p.S425, p.S428, p.S436) is indispensable to abscission (Fabbro et al., 2005; van der Horst et al., 2009). Recently homozygous truncating mutations in CEP55 have been identified as a cause of a lethal fetal condition, termed Meckel-like syndrome or MARCH syndrome (acronym for Multinucleated neurons, Anhydramnios, Renal dysplasia, Cerebellar hypoplasia, and Hydranencephaly, OMIM #236500), characterized by fetal loss with severe congenital malformations including hydranencephaly, renal dysplasia, oligohydramnios, and characteristic dysmorphisms (Bondeson et al., 2017; Frosk et al., 2017). To date only three studies have delineated this lethal CEP55-associated syndrome, though viable phenotypes linked to biallelic CEP55 variants have also been reported (Bondeson et al., 2017; Frosk et al., 2017; Rawlins et al., 2019; Barrie et al., 2020).
We herein present a Chinese family with a history of recurrent fetal loss, in which multiple fetuses present with complex brain and kidney malformations, oligohydramnios, and physical anomalies. Whole exome sequencing (WES) reveals compound heterozygous variants comprised of two nonsense mutations in the CEP55 gene, NM_018131.4(CEP55): c.190C > T(p.Arg64*) and c.208A > T(p.Lys70*). Given that the relevant studies are scarce, our case report expands the genotypic and phenotypic spectrum, thereby providing a better understanding of CEP55-associated lethal fetal syndrome.
The Chinese couple with five past pregnancies presented to our clinic with a history of recurrent pregnancy loss (Figure 1). In accordance to patient inquiries, they had no known consanguinity and history of antepartum teratogenic exposures. In the first pregnancy, nothing abnormal but oligohydramnios was documented and a healthy boy (II:1) was born by cesarean section in 2013. This 11-year-old boy has no any congenital defect or serious disease to date. The second pregnancy ended by a missed abortion due to embryonic demise in gestational week (GW) 13 + 3 in 2019 (II:2). No clinical or genetic material was available.
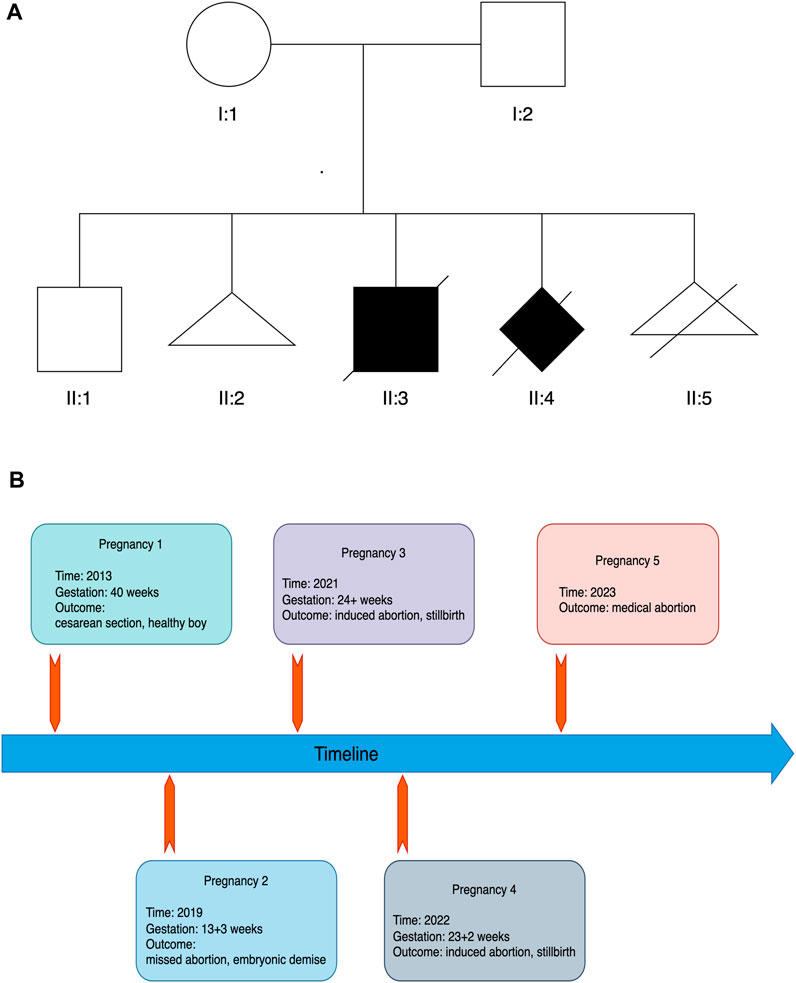
FIGURE 1. Pedigree (A) and timeline of pregnancies (B) in the family.
