 Aleksandra Mroczkowska-Bękarciak
Aleksandra Mroczkowska-Bękarciak Tomasz Wróbel
Tomasz Wróbel
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Genet., 08 September 2023
Sec. Cancer Genetics and Oncogenomics
Volume 14 - 2023 | https://doi.org/10.3389/fgene.2023.1241912
The classical BCR::ABL1-negative myeloproliferative neoplasms such as polycythemia vera (PV), essential thrombocythemia (ET), and myelofibrosis (MF) are clonal diseases with the presence of characteristic “driver mutations” in one of the genes: JAK2, CALR, or MPL. The search for mutations in these three genes is required for the diagnosis of MPNs. Nevertheless, the progress that has been made in the field of molecular genetics has opened a new era in medicine. The search for additional mutations in MPNs is helpful in assessing the risk stratification, disease progression, transformation to acute myeloid leukemia (AML), or choosing the right treatment. In some cases, advanced technologies are needed to find a clonal marker of the disease and establish a diagnosis. This review focuses on how the use of new technologies like next-generation sequencing (NGS) helps in the diagnosis of BCR::ABL1-negative myeloproliferative neoplasms.
The classical BCR::ABL1-negative myeloproliferative neoplasms (MPNs) are hematopoietic stem cell disorders with the clonal proliferation of one or more types of cells of the myeloid lineage. MPNs are associated with an increased risk of thrombosis and a tendency to transform into acute myeloid leukemia. According to the fifth edition of the World Health Organization (WHO) criteria, MPNs include chronic myeloid leukemia (CML), which is defined by the presence of the Philadelphia chromosome and BCR::ABL1 fusion gene, polycythemia vera (PV), essential thrombocythemia (ET), primary myelofibrosis (PMF), chronic neutrophilic leukemia (CNL), chronic eosinophilic leukemia (CEL), juvenile myelomonocytic leukemia, and myeloproliferative neoplasms, not otherwise specified. PV, ET, and PMF are referred to as classical BCR::ABL1-negative MPNs. CNL, CEL, juvenile myelomonocytic leukemia, and myeloproliferative neoplasms, not otherwise specified, are less common and called non-classical or atypical MPNs. The current criteria for the diagnosis of myeloproliferative neoplasms are provided by the fifth edition of the World Health Organization Classification of Haematolymphoid Tumours: Myeloid and Histiocytic/Dendritic Neoplasms, and the International Consensus Classification (ICC) of Myeloid Neoplasms and Acute Leukemias. The diagnosis still depends on the clinical picture, laboratory tests, evaluation of bone marrow biopsy, and molecular tests for the presence of mutations in the JAK2, CALR, or MPL genes. These mutations, called “driver mutations,” are crucial for diagnosis and have prognostic and therapeutic significance (Barbui et al., 2018; Arber et al., 2022; Khoury et al., 2022).
The diagnostic criteria for PV have been slightly refined. One of the major criteria, namely, increased red cell mass, has been removed as a diagnostic criterion according to the WHO classification. However, the ICC classification still retains this criterion (Table 1). The diagnostic criteria of ET have not changed (Table 2). In the diagnosis of primary myelofibrosis, it is necessary to not only differentiate prefibrotic PMF from ET and PV but also from fibrotic PMF. Serial monitoring of the spleen size and bone marrow fibrosis using repeatable and defined criteria is still important, especially for patients using JAK1/2 inhibitors (Table 3) (Levine et al., 2005; Khoury et al., 2022). The presence of JAK2V617F, JAK2 exon 12, MPL, and CALR mutations is included in the molecular diagnostic and prognostic algorithms for MPNs. All these mutations lead to the constitutive activation of the JAK-STAT signaling pathway. Furthermore, these genetic mutations are often considered mutually exclusive. The absence of driver mutations does not exclude diagnosis; these patients are referred to as triple-negative (TN) patients. Nevertheless, driver mutations are not the only mutations identified in patients with MPNs. The use of advanced techniques of molecular genetics, such as NGS, plays a very important role in hematooncology diagnosis. The development of high-throughput NGS methods diversified the mutational landscape and improved our understanding of pathophysiology. These molecular details also made it possible to improve the diagnosis, assign a better prognosis score, and track the effectiveness of treatments. Comprehensive gene sequencing of blood cancer patients is becoming more widely available. We wanted to share a graphical flowchart of stepwise molecular testing ending with NGS (Figure 1). Somatic additional mutations were revealed to affect a number of genes that were often altered throughout myeloid malignancies and MPNs. More than 50% of patients with MPNs have additional mutations in these myeloid cancer genes, which occur more frequently as people get older and most frequently in patients with PMF. The mutational profile in conjunction with cytogenetic, histopathological, hematological, and clinical variables plays an increasingly important role in the risk stratification of patients in terms of thrombosis, overall survival, and the rate of transformation to secondary myelofibrosis or secondary leukemia.
