 Xiaolong Qiu1†
Xiaolong Qiu1† Jinying Luo
Jinying Luo Guilin Li
Guilin Li Jinfu Zhou
Jinfu Zhou- 1Genetic Diagnosis and Therapy Center, Fujian Key Laboratory for Prenatal Diagnosis and Birth Defect, Fujian Maternity and Child Hospital, College of Clinical Medicine for Obstetrics & Gynecology and Pediatrics, Fujian Medical University, Fuzhou, China
- 2Obstetrics and Gynecology Department, Fujian Maternity and Child Hospital, College of Clinical Medicine for Obstetrics & Gynecology and Pediatrics, Fujian Medical University, Fuzhou, China
- 3Department of Preventive Medicine, School of Public Health, Fujian Medical University, Fuzhou, China
The estimated prevalence of tetrahydrobiopterin deficiency (BH4D) and the mutational spectrum of the causal 6-pyruvoyl-tetrahydropterin synthase (PTS) gene vary widely according to race and region. This study assessed the prevalence and genetic characteristics of BH4D in Fujian Province, southeastern China. A total of 3,204,067 newborns were screened between 2012 and 2022 based on the phenylalanine level and the phenylalanine/tyrosine ratio in dried blood spots. Differential diagnosis was determined by the urine purine spectrum, dihydropteridine reductase activity in red blood cells, and genetic testing. The PTS mutation spectrum and genotypes were determined by next-generation sequencing. A total of 189 newborns were diagnosed with hyperphenylalaninemia (HPA) over the study period, including 159 with phenylalanine hydroxylase deficiency and 30 with BH4D. Therefore, the prevalence of BH4D in Fujian was 9.36 per 1,000,000 live births (30/3,204,067) and the proportion of BH4D among patients with HPA was 15.87% (30/189). A total of 58 PTS alleles were identified in the 29 patients with PTS deficiency (PTPSD), and those alleles were composed of 10 different variants, including eight missense variants and two splice-site variants. The most prevalent variants were c.155A>G, p.Asn52Ser (44.83%); c.259C>T, p.Pro87Ser (39.66%); and c.84-291A>G, p.Tyr27Argfs*8 (3.45%). The predominant genotype was c [155A>G]; [259C>T] (11/29, 37.93%). The prevalence of BH4D and the spectrum of associated PTS mutations were successfully determined for the first time in Fujian Province, southeastern China. Since the mutation spectrum of PTS is region-specific, such data will facilitate molecular diagnosis and genetic counseling in PTPSD cases.
1 Introduction
Hyperphenylalaninemia (HPA) is the most common autosomal recessive metabolic disorder, characterized by increased levels of amino acids caused by a deficiency in phenylalanine hydroxylase (PAH) or its cofactor tetrahydrobiopterin (BH4) (Çıkı et al., 2021). Differential diagnosis of PAH deficiency (PAHD) and BH4 deficiency (BH4D) can be achieved through the urine purine spectrum, determination of dihydropteridine reductase (DHPR) activity in red blood cells, and genetic testing (Lin et al., 2022; Vela-Amieva et al., 2022). In addition to increasing the blood phenylalanine (Phe) level, BH4D can decrease the synthesis of neurotransmitters since BH4 is also a significant cofactor for tyrosine hydroxylase and tryptophan hydroxylase, the rate-limiting enzymes in dopamine and serotonin synthesis, respectively. Without effective treatment, BH4D can lead to the development of intellectual and movement disorders. Since the treatment methods used for PAHD and BH4D differ, timely differential diagnosis is particularly crucial. BH4D can be caused by a deficiency in one of the five enzymes responsible for BH4 synthesis and recycling, including GTP cyclohydrolase I, 6-pyruvoyl-tetrahydropterin synthase (PTS), septerin reductase, pterin-4-carbinolamine dehydratase, and DHPR (Wang et al., 2018; Yuan et al., 2021). Among the enzymes, PTS deficiency (PTPSD) is the most common form of BH4D, which is caused by mutations in the PTS gene.
Newborn screening (NBS) programs for HPA have been implemented in many countries, resulting in significant improvements in BH4D early diagnosis and treatment (Shintaku and Ohwada, 2013; Khani et al., 2021). BH4D incidence is approximately 1%–3% among patients with HPA worldwide (Dhondt and Hayte, 2002; de Baulny et al., 2007; Dhondt, 2010; Wang et al., 2021). However, its incidence is higher in East Asia (Chiu et al., 2012). Based on a national cross-sectional survey of 107 million cases subject to NBS in mainland China from 2013 to 2019, the incidence of BH4D was estimated to be 3.8 per 1,000,000 live births, although the incidence rate varied greatly in different geographical locations (Yuan et al., 2021). Moreover, over 95% of PTPSD cases in mainland Chinese patients with BH4D are caused by PTS gene mutation (Ye et al., 2013; Han et al., 2015; Li et al., 2018). Although a single-center study on BH4D was conducted in Quanzhou (Lin et al., 2022) and Xianmen (Wang et al., 2019), Fujian Province, southeastern China, the overall prevalence and mutation spectrum of BH4D in Fujian have not been reported.