In the third pregnancy, a first trimester ultrasound scan (GW 12 + 2) revealed soft tissue swelling throughout the fetus (II:3) with an increased thickness of nuchal translucency (2.7 mm). A second trimester ultrasound scan was then performed at GW 21 + 4 and showed that fetus II:3 presented with hydrops, severe hydrocephalus (Figure 2A), ascites (Figure 2B), pleural effusion (Figure 2C), oligohydramnios, polycystic dysplasia of bilateral kidneys (Figures 2D, E), clubfoot (Figure 2F) and endocardial cushion defects (Figure 2G). Of note, a clear view of detailed brain structures by ultrasound scan was unavailable due to the fetal position, and thus we could not rule out the possibility of hydranencephaly. Owing to the fetal anomalies and the predicted lethal outcome, the pregnancy was terminated by an induced abortion at GW 24+ in 2021. A stillborn male fetus weighing 680g was delivered, displaying neck swelling, generalized edema, clubbed feet and syndactyly.
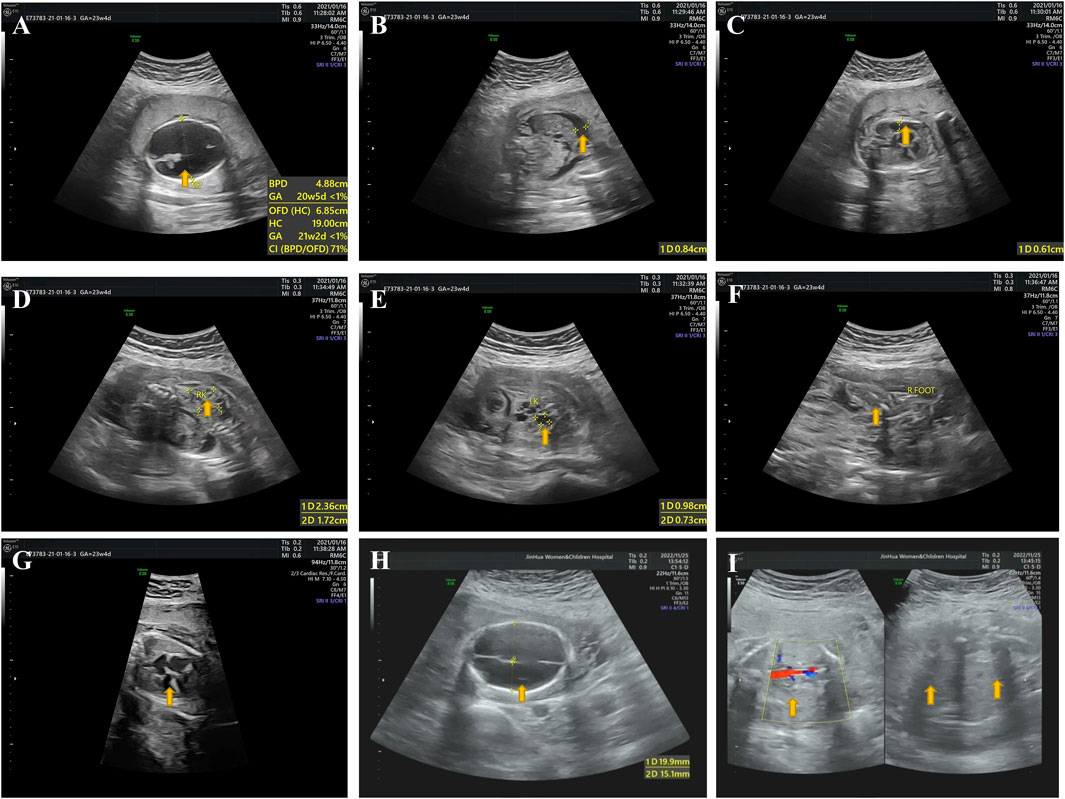
FIGURE 2. Ultrasound pictures from the third and fourth pregnancy (fetuses II:3 and II:4). (A) II:3: hydrocephalus. (B) II:3: ascites. (C) II:3: pleural effusion. (D) II:3: polycystic dysplasia of right kidney. (E) II:3: polycystic dysplasia of left kidney. (F) II:3: clubbed right foot. (G) II:3: endocardial cushion defects. (H) II:4: hydranencephaly. (I) II:4: bilateral renal agenesis. Yellow arrows indicate where the anomalies occur.