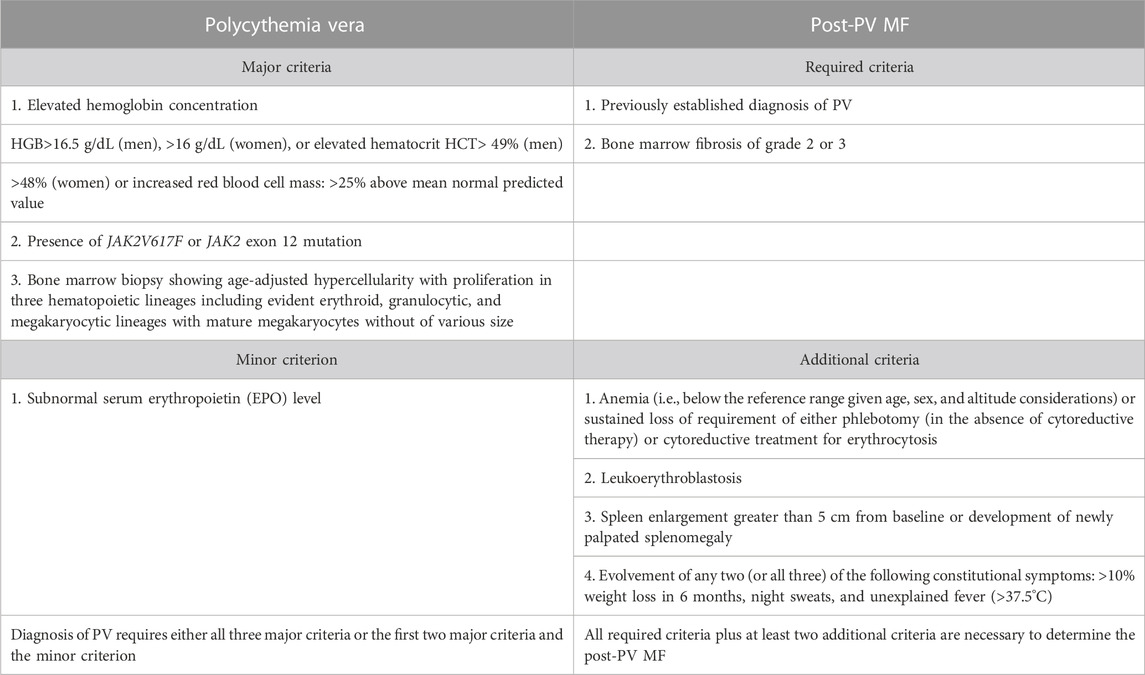
TABLE 1. Diagnostic criteria for polycythemia vera (PV) by Khoury et al. (2022) and Arber et al. (2022).
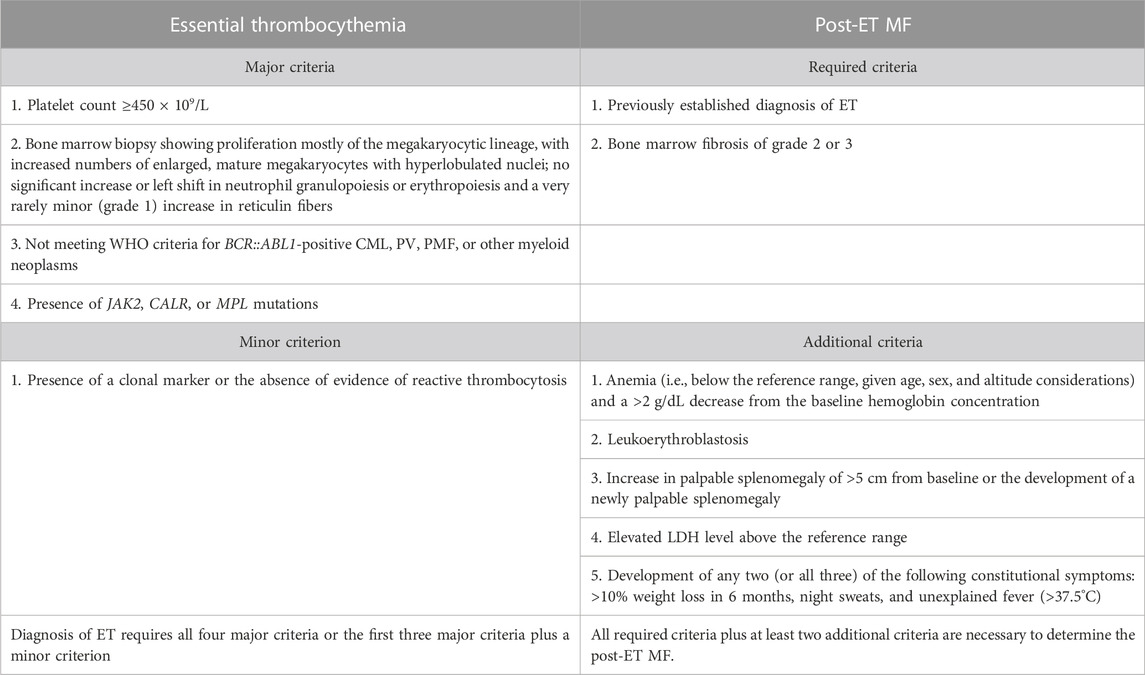
TABLE 2. Diagnostic criteria for essential thrombocythemia (ET) by Khoury et al. (2022) and Arber et al. (2022).