In the present study, the mutation prevalence and spectrum of PTS in patients diagnosed with BH4D were evaluated based on a multi-center and large-scale NBS program in Fujian Province, southeastern China. The biochemical features of patients with BH4D were also analyzed.
2 Methods
2.1 Study population
A total of 3,204,067 newborns (1,490,053 females and 1,714,014 males) screened across NBS centers in Fujian Province from January 2012 to December 2022 were included in the present study. The research protocol was approved by the Ethics Committee of Fujian Provincial Maternity and Child Hospital (permission no. 2017-037). Written informed consent was obtained from the guardians of all participants.
2.2 Newborn screening and biochemical analysis
The NBS workflow was based on a previously described procedure (Zhou et al., 2022). In brief, dried blood spot (DBS) samples were collected and transported via a cold-chain transportation system to the corresponding NBS center. The Phe concentration in the DBS samples was quantified using a DEFIA-1420 semi-auto time-resolved fluoroimmunoassay analyzer (Wallac, Finland) or an ACQUITY TQD tandem mass spectrometry (MS/MS) system (Waters, USA) with a Neonatal Phenylalanine kit (PerkinElmer, Turku, Finland). The cutoff value of Phe was set to 2.0 mg/dL (fluorometric method) or 120 μmol/L (MS/MS). When the Phe levels of the newborns exceeded the threshold, the families were contacted to undergo repeated testing using MS/MS. A consistent Phe level >120.0 μmol/L and Phe/tyrosine (Tyr) ratio >2.0 was used to establish a diagnosis of HPA.
2.3 Classification of HPA and differential diagnosis
Analysis of the urine purine spectrum, determination of DHPR activity in red blood cells, and genetic testing were performed to differentiate between PAHD and BH4D among newborns diagnosed with HPA. Biopterin (B) and neopterin (N) levels in the urine, and DHPR activity in DBS samples were measured simultaneously at the Xin Hua Hospital affiliated with Shanghai Jiao Tong University Medical School.
2.4 Urine purine spectrum analysis
Firstly, 100 mg ascorbic acid was added to 10 mL fresh urine to keep BH4 in its reduced form. After centrifugation, 50 µL of the supernatant was mixed with 450 µL of H2O. Then, 50 µL of 0.1 mol/L HCl and 10 mg MnO2 was added to the diluted urine and it was allowed to incubate for 15 min at room temperature. The mixture was centrifuged at 13,000 × g for 10 min. The supernatant was used for the measurement of B and N by high-performance liquid chromatography.
2.5 DHPR activity test
The DHPR activity was measured based on spectrophotometric monitoring of the formation of ferrocytochrome C in a coupled reaction, as describedin a previously published study (Arai et al., 1982). Quality assurance of the DHPR assay was confirmed by monitoring the dried blood spot elutes from a normal adult and a DHPR deficient patient.
2.6 Genotype analysis
Genomic DNA was isolated from DBS samples using a QIAamp DNA Mini Kit (Tiangen Biotech, China) according to the manufacturer’s instructions. Targeted next-generation sequencing was used to detect a target sequencing panel of 94 genes (including PAH, PTS, GCH1, QDPR, and PCBD1) related to inborn metabolic errors for the patients and their parents using the Biosan (Zhejiang, China) platform, as describedin a previous study (Zhou et al., 2022). Genetic variants were classified according to the American College of Medical Genetics and Genomics guidelines (https://clinicalgenome.org).
3 Results
3.1 Newborn screening for BH4D
Among the 3,204,067 newborns screened over 11 years, 2,253 had elevated concentrations of Phe at the initial NBS, yielding a positivity rate of 0.07%. Subsequently, 2,195 newborns with elevated Phe levels were successfully recalled, and 205 newborns with positive results confirmed using MS/MS were subjected to analysis of the urine purine spectrum, determination of DHPR activity, and genetic testing. Finally, 189 newborns were diagnosed with HPA, with a positive predictive value of 8.61% (189/2195) (Figure 1). Therefore, the estimated HPA incidence in Fujian Province is 1 in 16,952 newborns. Among the patients with HPA, 159 had PAHD and 30 had BH4D. Therefore, the prevalence of BH4D in Fujian Province was 9.36 per 1,000,000 live births (30/3,204,067) and the ratio of BH4D among patients with HPA was 15.87% (30/189).