In the fourth pregnancy, a second trimester ultrasound scan (GW 23 + 1) revealed a fetus (II:4) with hydranencephaly (fluid-filled sacs and missing cerebral tissues, preservation of cerebral midline and thalamus, non-visualized cerebellum and posterior fossa structures; Figure 2H), anhydramnios (amniotic fluid index: 0 cm) and bilateral renal agenesis (Figure 2I). Face and limbs could not be identified. Also, ultrasound measurements indicated a fetal age at GW 20 + 1, suggesting an intrauterine fetal growth restriction (IUGR). Like the third pregnancy, this pregnancy ended by an induced abortion at GW 23 + 2 in 2022, leading to a stillbirth of a fetus of unknown gender manifesting syndactyly and clubfoot. Postmortem histological examinations were not performed on fetuses II:3 and II:4 owing to severe autolysis.
The fifth pregnancy accidentally occurred in 2023. After a genetic consultation, the couple voluntarily abandoned this pregnancy and a medical abortion was thus administered.
To uncover genetic factors contributing to the fetal malformations, chromosomal microarray and WES were performed on fetus II:4 with the product of conception (POC). The chromosomal microarray analysis revealed no genetic abnormalities, excluding the possibility of pathogenic copy number variations. The WES analysis identified compound heterozygous nonsense mutations, NM_018131.4(CEP55): c.190C > T(p.Arg64*) and c.208A > T(p.Lys70*), both of which were predicted to result in a premature stop and produce a truncated CEP55 protein lacking the TSG101 and ALIX binding domain (Figure 3A). The nonsense CEP55 variant p.Arg64* (ClinVar ID: 1065435) is classified as likely pathogenic in the ClinVar database. This variant in the compound heterozygous state has previously been reported in a case of nonimmune hydrops fetalis (NIHF) with a second missense CEP55 variant p.His458Arg (Sparks et al., 2020). The nonsense CEP55 variant p.Arg70* is not included in the ClinVar or HGMD databases. Both variants are listed in gnomeAD, but the lack of homozygotes and extremely low allele frequencies (1.208e−5 for p.Arg64* and 1.595e−5 for p.Lys70*) do not argue against causality for an autosomal recessive disease. Moreover, Sanger sequencing confirmed that both variants were located in trans and segregated with disease in the family (Figure 3B). According to the ACMG guidelines (Richards et al., 2015), we have classified these two variants as pathogenic. No other pathogenic variants were identified. Based on the clinical and genetic findings, fetus II:4 has been diagnosed with MARCH syndrome (or Meckel-like syndrome).
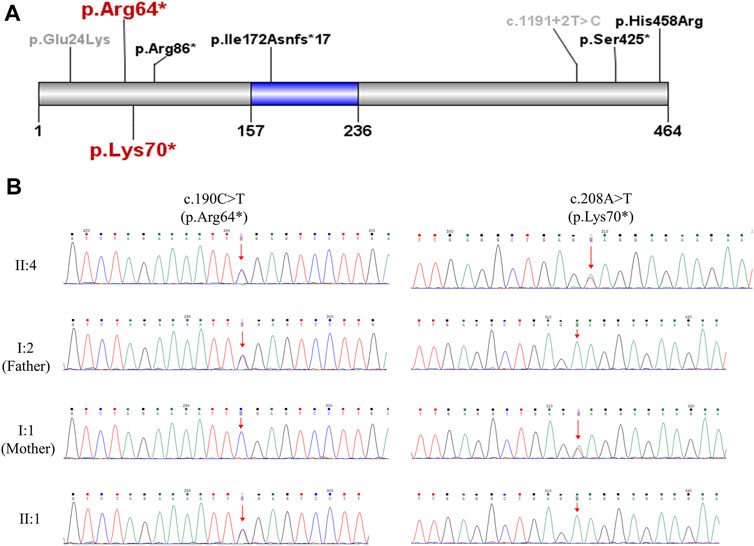
FIGURE 3. Genetic analysis of the CEP55 variants. (A) Schematic representation of CEP55 protein indicating the position of the disease-associated variants. The region marked in blue (residue 157–236) is tumour suppressor gene 101 (TSG101) and apoptosis-linked gene 2 interacting protein X (ALIX) binding domain. The two variants p.Arg64* and p.Lys70* identified in the current study are highlighted in red. The variants that caused perinatal deaths in previous studies (Bondeson et al., 2017; Frosk et al., 2017; Rawlins et al., 2019; Sparks et al., 2020) are marked in black. The variants identified in viable cases (Barrie et al., 2020) are marked in gray. (B) Sanger sequencing validation of the two CEP55 variants in the family.