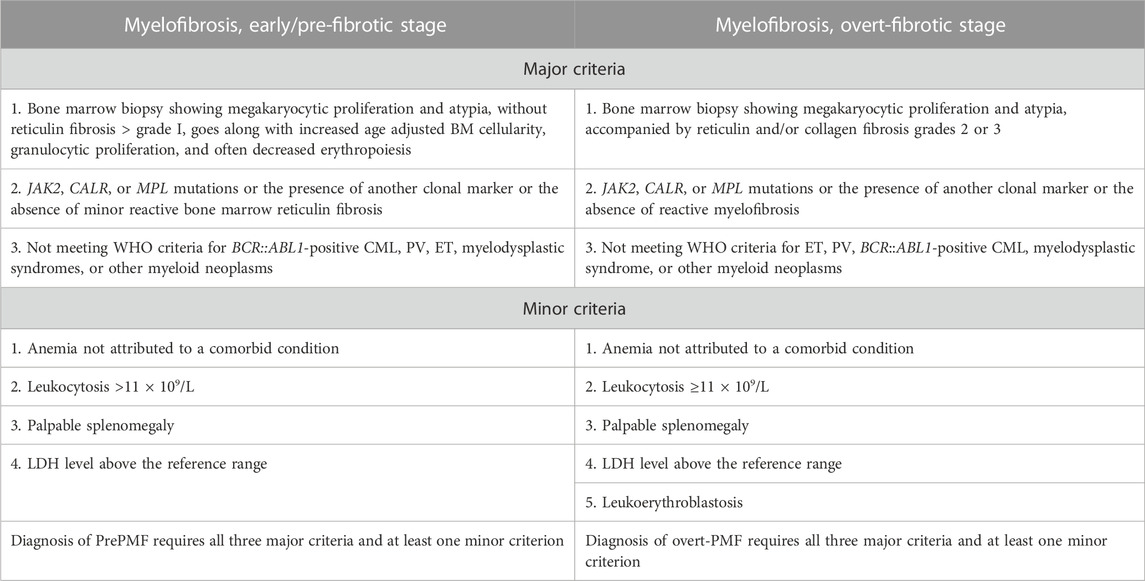
TABLE 3. Diagnostic criteria for primary myelofibrosis (PMF) by Khoury et al. (2022) and Arber et al. (2022).
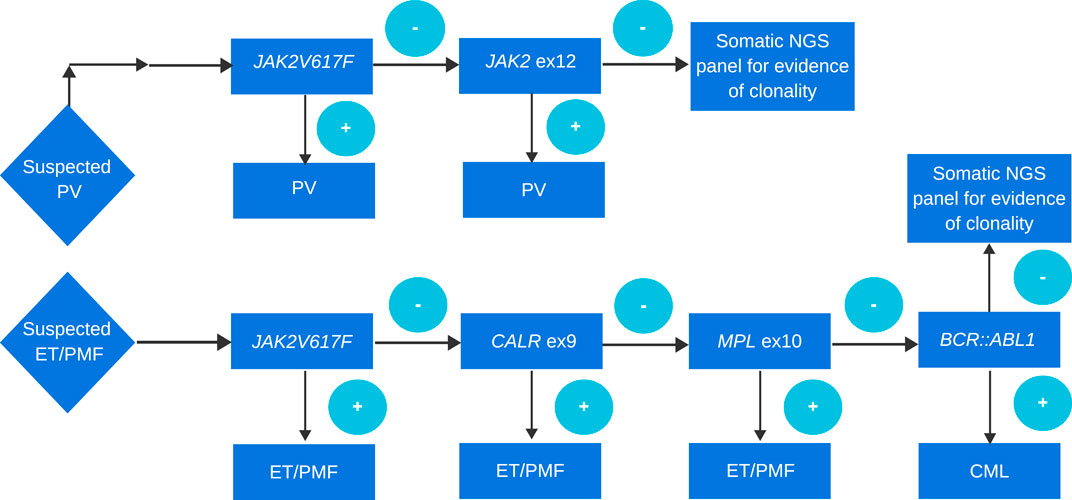
FIGURE 1. Approach to genetic diagnosis of PV, ET, and PMF. A cascade approach is routinely used in many laboratories. Although more and more often if the probability of MPN is high in order to simultaneously search for driver mutation, other evidence of clonality, and search for mutations in genes of prognostic relevance, a panel testing approach is performed.