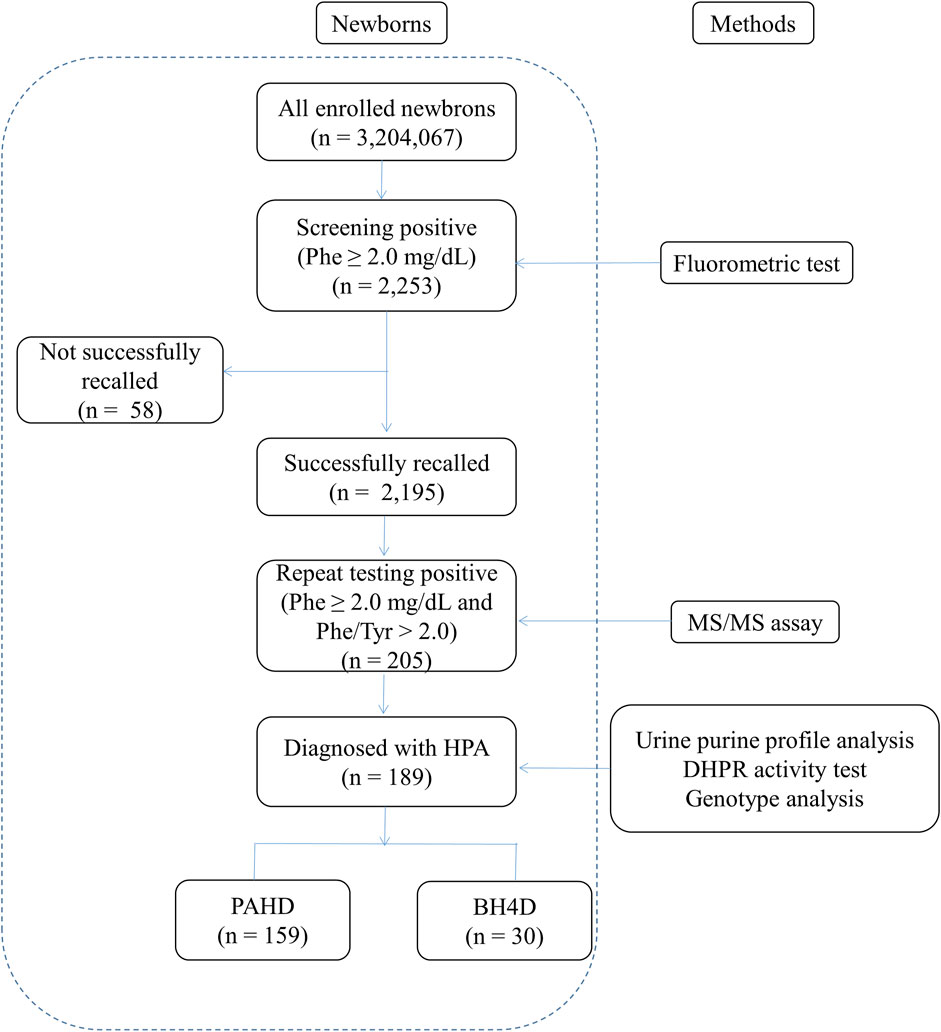
FIGURE 1. Workflow of screening of newborns for hyperphenylalaninemia. Phe, phenylalanine; Tyr, tyrosine; HPA, hyperphenylalaninemia; PAHD, phenylalanine hydroxylase deficiency; BH4D, tetrahydrobiopterin deficiency.
3.2 Biochemical characteristics of patients with BH4D
The 30 patients with BH4D, including 29 with PTPSD (96.67%) and one with DHPR deficiency (3.33%), were further investigated. The mean Phe level and Phe/Tyr ratio among patients with PTPSD were 898.5 ± 454.3 μmol/L and 12.91 ± 7.62, respectively. All 25 patients with PTPSD who underwent urine purine spectrum and DHPR activity analyses had low or nearly no B, with an overall very low percentage of B [B/(B + N)%, B%] (<5%), and normal DHPR activity (Table 1). The patient with DHPR deficiency had a normal B concentration (2.89 mmol/molCr) and B% (45.64%), but very low DHPR activity (5.5% that of the control).