To our knowledge, this is the fifth described family with perinatal deaths caused by biallelic CEP55 mutations (Table 1). It is with regret that we did not obtain materials to perform WES on fetus II:3 to consolidate our results because the third pregnancy was not followed up in our hospital. However, we believe that fetuses II:3 and II:4 harbor the same genetic aetiology given their high phenotypical similarities, including features of renal dysplasia, oligohydramnios, clubfoot, and central nervous system (CNS) abnormalities.
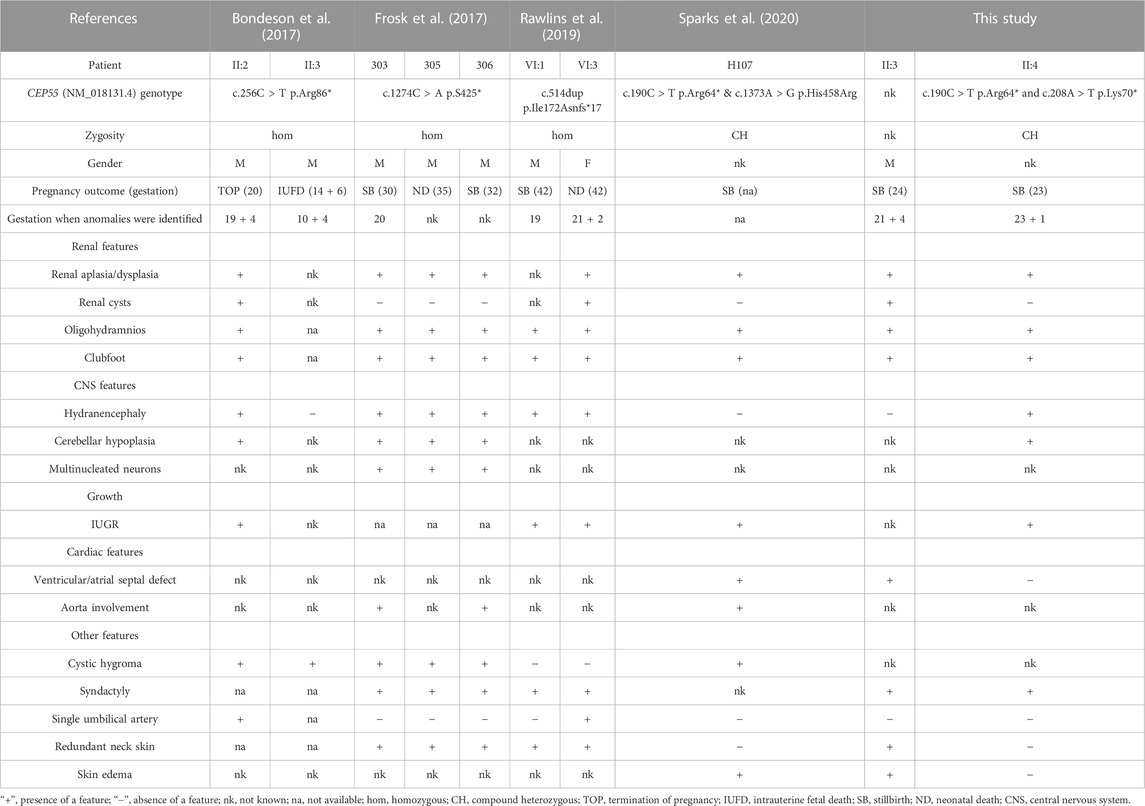
TABLE 1. Comparison of perinatal deaths due to biallelic CEP55 variants.