JAK2 is a non-receptor tyrosine kinase that is involved in hematopoietic cytokine signaling through erythropoietin, thrombopoietin, and granulocyte colony-stimulating growth factor receptors. The normal function of this signaling pathway ensures proper regulation of cell proliferation, survival, differentiation, and immune response. JAK2 gene mutations lead to constitutive activation of the STAT signaling pathway and uncontrolled cell proliferation. The most common mutation in myeloproliferative neoplasms, which was discovered in 2005, is the G to T transversion in exon 14, resulting in a valine to phenylalanine substitution at codon 617 (Baxter et al., 2005; James et al., 2005; Kralovics et al., 2005; Levine et al., 2005; Guglielmelli et al., 2017; Loscocco et al., 2021a). The JAK2V617F mutation is detected in approximately 98% of patients with PV and 50%–60% of patients with ET and PMF. Almost 2%–3% of patients with PV who are JAKV617F-negative may harbor rare insertions or deletions in exon 12 of the JAK2 gene. The JAK2V617F mutation is usually detected by allele-specific PCR (Baxter et al., 2005; Scott et al., 2007; Scott, 2011; Tefferi et al., 2016a). Quantitative PCR methods such as RQ-PCR, ddPCR (droplet digital PCR), and NGS (next-generation sequencing) can also be used to support the diagnosis of PV, ET, or MF to separate triple-negative cases and monitor MRD (minimal residual disease) (Haslam and Langabeer, 2016; Link-Lenczowska et al., 2018; Arber et al., 2022). Peripheral blood counts, in patients with this mutation, show erythrocytosis, thrombocytosis, and/or leukocytosis. The JAK2 mutation status is widely known as the thrombosis risk factor and disease risk progression in patients with MPNs (Zhang et al., 2020). PMF patients with JAK2 mutations have been associated with older age, higher hemoglobin levels, lower platelet counts, and a higher level of leukocytes (Vannucchi et al., 2013). Mutations in JAK2V617F are one of the four risk factors in the International Prognostic Score of thrombosis for ET (IPSET-thrombosis) (Barbui et al., 2012). Additionally, JAK2V617F allele burden can provide important prognostic information, which has been associated with hematologic parameters and clinical outcomes. Patients with PV and higher JAK2V617F allele burdens have been associated with a higher hemoglobin level, increased age, pruritus, presence of splenomegaly, a higher risk of thrombotic events, and disease progression (Tefferi et al., 2016a; Sazawal et al., 2019; Guglielmelli et al., 2021a; Lee et al., 2021; Regimbeau et al., 2022). In ET patients, the JAK2V1617 allele burden level has been correlated with older age and a higher hemoglobin level, which is associated with risk factors for thrombotic events (Lee et al., 2021). High JAK2V617F allele burden is associated with disease progression in PMF cases. Studies have shown that allele burden may vary across different MPN phenotypes. The lowest allele burden has been associated with ET cases, and a higher allele burden has been observed in PV and PMF cases (Park et al., 2013; Vannucchi et al., 2013; Barosi et al., 2017; Lee et al., 2021; Loomila et al., 2022). The quantification of JAK2V617F allele burden is a helpful marker of treatment response and a measure of MRD after stem cell transplantation. However, there is a certain degree of overlap, and allelic burden cannot be considered pathognomonic of disease (Rumi et al., 2020; Sørensen et al., 2020; Stegelmann et al., 2023).
CALR is a highly conserved protein that is localized in the endoplasmic reticulum. It is involved in the quality control of N-glycosylated proteins and the modulation of calcium ion homeostasis. Mutations in this gene are the second most common genetic abnormality in myeloproliferative neoplasms after JAK2V617F gene mutations. This type of mutation is not found in patients with PV. CALR mutations are detected in about 20%–25% ET cases and 35% PMF (Constantinescu and Pecquet, 2020). The first studies about calreticulin date back to 2013 (Klampfl et al., 2013; Nangalia et al., 2013). The two most prevalent mutations, detected in exon 9 and corresponding to 52-bp deletion (p.Leu367ThrfsTer46), are recognized as type 1, and that with a 5-bp insertion (p.Lys385AsnfsTer47) is called type 2. Other insertions and deletions occurring at a lower frequency are reported as type like-1 or type like-2 mutations. Although type 1 and type 2 mutations account for 80% of all mutations, type 1 CALR mutation is much more common than type 2. Among ET patients, type 1 occurs in approximately 55% of cases and type 2 occurs in approximately 35% of cases; however, in PMF, type 1 definitely predominates and occurs in approximately 75% of cases, whereas type 2 occurs in only 15% of cases (Cabagnols et al., 2015; Vainchenker and Kralovics, 2017; How et al., 2019). Studies have shown that CALR mutations can activate the thrombopoietin receptor MPL and afterward the JAK-STAT signaling pathway (Chachoua et al., 2016; Nivarthi et al., 2016). Clinical characteristics and outcomes are different between patients who have CALR mutations and patients with JAK2 mutation, even though they both