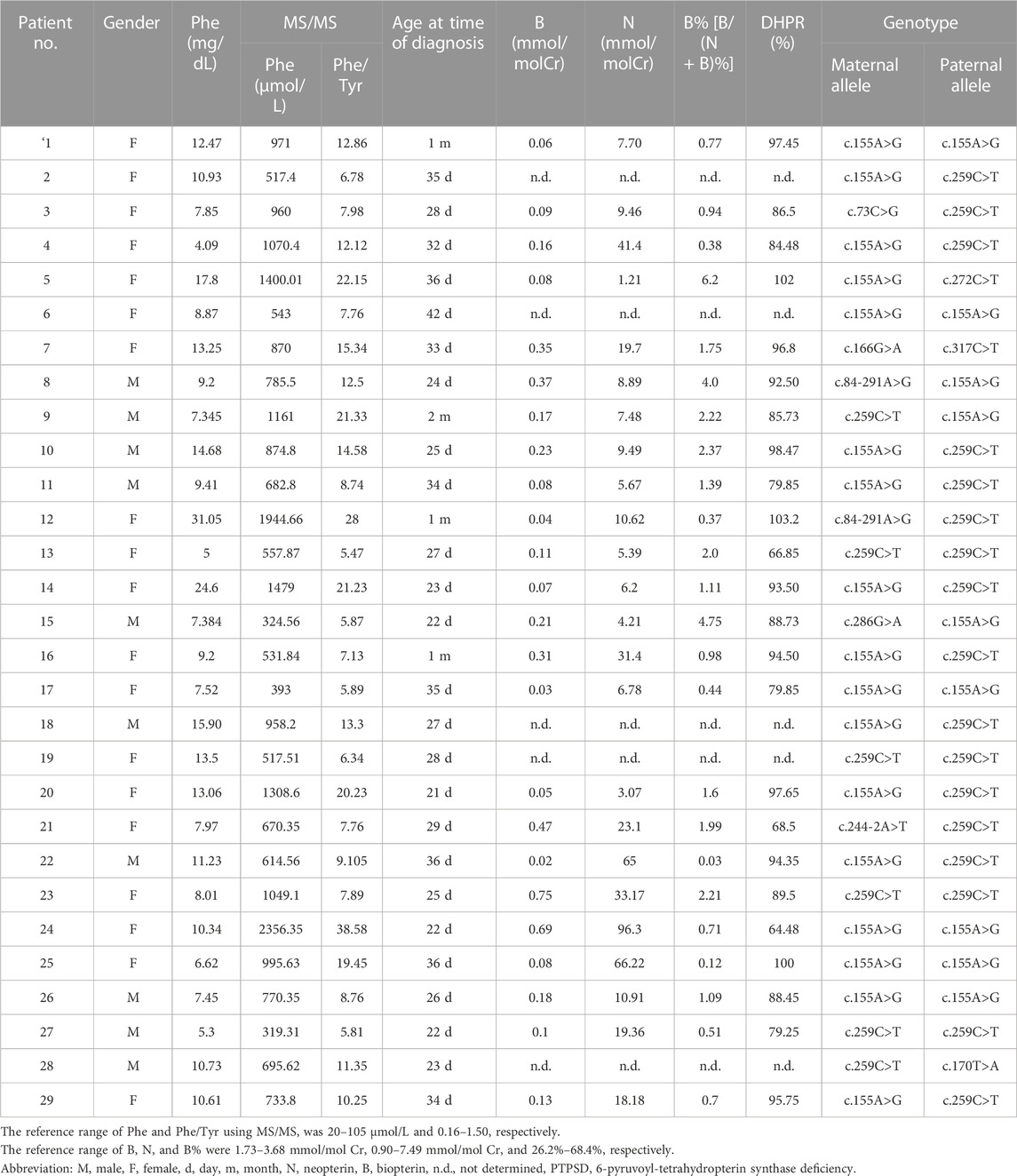
TABLE 1. Biochemical and genetic features of 29 PTPSD patients.
3.3 Variant spectrum of the PTS gene
A total of 58 PTS alleles were identified in the 29 patients with PTPSD, and those alleles were composed of 10 different variants, including eight (80%) missense variants and two (20%) splice-site variants (Table 2). At the amino acid sequence level, the three most prevalent variants accounted for 87.93% of the total: c.155A>G, p.Asn52Ser (44.83%); c.259C>T, p.Pro87Ser (39.66%); and c.84-291A>G, p.Tyr27Argfs*8 (3.45%). Only one novel variant, c.170T>A, p.Val57Glu in the PTS gene was identified in this study, the other variants observed in this study have been reported before. Excluding exon 4, the variants were relatively evenly distributed along the PTS gene (Figure 2).
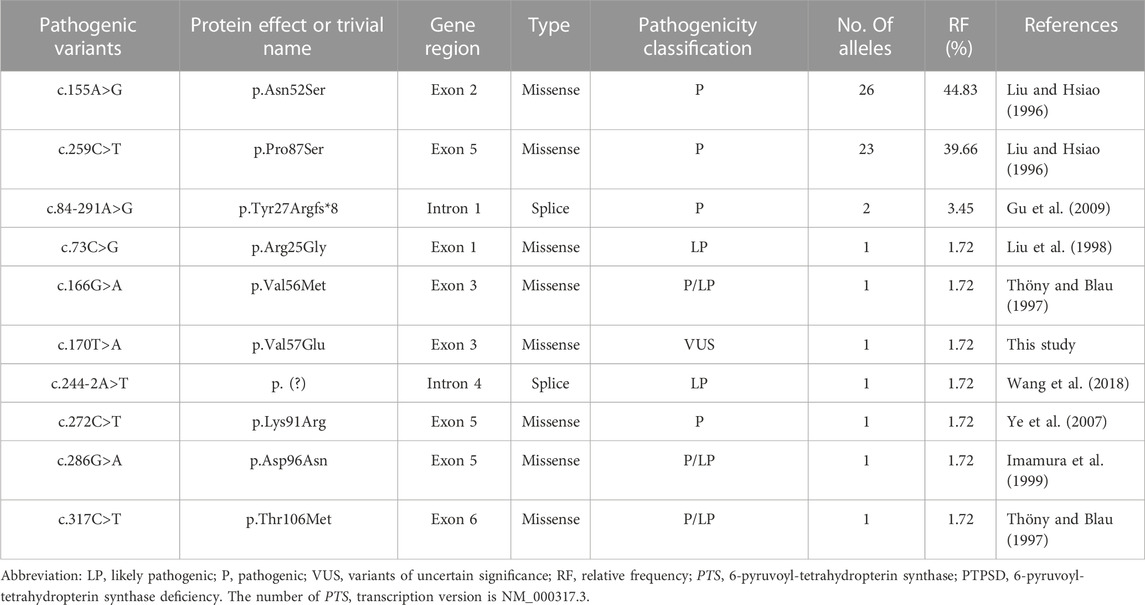
TABLE 2. PTS gene mutations in the PTPSD patients from Fujian province.