The CEP55-associated lethal fetal syndrome was firstly defined by Bondeson et al. (2017) and Frosk et al. (2017) in 2017. Bondeson et al. (2017) termed this syndromic phenotype Meckel-like syndrome because phenotypic overlap with Meckel syndrome (MKS) was observed in CEP55 fetuses. MKS is a lethal autosomal recessive ciliopathy characterized by a classic triad of renal cystic dysplasia, CNS anomalies and polydactyly. Of note, polydactyly was absent in all CEP55 fetuses. With regard to CNS abnormality, encephalocele dominates MKS cases while hydranencephaly is present in most of CEP55 fetuses (Barisic et al., 2015). In addition, hepatic ductal plate malformation is also a frequent feature of MKS on postmortem histological examinations. To date only three CEP55 fetuses have been autopsied, none of whom displays this anomaly (Frosk et al., 2017). These phenotypic disparities may result from pathomolecular differences. In our view, the CEP55-associated lethal fetal condition could be considered a relatively severe form of MKS given the high incidence of hydranencephaly and renal agenesis. Indeed, CEP55-knockout mouse mutants exhibited microcephaly instead of hydranencephaly (Tedeschi et al., 2020; Little et al., 2021; Zhang et al., 2021), possibly due to interspecies differences concerning brain development between human and mouse.
The work by Frosk et al. (2017) complemented the anatomical and histological characteristics and potential pathogenesis of CEP55-associated lethal fetal syndrome via autopsy and in vitro and in vivo experiments. In addition to the phenotypes mentioned above, the three siblings described by Frosk et al. (2017) with a homozygous downstream nonsense mutation c.1274C > A (p.S425*) displayed multinucleated neurons on autopsy, leading the authors to term this condition MARCH syndrome. This cerebral histological abnormality resulted from abscission failure due to defective localisation of truncated protein to the midbody caused by CEP55 downstream truncating variants (Frosk et al., 2017). Compared with nonsense variants near the end of the gene, all CEP55 truncating variants reported in other relevant studies (Bondeson et al., 2017; Rawlins et al., 2019; Sparks et al., 2020), including ours, were located upstream or within the TSG101 and ALIX binding domain, suggesting a failed recruitment of ESCRT machinery during cytokinesis. Therefore, the pathomechanisms of upstream and downstream truncating variants in the CEP55 gene were likely to be different. Moreover, qPCR on heterozygous parents in previous studies revealed almost equal levels of wild-type and truncated transcript, regardless of upstream or downstream truncation, indicating that CEP55 truncating variants were not targeted by nonsense-mediated mRNA decay pathway (Bondeson et al., 2017; Frosk et al., 2017).
As shown in Table 1, all CEP55-associated dead fetuses harbor biallelic truncating variants except one case with a truncating mutation and a non-truncating (missense) mutation c.1373A > G (p.His458Arg) (Sparks et al., 2020). The exceptional case was described to have hydrocephaly, renal dysplasia, skin edema, clubfoot, oligohydramnios, and complex congenital heart disease, which was consistent with the phenotype of fetus II:3 in our study (Sparks et al., 2020). In view that the missense mutation c.1373A > G (p.His458Arg) is located within the last 40 amino acids, a region essential for localisation to the midbody during cytokinesis (Frosk et al., 2017), we speculate that it might also disrupt localisation of CEP55 to the midbody of dividing cells.
Furthermore, Barrie et al. (2020) depicted a viable phenotype spectrum of CEP55-associated disease in seven cases, all of whom survived the perinatal period and displayed microcephaly, intellectual disability, and mild skeletal abnormalities without renal involvements. Of these patients, four carried compound heterozygous CEP55 variants comprised of a common missense mutation c.70G > A p.(Glu24Lys) and a nonsense mutation, while the other three siblings had a homozygous downstream splice site variant c.1191 + 2T > C. The effect of non-truncating variants on CEP55 function remains unclear.
It was previously established that depletion of CEP55 caused abscission failure in most human cells (such as HeLa cells) in vitro (Fabbro et al., 2005; Lee et al., 2008; van der Horst et al., 2009). However, recent in vivo evidence from CEP55-knockout mice has shown that CEP55 is dispensable for abscission in most cell types except neural stem cells (NSCs), explaining binucleation only in neurons but not other tissues in dead fetuses with CEP55 variants. Even in NSCs where CEP55 and ESCRT are required for survival and abscission, CEP55 just ensures the speed and success of abscission but is not absolutely necessary (Tedeschi et al., 2020; Little and Dwyer, 2021; Little et al., 2021). Further in vivo experiments are needed to elucidate an alternative CEP55-independent pathway for midbody ESCRT recruitment or even an ESCRT-independent cell division mechanism. Additionally, CEP55 has been proposed to function as a key regulator of cilia disassembly through stabilizing Aurora A kinase (Zhang et al., 2021).