activate the same JAK/STAT signaling pathway. CALR mutant patients have lower leukocyte counts, lower hemoglobin levels, higher platelet counts, lower thrombosis risks, and better survival than JAK2 mutant patients (Klampfl et al., 2013; Rotunno et al., 2014). The presence of mutations in the CALR gene in ET patients is associated with a lower risk of thrombosis (Tefferi et al., 2014a; Rotunno et al., 2014; Pei et al., 2016; Guglielmelli et al., 2021b; Zulkeflee et al., 2021). It seems that both CALR and JAK2 mutations have a similar risk of transformation to post-ET myelofibrosis (Tefferi et al., 2014b; Rotunno et al., 2014; Rumi et al., 2014). However, some studies have shown higher risks of fibrotic progression in CALR-mutated than JAK2-mutated patients with ET (Al Assaf et al., 2015; Grinfeld et al., 2018). More detailed analyses have shown that type 1 of the CALR mutation is associated with an increased risk of transformation to MF (Pietra et al., 2016; Loscocco et al., 2021b; Weir et al., 2023). Little information is accessible about the allele burden of CALR mutations. A recent paper about correlations of the CALR mutation variant allele frequency (VAF) in patients with myelofibrosis showed an association with a higher VAF of CARL, a lower hemoglobin level and platelet count, higher peripheral blood CD34+ cell counts, the need for cytoreduction therapy, and shorter leukocytosis-free survival. In addition, the authors emphasize that high-molecular risk mutations (HMR) were more frequent in CALR patients with high VAF (Guglielmelli et al., 2023). Other studies have focused on various phenotypic impacts of the CALR allele burden, according to the type of CALR mutant. They have found that in patients with type 1-like CALR mutation, CALR mutant burden was negatively correlated with the hemoglobin level and platelet count, whereas CALR mutant burden was positively correlated with absolute neutrophil count and platelet count with type 2-like CALR-mutated patients. Nevertheless, the study was conducted on a small number of patients (Kim et al., 2022). There are several methods for detecting the CALR mutation. The most common methods are fragment analysis PCR and high-resolution melting-curve (HRM) analysis. Sanger sequencing can be useful in confirming the type of mutation; however, this technique has limited sensitivity. Next-generation sequencing (NGS) has the lowest limit of detection (LOD), but it is quite expensive for routine diagnostics (Jones et al., 2015; Giannopoulos et al., 2019).
The myeloproliferative leukemia virus oncogene (MPL) encodes a receptor for thrombopoietin that regulates megakaryopoiesis and platelet production, and is also crucial in the self-renewal of hematopoietic stem cells. Mutations in the MPL gene activate JAK2 and the thrombopoietin pathway. The first studies on the MPL mutant in ET and MF patients were published in 2006. MPL mutations are detected in 5%–10% of all myelofibrosis patients and in 1%–4% of patients with essential thrombocythemia. All mutations are located in exon 10. The most common mutations are substitutions at the Trp515 position, W515L, and W515K. Other mutations in that position are very rare (Pikman et al., 2006; Defour et al., 2016; Guglielmelli and Calabresi, 2021). Patients with ET or MF and MPL mutations have lower hemoglobin and hematocrit values in relation to higher EPO levels. The presence of this mutation in ET patients is associated with a higher risk of transformation to MF or AML (Asp et al., 2016). Mutations in the MPL gene can be detected in several ways, such as an allele-specific PCR-based strategy, Sanger sequencing, real-time qPCR assay, ARMS-PCR, HRM curve, and NGS. In recent years, most of the methods for detecting mutations in the MPL gene have been improved. Most of these techniques are sensitive, efficient, and cost-effective (Zhuge et al., 2010; Furtado et al., 2013; Arunachalam et al., 2018; Ullah et al., 2022).
JAK2, CALR, and MPL can be seen in about 90% of MPN cases; nevertheless, in 2% of PV, 15% of ET patients, and approximately 8%–10% of MF cases, driver mutations are not detected; such patients are termed TN patients. Some patients may have rare mutations in the JAK2, MPL, and CALR genes that are not detected by routine diagnostics. In other cases, the mutation may have been at a very low mutant allele burden and can be detected only by applying a more sensitive methodology. Implementation of more advanced techniques of molecular biology, like next-generation sequencing, allowed to improve the understanding of triple-negative patients and revealed the presence of non-driver mutations. NGS helps in finding a clonal marker to confirm the diagnosis. Triple-negative PMF patients were associated with poor survival and increased leukemic transformation. These patients tend to have lower Hb levels, platelet and leukocyte counts, and a higher IPSS (International Prognostic Scoring System) risk. TN-ET patients are associated with a low vascular event rate and a lower risk of leukemic transformation compared to TN-MF patients (Tefferi et al., 2018a; Szuber and Tefferi, 2018; Cattaneo et al., 2021; Michail et al., 2021).