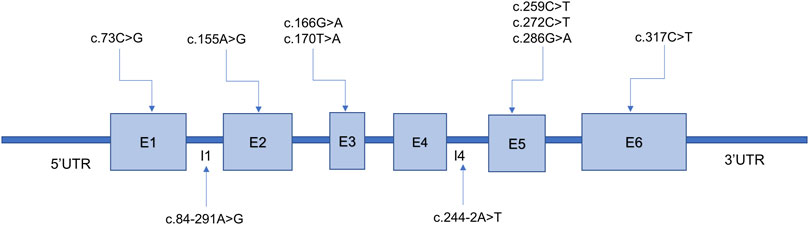
FIGURE 2. Ten variants identified in the 6-pyruvoyl-tetrahydropterin synthase (PTS) gene among patients with PTS deficiency from Fujian Province. E, exon; I, intron. The PTS transcript version access number is NM_000317.3.
3.4 Genotype distribution of patients with BH4D
All patients with PTPSD carried biallelic variants and were either compound heterozygous (n = 19) or homozygous (n = 10), with one variant originating from the mother and the other one originating from the father (Table 1). At the amino acid level, 11 distinct combinations were found in the 29 patients, and the most abundant genotypes observed were c.[155A>G]; [259C>T] (11/29, 37.93%), c.[155A>G]; [155A>G] (6/29, 20.69%), and c.[259C>T]; [259C>T] (4/29, 13.79%), as shown in Table 1. The patient with DHPR deficiency had a compound heterozygous mutation in the DHPR gene, and the genotype was c.[508G>C][523_525GCA>AGA].
4 Discussion
Early detection of HPA through NBS is crucial for timely intervention and prevention of adverse clinical symptoms in affected individuals. With the widespread application of the urine purine spectrum, DHPR activity in red blood cells, and next-generation sequencing, PAHD and BH4D can now be identified in a timely manner.
Only a few studies to date have investigated the prevalence of BH4D, with a global incidence of approximately 1 in 500,000 live births (Blau et al., 2018); however, wide variation in the estimated prevalence has been reported depending on the geographical location and ethnic composition of the different provinces in China (Wang et al., 2021; Xie et al., 2022). The prevalence of BH4D in the northern regions (4.1 per 1,000,000) of China is reportedly higher than that in the southern regions (1.6 per 1,000,000) (Yuan et al., 2021). The prevalence is up to 12.5 per 1,000,000 live births in Jiangxi Province, South China (Xie et al., 2022), and estimated at 15.5 and 9.44 per 1,000,000 live births in Quanzhou city (Lin et al., 2022) and Xianmen city (Wang et al., 2019) of Fujian Province, respectively. In the present study, 30 patients with BH4D were identified among 3.2 million NBS results, representing an incidence of 9.36 per 1,000,000 live births in Fujian Province. The incidence was much higher than the national average in China and is similar to that reported in Jiangxi Province, South China. The proportion of BH4D in HPA cases varies greatly in different regions of the world, with an incidence rate of 0.5% in Russia (Gundorova et al., 2021) and an incidence rate as high as 30% in Jordan (Carducci et al., 2020). A previous study estimated the proportion of BH4D among patients with HPA in mainland China to be 8.55% (Ye et al., 2009). However, significant regional differences in the proportion have also been observed, with a higher proportion in the southern region (Xie et al., 2022) and a lower proportion in the northern region (Han et al., 2015). In the present study, BH4D accounted for 15.87% of patients with HPA, which is similar to the proportion reported for Taiwan (Niu et al., 2010). In addition, 29 out of the 30 (96.67%) patients with BH4D in our study had PTPSD, which is consistent with previous studies (Ye et al., 2013; Han et al., 2015; Xie et al., 2022).
The human PTS gene is located on chromosome 11q22.3 and contains six exons encoding more than 500 nucleotides. More than 700 confirmed pathogenic PTS variants have been detected (http://www.hgmd.cf.ac.uk; data collected on 17 July 2023). The high-frequency regions of variants and the mutational spectrum of PTS vary widely by race and region. A previous study showed that exon 6 was the most frequently mutated region in Italy (Manti et al., 2020), whereas another study indicated that exon 5 was the most commonly mutated region in Chinese patients (Li et al., 2022). However, excluding exon 4, the variants were found to be relatively evenly distributed along the PTS gene in the newborns screened in the present study. The c.238A>G (p. Met80Val) mutation is highly prevalent in the Arab population, accounting for 33% (Almannai et al., 2019), whereas c.317C>T (p.Thr106Met) is the most common variant in Russia (Gundorova et al., 2021). In East Asian populations, the three most prevalent variants accounted for 87.93% of all variants: c.259C>T (p.Pro87Ser) (39.66%), c.155A>G (p.Asn52Ser) (15.62%), and c.286G>A (p.Asp96Asn) (10.51%) (Chiu et al., 2012). Some studies have reported that c.259C>T is the most prevalent variant in mainland China (Ye et al., 2013; Han et al., 2015; Xie et al., 2022). Ten distinct variants were detected in the present study, more than two-thirds of which were identified only once, revealing a high degree of genetic heterogeneity among patients with PTPSD in Fujian Province. The following three variants accounted for 87.93% of the total variants among the patients with PTPSD identified in our study: c.155A>G (p.Asn52Ser) (44.83%), c.259C>T (p.Pro87Ser) (39.66%), and c.84-291A>G (p.Tyr27Argfs*8) (3.45%), which is consistent with the mutation spectrum identified in Taiwan (Chiu et al., 2012). In addition, one novel variant, c.170T>A, p.Val57Glu in the PTS gene was identified in this study, which enriched the human genetic variation database.