Our case report elaborately presents the phenotypes of two Chinese stillborn babies linked to CEP55 variants. This is the first report of CEP55-associated lethal fetal syndrome in China, and endocardial cushion defects observed in fetus II:3 is firstly described in such patients. However, there are some limitations in our study: 1) as mentioned above, WES was not performed on fetus II:3 owing to the unavailability of samples; 2) autopsy was not conducted due to autolysis; 3) functional investigations were absent.
We describe a Chinese family with multiple stillbirths due to biallelic nonsense CEP55 variants. Our findings extend and summarize the genotypic and phenotypic features of CEP55-associated lethal fetal syndrome, equipping clinicians with crucial insights when encountering a family with stillbirths displaying complex brain and kidney malformations.
The datasets for this article are not publicly available due to concerns regarding participant/patient anonymity. Requests to access the datasets should be directed to the corresponding authors.
The studies involving humans were approved by the Ethics Committee of the Jinhua Maternity and Child Health Care Hospital (Approval No. 2023QT026). The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants’ legal guardians/next of kin. Written informed consent was obtained from the individual(s), and minor(s)’ legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
YW: Conceptualization, Data curation, Funding acquisition, Project administration, Writing–original draft. FS: Data curation, Investigation, Methodology, Resources, Writing–original draft. LY: Data curation, Formal Analysis, Investigation, Writing–original draft. QL: Formal Analysis, Visualization, Writing–original draft. ZY: Formal Analysis, Validation, Writing–original draft. KW: Supervision, Writing–review and editing. HW: Conceptualization, Data curation, Supervision, Writing–original draft, Writing–review and editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. Key social development projects of the Jinhua science and technology plan project, Grant No. 2022-3-128; Research and cultivation fund project of the Jinhua Maternity and Child Healthcare Hospital, Grant No. JHFB 2021-2-03.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Barisic, I., Boban, L., Loane, M., Garne, E., Wellesley, D., Calzolari, E., et al. (2015). Meckel-Gruber Syndrome: a population-based study on prevalence, prenatal diagnosis, clinical features, and survival in Europe. Eur. J. Hum. Genet. 23 (6), 746–752. doi:10.1038/ejhg.2014.174
Barrie, E. S., Overwater, E., van Haelst, M. M., Motazacker, M. M., Truxal, K. V., Crist, E., et al. (2020). Expanding the spectrum of CEP55-associated disease to viable phenotypes. Am. J. Med. Genet. A 182 (5), 1201–1208. doi:10.1002/ajmg.a.61512
Bondeson, M. L., Ericson, K., Gudmundsson, S., Ameur, A., Pontén, F., Wesström, J., et al. (2017). A nonsense mutation in CEP55 defines a new locus for a Meckel-like syndrome, an autosomal recessive lethal fetal ciliopathy. Clin. Genet. 92 (5), 510–516. doi:10.1111/cge.13012
Fabbro, M., Zhou, B. B., Takahashi, M., Sarcevic, B., Lal, P., Graham, M. E., et al. (2005). Cdk1/Erk2-and Plk1-dependent phosphorylation of a centrosome protein, Cep55, is required for its recruitment to midbody and cytokinesis. Dev. Cell 9 (4), 477–488. doi:10.1016/j.devcel.2005.09.003
Frosk, P., Arts, H. H., Philippe, J., Gunn, C. S., Brown, E. L., Chodirker, B., et al. (2017). A truncating mutation in CEP55 is the likely cause of MARCH, a novel syndrome affecting neuronal mitosis. J. Med. Genet. 54 (7), 490–501. doi:10.1136/jmedgenet-2016-104296
Gregory, E. C., Valenzuela, C. P., and Hoyert, D. L. (2022). Fetal mortality: united States, 2020. Natl. Vital Stat. Rep. 71 (4), 1–20. doi:10.15620/cdc:118420