The amazing progress of genetics over the last decade has changed the oncological diagnosis. Thanks to the development of advanced techniques in molecular biology, we have learned the pathomechanisms of numerous cancers. Indeed, knowledge about genetic changes in cancer has allowed the creation of diagnostic and prognostic models. NGS is a technique based on reading the genetic sequence nucleotide by nucleotide. This method allows the transition from the analysis of small gene fragments by traditional methods to the use of techniques capable of obtaining the complete sequence of many genes or even the entire genome. There are many NGS platforms with different sequencing chemistry and signal detection methods. The use of advanced techniques of molecular genetics such as NGS plays a very important role in hematooncology diagnostics. Despite the fact that the routine clinical usage of targeted or complete gene sequencing of hematological cancer patients is becoming more and more accessible, there are still numerous unresolved issues. The rapid expansion of the number of genes under consideration, the prevalence of variants of uncertain significance (VUSs) in less well-studied genes, the need to distinguish between germline and somatic mutations, and the lack of established guidelines regarding which patients and which genes should be tested are all challenges to clinical testing for these neoplasms. The most important aspect is data analysis, where bioinformatic tools are needed for mapping the reads to the human reference genome. Clarifying the somatic vs. germline origin of the mutations is one of the biggest problems in using and interpreting NGS data. Filtering NGS results would require a reliable method to distinguish between somatic and germline mutations. Variant categorization and annotation, including classifying variants into distinct “tiers” based on pathogenicity or clinical importance, may differ between laboratories in addition to interlaboratory variations in VAFs and the assignment of somatic status to variants. In general, it is recommended that interpretations follow specialist guidelines, such as those provided by the College of American Pathologists, the American Society for Clinical Oncology, and the Association for Molecular Pathology (Li et al., 2017). Despite all these disadvantages, genome profiling becomes the basis for prognosis prediction that is specifically tailored to each patient, but it is important to realize that the practical application of the latest scientific developments is still in progress. Over the past decade, tremendous progress has been made in understanding the pathogenesis of BCR::ABL1-negative myeloproliferative neoplasms. The dynamic development of genetics and the use of NGS in diagnostics have opened a new era in medicine. Additional mutations occur in more than 50% of patients with PV/ET and 80% of PMF cases. However, these mutations are not unique to MPN but are characteristic of all hematological malignancies. Most often these additional mutations occur in genes related to epigenetic regulation (TET2, DNMT3A, IDH1/2, ASXL1, and EZH2), mRNA splicing (SF3B1, SRSF2, U2AF1, and ZRSR2), signaling pathways (NRAS, KRAS, CBL, NF1, SH2B3, and PTPN11), and transcription factors (RUNX1 and TP53) (Tefferi et al., 2016a; Tefferi et al., 2016b; Luque Paz et al., 2023). The approximate frequency of additional MPN somatic mutations is shown in Table 4. The main target of myeloproliferative neoplasm therapy is based on the treatment of related symptoms, avoiding thrombosis and bleeding, improving the quality of life, and minimizing the risk of transformation to AML and post-PV/ET myelofibrosis. Until recently, information obtained from studies using advanced molecular techniques, such as NGS, contributed to improving the diagnosis and identification of new molecular biomarkers, establishing more accurate risk assessments, and selecting more individual therapeutic interventions. Testing for additional mutations has an impact on the therapeutic decision-making process. Data obtained from the NGS enable the creation of prognostic scoring systems based on genetic changes. An example is the GIPSS scale used in the assessment of prognostic risks in PMF. However, Grinfeld et al. created multi-stage prognostic models for specific individuals based on the genomic classification for all myeloproliferative neoplasms. This model identifies patients with chronic-phase MPN who are at a high risk of disease progression. Such patients might be taken into account for clinical trials of novel therapeutics. This model also identifies the majority of patients who initially appeared to have a good prognosis. Among these patients, a conservative therapeutic approach focusing on cytoreduction and vascular risk reduction will be sufficient. NGS opens up the possibility of using targeted therapies due to the presence of a mutation in a specific gene (Grinfeld et al., 2018; Loscocco et al., 2020; Luque Paz et al., 2023).