The most prevalent genotypes among the patients with BH4D identified in the present study were c.[155A>G]; [259C>T] (11/29, 37.93%), c.[155A>G]; [155A>G] (6/29, 20.69%), and c.[259C>T]; [259C>T] (4/29, 13.79%), which is inconsistent with previous findings in other provinces in China. The most prevalent genotypes of patients with BH4D in Beijing and Shandong were c.[ 84-291A>G]; [286G>A] (4/11, 36.36%) and c.[259C>T]; [259C>T] (3/11, 27.27%), respectively. The results further highlight the regional and ethnic heterogeneity of variants contributing to BH4D in China.
All PTPSD patients received treatment with BH4, L-dopa, and 5-HTP. The median minimal starting dose of BH4 was 1 mg/kg/day, and adjusted according to Phe concentrations to achieve the target Phe concentration for the corresponding ageof the patient (Yang et al., 2014). When the Phe concentration of the patients with BH4 treatment is not well controlled, diet therapy will be used. The median minimal starting doses of L-dopa and 5-HTP were also 1 mg/kg/day, with increments of 1 mg/kg every 5–7 days as required to achieve target concentrations for the corresponding age and adjusted according to clinical symptoms and blood prolactin concentrations. In addition, the clinical outcomes of some patients, including physical development status and intelligence, are being evaluated and collected. We hope to gather data on and show the clinical outcomes for all patients and conduct an analysis of gene-phenotype correlation in the future.
5 Conclusion
The biochemical and molecular features of 30 non-related patients with BH4D were investigated. The estimated incidence of BH4D in Fujian Province is 9.36 per 1,000,000 live births. The prevalence of PTPSD among patients with BH4D was 96.67%. We successfully established the PTS mutation spectrum in Fujian Province for the first time and found that the c.155A>G variant was the most prevalent PTS variant. The most dominant genotype was c.[155A>G]; [259C>T] (37.93%). The mutation spectrum is region-specific and therefore should facilitate the improvement of the molecular diagnosis and genetic counseling of families affected by PTPSD.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary material, further inquiries can be directed to the corresponding authors.
Ethics statement
The study was conducted in accordance with the guidelines of the Declaration of Helsinki and approved by the Ethics Review Committee of Fujian Provincial Maternity and Child Hospital (permission no. 2017-037). Written informed consent was obtained from the legal guardians of all participants for the publication of any potentially identifiable data included in this article.
Author contributions
XQ: Conceptualization and writing of the original draft. PZ: Data curation and formal analysis. JL: Methodology and Visualization. GL: Investigation and Software. LD: Supervision and Validation. YZ: Investigation and Validation. LX: Conceptualization and Writing, review, and editing. JZ: Project administration, writing, review, and editing. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the Key Project on Science and Technology Program of the Fujian Health Commission (Grant No. 2021ZD01002), Natural Science Foundation of Fujian Province (Grant No. 2020J01327), Fujian Provincial Health Technology Project (Grant Nos 2020GGB017 and 2022CXA033), and Joint Funds for the Innovation of Science and Technology, Fujian Province (Grant No. 2020Y9143).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Almannai, M., Felemban, R., Saleh, M. A., Faqeih, E. A., Alasmari, A., AlHashem, A., et al. (2019). 6-Pyruvoyltetrahydropterin synthase deficiency: review and report of 28 Arab subjects. Pediatr. Neurol. 96, 40–47. doi:10.1016/j.pediatrneurol.2019.02.008
Arai, N., Narisawa, K., Hayakawa, H., and Tada, K. (1982). Hyperphenylalaninemia due to dihydropteridine reductase deficiency: diagnosis by enzyme assays on dried blood spots. Pediatrics 70, 426–430. doi:10.1542/peds.70.3.426
Blau, N., Martinez, A., Hoffmann, G. F., and Thöny, B. (2018). DNAJC12 deficiency: a new strategy in the diagnosis of hyperphenylalaninemias. Mol. Genet. Metab. 123, 1–5. doi:10.1016/j.ymgme.2017.11.005
Carducci, C., Amayreh, W., Ababneh, H., Mahasneh, A., Al Rababah, B., Al Qaqa, K., et al. (2020). Molecular genetics of phenylketonuria and tetrahydrobiopterin deficiency in Jordan. JIMD Rep. 55, 59–67. doi:10.1002/jmd2.12130
Chiu, Y.-H., Chang, Y.-C., Chang, Y.-H., Niu, D.-M., Yang, Y.-L., Ye, J., et al. (2012). Mutation spectrum of and founder effects affecting the PTS gene in East Asian populations. J. Hum. Genet. 57, 145–152. doi:10.1038/jhg.2011.146
Çıkı, K., Akar, H. T., Özgül, R. K., Gülbakan, B., and Yıldız, Y. (2021). Perplexing etiology of hyperphenylalaninemia in an infant referred via newborn screening. Clin. Chem. 67, 1428–1431. doi:10.1093/clinchem/hvab106
de Baulny, H. O., Abadie, V., Feillet, F., and de Parscau, L. (2007). Management of phenylketonuria and hyperphenylalaninemia. J. Nutr. 137, 1561S–1563S; discussion 1573S-1575S. doi:10.1093/jn/137.6.1561S
Dhondt, J. (2010). Lessons from 30 years of selective screening for tetrahydrobiopterin deficiency. J. Inherit. Metab. Dis. 33, 219–223. doi:10.1007/s10545-010-9091-9
Dhondt, J. L., and Hayte, J. M. (2002). Screening of tetrahydrobiopterin deficiency among hyperphenylalaninemic patients. Ann. Biol. Clin. Paris. 60, 165–171.