Lee, H. H., Elia, N., Ghirlando, R., Lippincott-Schwartz, J., and Hurley, J. H. (2008). Midbody targeting of the ESCRT machinery by a noncanonical coiled coil in CEP55. Science 322 (5901), 576–580. doi:10.1126/science.1162042
Little, J. N., and Dwyer, N. D. (2021). Cep55: abscission boss or assistant? Trends Cell Biol. 31 (10), 789–791. doi:10.1016/j.tcb.2021.07.006
Little, J. N., McNeely, K. C., Michel, N., Bott, C. J., Lettieri, K. S., Hecht, M. R., et al. (2021). Loss of coiled-coil protein Cep55 impairs neural stem cell abscission and results in p53-dependent apoptosis in developing cortex. J. Neurosci. 41 (15), 3344–3365. doi:10.1523/JNEUROSCI.1955-20.2021
Rawlins, L. E., Jones, H., Wenger, O., Aye, M., Fasham, J., Harlalka, G. V., et al. (2019). An Amish founder variant consolidates disruption of CEP55 as a cause of hydranencephaly and renal dysplasia. Eur. J. Hum. Genet. 27 (4), 657–662. doi:10.1038/s41431-018-0306-0
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American college of medical genetics and Genomics and the association for molecular pathology. Genet. Med. 17 (5), 405–424. doi:10.1038/gim.2015.30
Robinson, G. E. (2014). Pregnancy loss. Best. Pract. Res. Clin. Obstet. Gynaecol. 28 (1), 169–178. doi:10.1016/j.bpobgyn.2013.08.012
Sahlin, E., Gréen, A., Gustavsson, P., Liedén, A., Nordenskjöld, M., Papadogiannakis, N., et al. (2019). Identification of putative pathogenic single nucleotide variants (SNVs) in genes associated with heart disease in 290 cases of stillbirth. PLoS One 14 (1), e0210017. doi:10.1371/journal.pone.0210017
Shamseldin, H. E., Kurdi, W., Almusafri, F., Alnemer, M., Alkaff, A., Babay, Z., et al. (2018). Molecular autopsy in maternal-fetal medicine. Genet. Med. 20 (4), 420–427. doi:10.1038/gim.2017.111
Smith, R., Dedman, L., Sultana, Z., Banney, D., and Maiti, K. (2022). Insights into fetal death-a patient resource. Am. J. Obstet. Gynecol. 226 (6), 761–763. doi:10.1016/j.ajog.2022.02.029
Sparks, T. N., Lianoglou, B. R., Adami, R. R., Pluym, I. D., Holliman, K., Duffy, J., et al. (2020). Exome sequencing for prenatal diagnosis in nonimmune hydrops fetalis. N. Engl. J. Med. 383 (18), 1746–1756. doi:10.1056/NEJMoa2023643
Stanley, K. E., Giordano, J., Thorsten, V., Buchovecky, C., Thomas, A., Ganapathi, M., et al. (2020). Causal genetic variants in stillbirth. N. Engl. J. Med. 383 (12), 1107–1116. doi:10.1056/NEJMoa1908753
Tedeschi, A., Almagro, J., Renshaw, M. J., Messal, H. A., Behrens, A., and Petronczki, M. (2020). Cep55 promotes cytokinesis of neural progenitors but is dispensable for most mammalian cell divisions. Nat. Commun. 11 (1), 1746. doi:10.1038/s41467-020-15359-w
van der Horst, A., Simmons, J., and Khanna, K. K. (2009). Cep55 stabilization is required for normal execution of cytokinesis. Cell Cycle 8 (22), 3742–3749. doi:10.4161/cc.8.22.10047
Zhang, Y. C., Bai, Y. F., Yuan, J. F., Shen, X. L., Xu, Y. L., Jian, X. X., et al. (2021). CEP55 promotes cilia disassembly through stabilizing Aurora A kinase. J. Cell Biol. 220 (2), e202003149. doi:10.1083/jcb.202003149
Keywords: CEP55, stillbirth, Meckel syndrome, case report, fetal loss
Citation: Wang Y, Sheng F, Ying L, Lou Q, Yu Z, Wang K and Wang H (2023) CEP55-associated lethal fetal syndrome: a case report of a Chinese family. Front. Genet. 14:1267241. doi: 10.3389/fgene.2023.1267241
Received: 07 August 2023; Accepted: 10 October 2023;
Published: 20 October 2023.
Edited by:
Anupam Basu, National Institute of Biomedical Genomics (NIBMG), IndiaReviewed by:
Xinyu Shao, Suzhou Dushu Lake Hospital, ChinaCopyright © 2023 Wang, Sheng, Ying, Lou, Yu, Wang and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Haoyi Wang, d2h5bWFza2VyQDE2My5jb20=; Kaixuan Wang, a2FpeHVhbjEwMjY4QDEyNi5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.