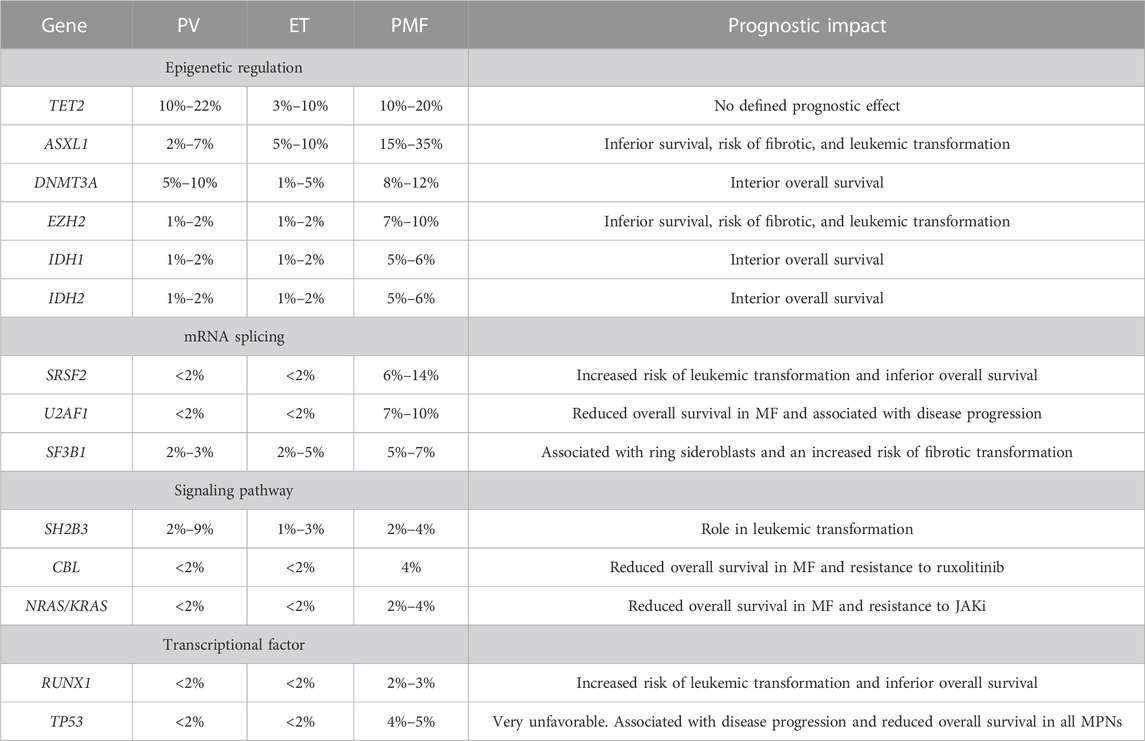
TABLE 4. Approximate frequency of additional MPN somatic mutations by Tefferi et al. (2016a), Tefferi et al. (2016b), Senín et al. (2018), and Vannucchi et al. (2013).
In recent years, research has focused on the significance of additional mutations in MPN patients. Using a targeted sequencing panel of 27 genes, Tefferi et al. observed that the most common mutations among ET patients were TET2 and ASXL1 genes. In addition, mutations in TET2 and SF3B1 were associated with an older age, SF3B1 with higher platelet counts, and ASXL1 with palpable splenomegaly. Mutations in the IDH2, EZH2, and SH2B3 genes were crucial as risk factors for survival in ET patients. Leukemia-free survival was associated with mutations in TP53, EZH2, SRSF2, and IDH2, while myelofibrosis-free survival was associated with SF3B1 and U2AF1 mutations. This study identified SH2B3, IDH2, U2AF1, SF3B1, EZH2, and TP53 variants as being “adverse” in patients with ET. The most common mutations among PV patients were also TET2 and ASXL1 genes. A phenotypic correlation in PV was observed only between SH2B3 and palpable splenomegaly. In PV patients, mutations in SRSF2, ASXL1, and IDH2 were associated with inferior survival. Significant association was also noted between SRSF2, IDH2, and RUNX1 for leukemia-free survival, and SRSF2 and RUNX1 for myelofibrosis-free survival. Considering overall, leukemia-free or myelofibrosis-free survival, ASXL1, SRSF2, and IDH1 mutations were related as “adverse.” Through the use of next-generation sequencing, this work has defined mutations that were associated with poorer overall survival (Tefferi et al., 2016a). Another study has shown that there is an association between additional mutations and clinical outcomes in patients with PV and ET. Using a panel of 51 genes, Senin et al. analyzed mutational profiles in 100 patients. The most common mutations occurred in the following genes: TET2, DNMT3A, TP53, and ASXL1. Shorter survival was associated with mutations in DNMT3A, SRSF2, SF3B1, IDH1/2, and RUNX1. The presence of additional mutations was associated with patients who transformed into AML or MF. These patients have developed new mutations more frequently during the time of transformation, which suggests genetic instability. Patients with mutations in SF3B1 and IDH1/2 had a higher risk of transformation to MF. Mutations in the ASXL1, TP53, IDH1/2, SRSF2, and RUNX1 genes have been associated with the risk of transformation to AML. They also noticed that the VAF for some mutations of the observed patients increased compared to the VAF from the time of diagnosis (Senín et al., 2018). Similarly, Luque Paz et al. observed that the most frequent mutations in patients with PV and ET were also TET2, ASXL1, IDH1/2, and DNMT3A. They observed that patients with additional mutations had evidence of progression at 3 years. In these kinds of patients, they noticed mutations in the ASXL1, IDH1/2, and SRSF2 genes. Studies also revealed that allele burden increased between diagnosis and follow-up. Furthermore, next-generation sequencing helped establish that some of the additional mutations observed during the follow-up also existed at the time of diagnosis in VAF <2% (Luque Paz et al., 2017). In another study, Laque Paz et al. investigated the time of leukemic transformation in PV and ET patients using a panel of 52 genes. ET patients with CALR mutation showed late transformation. The most recurrent mutated genes were TP53, TET2, RUNX1, ASXL1, and EZH2. Mutations in the following genes were associated with late transformation: TP53, BCORL1, and NRAS. U2AF1, IDH1/2, EZH2, and DNMT3 were linked to an earlier time of transformation (Luque Paz et al., 2020). Many other publications confirm that additional mutations in PV and ET are frequent and are associated with inferior overall survival and increased risks of transformation to MF or AML (Lasho et al., 2018; Pasca et al., 2022; Morishita et al., 2023).