Gu, M., Ye, J., Qiu, W., Han, L., Zhang, Y., and Gu, X. (2009). Mutational analysis of patients with 6-pyruvoyltetrahydrobiopterin synthesis deficiency. Zhonghua Yi Xue Yi Chuan Xue Za Zhi 26, 183–186. doi:10.3760/cma.j.issn.1003-9406.2009.02.014
Gundorova, P., Kuznetcova, I. A., Baydakova, G. V., Stepanova, A. A., Itkis, Y. S., Kakaulina, V. S., et al. (2021). BH4-deficient hyperphenylalaninemia in Russia. PLoS One 16, e0249608. doi:10.1371/journal.pone.0249608
Han, B., Zou, H., Han, B., Zhu, W., Cao, Z., and Liu, Y. (2015). Diagnosis, treatment and follow-up of patients with tetrahydrobiopterin deficiency in Shandong province, China. Brain Dev. 37, 592–598. doi:10.1016/j.braindev.2014.09.008
Imamura, T., Okano, Y., Shintaku, H., Hase, Y., and Isshiki, G. (1999). Molecular characterization of 6-pyruvoyl-tetrahydropterin synthase deficiency in Japanese patients. J. Hum. Genet. 44, 163–168. doi:10.1007/s100380050134
Khani, S., Barzegari, M., Esmaeilizadeh, Z., Farsian, P., Alaei, M., Salehpour, S., et al. (2021). The treatment and clinical follow-up outcome in Iranian patients with tetrahydrobiopterin deficiency. J. Pediatr. Endocrinol. Metabolism 34, 1157–1167. doi:10.1515/jpem-2021-0155
Li, L., Yang, H., Zhao, J., Yang, N., Gong, L., Tang, Y., et al. (2022). Identification and molecular analysis of 11 cases of the PTS gene variants associated with tetrahydrobiopterin deficiency. Front. Genet. 13, 919209. doi:10.3389/fgene.2022.919209
Li, N., Yu, P., Rao, B., Deng, Y., Guo, Y., Huang, Y., et al. (2018). Molecular genetics of tetrahydrobiopterin deficiency in Chinese patients. J. Pediatr. Endocrinol. Metabolism 31, 911–916. doi:10.1515/jpem-2018-0037
Lin, Y., Lin, W., Su, R., Zheng, Z., Fu, Q., and Wang, G. (2022). Newborn screening and genetic features of patients with hyperphenylalaninemia in a southern Chinese population. Clin. Chim. Acta 535, 13–18. doi:10.1016/j.cca.2022.08.009
Liu, T.-T., and Hsiao, K.-J. (1996). Identification of a common 6-pyruvoyl-tetrahydropterin synthase mutation at codon 87 in Chinese phenylketonuria caused by tetrahydrobiopterin synthesis deficiency. Hum. Genet. 98, 313–316. doi:10.1007/s004390050213
Liu, T.-T., Hsiao, K.-J., Lu, S.-F., Wu, S.-J., Wu, K.-F., Chiang, S.-H., et al. (1998). Mutation analysis of the 6-pyruvoyl-tetrahydropterin synthase gene in Chinese hyperphenylalaninemia caused by tetrahydrobiopterin synthesis deficiency. Hum. Mutat. 11, 76–83. doi:10.1002/(SICI)1098-1004(1998)11:1<76:AID-HUMU12>3.0.CO;2-W
Manti, F., Nardecchia, F., Banderali, G., Burlina, A., Carducci, C., Carducci, C., et al. (2020). Long-term clinical outcome of 6-pyruvoyl-tetrahydropterin synthase-deficient patients. Mol. Genet. Metab. 131, 155–162. doi:10.1016/j.ymgme.2020.06.009
Niu, D., Chien, Y., Chiang, C., Ho, H., Hwu, W., Kao, S., et al. (2010). Nationwide survey of extended newborn screening by tandem mass spectrometry in Taiwan. J. Inherit. Metab. Dis. 33, 295–305. doi:10.1007/s10545-010-9129-z
Shintaku, H., and Ohwada, M. (2013). Long-term follow-up of tetrahydrobiopterin therapy in patients with tetrahydrobiopterin deficiency in Japan. Brain Dev. 35, 406–410. doi:10.1016/j.braindev.2012.06.010
Thöny, B., and Blau, N. (1997). Mutations in the GTP cyclohydrolase I and 6-pyruvoyl-tetrahydropterin synthase genes. Hum. Mutat. 10, 11–20. doi:10.1002/(SICI)1098-1004(1997)10:1<11:AID-HUMU2>3.0.CO;2-P
Vela-Amieva, M., Alcántara-Ortigoza, M. A., Ibarra-González, I., González-del Angel, A., Fernández-Hernández, L., Guillén-López, S., et al. (2022). Genotypic spectrum underlying tetrahydrobiopterin metabolism defects: experience in a single Mexican reference center. Front. Genet. 13, 993612. doi:10.3389/fgene.2022.993612
Wang, R., Shen, N., Ye, J., Han, L., Qiu, W., Zhang, H., et al. (2018). Mutation spectrum of hyperphenylalaninemia candidate genes and the genotype-phenotype correlation in the Chinese population. Clin. Chim. Acta 481, 132–138. doi:10.1016/j.cca.2018.02.035
Wang, X., He, Y., Jiang, Y., Feng, X., Zhang, G., Xia, Z., et al. (2019). Screening and mutation analysis of hyperphenylalaninemia in newborns from Xiamen, China. Clin. Chim. Acta 498, 161–166. doi:10.1016/j.cca.2019.08.021
Wang, X., Wang, Y., Ma, D., Zhang, Z., Li, Y., Yang, P., et al. (2021). Neonatal screening and genotype-phenotype correlation of hyperphenylalaninemia in the Chinese population. Orphanet J. Rare Dis. 16, 214. doi:10.1186/s13023-021-01846-w
Xie, K., Zeng, B., Zhang, L., Chen, S., Zou, Y., Yuan, H., et al. (2022). Mutation spectrum of PTS gene in patients with tetrahydrobiopterin deficiency from jiangxi province. Front. Genet. 13, 1077729. doi:10.3389/fgene.2022.1077729
Yang, Y., and Ye, Y.Subspecial Group of EndocrineNewborn Screening Committee of Professional Society of Birth Defect Prevention and ControlChinese Assocation of Preventive MedicalChinese Assocation of Preventive Medical (2014). Consensus about the diagnosis and treatment of hyperphenylalaninemia. Zhonghua Er Ke Za Zhi 52, 420–425.
Ye, J., Qiu, W., Han, L., Zhang, Y., Zhou, J., Zhang, Y., et al. (2007). Diagnosis, treatment and long-term following up of 223 patients with hyperphenylalaninemia detected by neonatal screening programs. Zhonghua Yu Fang. Yi Xue Za Zhi 41, 189–192.
Ye, J., Qiu, W., Han, L., Zhou, J., Gao, X., and Gu, X. (2009). The investigation of differential diagnostic development and incidence of tetrahydrobiopterin deficiency. Zhonghua Yu Fang. Yi Xue Za Zhi 43, 128–131.
Ye, J., Yang, Y., Yu, W., Zou, H., Jiang, J., Yang, R., et al. (2013). Demographics, diagnosis and treatment of 256 patients with tetrahydrobiopterin deficiency in mainland China: results of a retrospective, multicentre study. J. Inherit. Metab. Dis. 36, 893–901. doi:10.1007/s10545-012-9550-6
Yuan, X., Zhu, J., Liu, H., Xiang, L., Yao, Y., Li, Q., et al. (2021). Birth prevalence of tetrahydrobiopterin deficiency in China: data from the national newborn screening program, 2013–2019. J. Pediatr. Endocrinol. Metabolism 34, 835–841. doi:10.1515/jpem-2021-0077
Zhou, J., Zeng, Y., Qiu, X., Lin, Q., Chen, W., Luo, J., et al. (2022). Characterization of phenylalanine hydroxylase gene variants and analysis of genotype–phenotype correlation in patients with phenylalanine hydroxylase deficiency from Fujian Province, Southeastern China. Mol. Biol. Rep. 49, 10409–10419. doi:10.1007/s11033-022-07579-8
Keywords: tetrahydrobiopterin deficiency, newborn screening, 6-pyruvoyl-tetrahydropterin synthase, variant spectrum, southeastern China
Citation: Qiu X, Zhao P, Luo J, Li G, Deng L, Zeng Y, Xu L and Zhou J (2023) Biochemical and molecular features of tetrahydrobiopterin deficiency in Fujian Province, southeastern China. Front. Genet. 14:1250568. doi: 10.3389/fgene.2023.1250568
Received: 30 June 2023; Accepted: 04 August 2023;
Published: 11 August 2023.
Edited by:
Hui-Qi Qu, Children’s Hospital of Philadelphia, United StatesReviewed by:
Cynthia Fernandez-Lainez, National Institute of Pediatrics, MexicoYongyi Zou, Jiangxi Maternal and Child Health Hospital, China
Copyright © 2023 Qiu, Zhao, Luo, Li, Deng, Zeng, Xu and Zhou. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jinfu Zhou, emhvdTgxMTIwM0Bmam11LmVkdS5jbg==; Liangpu Xu, eGlsaWFuZ3B1QGZqbXUuZWR1LmNu
†These authors have contributed equally to this work