A few studies have shown associations between treatment response and high-risk mutations. Intolerance to hydroxyurea (HU) occurs in 20%–30% of PV patients, and this has been correlated with an increased risk of thrombosis, disease progression, and shorter survival. The possibility of evolving resistance to HU treatment has been estimated at 64% in patients with the TP53 mutation and at 49% in patients with spliceosome or chromatin gene mutations (Alvarez-Larrán et al., 2021). Some of the additional mutations were correlated with an increased risk of developing cytopenias during HU treatment (Senín et al., 2018). Similar observations were made in the response of ET patients with CALR mutations to interferon alpha. The presence of TET2, IDH2, ASXL1, and TP53 mutations was associated with a poorer response to therapy (Verger et al., 2015). The presence of additional mutations in the TET2, ASXL1, DNMT3A, IDH, and EZH2 genes in PV and ET patients was associated with failure to achieve a complete molecular response during treatment with pegylated interferon alpha (Quintás-Cardama et al., 2013). Another study has revealed that failure to achieve a complete molecular response is associated with DNMT3A mutations (Knudsen et al., 2022).
Tefferi et al. observed that as for a phenotypic correlation, patients with ET and TET2 mutations were associated with an increased risk of thrombosis, which was independent of driver mutation status and age. Nevertheless, such an association was not noticed in a cohort of Italian patients with ET (Tefferi et al., 2016a). Another study also highlighted that there may be an association between the history of thrombosis and the presence of TET2, DNMT3A, or ASXL1 mutations. However, different studies on 587 patients did not find a significant influence on either arterial or venous events (Cerquozzi et al., 2017; Segura-Díaz et al., 2020).
Among patients with myelofibrosis, additional mutations are more frequent than those in other myeloproliferative neoplasms. These are mutations in genes involving DNA methylation, chromatin modification, RNA splicing, or DNA repair.
According to the latest recommendations, in the absence of driver mutations, the detection of other mutations in genes such as ASXL1, EZH2, IDH1, IDH2, SF3B1, SRSF2, and TET2 supports the clonal nature of the disease. This information is included as a major criterion for pre-PMF and over-fibrotic PMF stages (Arber et al., 2022). Additional mutations in myelofibrosis have been studied for several years because of the worse prognosis and higher risk of transformation to AML than PV or ET. The first publication about additional mutations in myelofibrosis appeared in 2013.
Vannucchi et al. investigated 879 patients. A study revealed that in the European cohort, ASXL1, EZH2, and SRSF2 were associated with shorter survival. This has been confirmed in another Mayo Clinic group where mutations in the ASXL1, SRSF2, and EZH2 genes were linked to poor survival. Increased risks of leukemic transformation were also associated with mutations in ASXL1, SRSF2, and IDH1/2 genes in the European group. In the Mayo Clinic group, leukemia-free survival was associated with IDH1 and SRSF2 mutations. Mutations in the ASXL1, SRSF2, EZH2, and IDH1/2 genes, referred to as high-risk mutations (HRM), identify patients who have a higher risk of transformation to AML or a higher risk of death (Vannucchi et al., 2013). Guglielmelli et al. searched for a codependence between the number of HMR mutations and their effects on OS and leukemia-free survival (LFS). Patients with two or more additional high-risk mutations (ASXL1, EZH2, SRSF2, and IDH1/2) had decreased OS and LFS compared to patients with only one additional mutation or those without any (Guglielmelli et al., 2014). Tefferi et al. showed that the most frequent genes in patients with MF were ASXL1, TET2, SRSF2, U2AF1, ASXL1, SRSF2, CBL, KIT, RUNX1, CEBPA, and SH2B3, which have been associated with poorer overall survival and leukemia-free survival. Studies have confirmed that the number of additional mutations matters; however, analyses have shown that patients with one or two adverse mutations have similar survival but inferior survival compared to patients who had no additional mutations and superior to those who had three or more (Tefferi et al., 2016b). Studies conducted using a targeted panel of 137 genes on the cohort of 259 patients with pre-PMF, overt-PMF, and PMF-AP/BP have shown that the most recurrent additional mutations were in ASXL1, TET2, SRSF2, U2AF1, and SETBP1. Mutations in ASXL1 and U2AF1 were more frequent in overt-PMF than those in pre-PMF. Mutations in ASXL1, SRSF2, RUNX1, SETBP1, NOTCH2, NRAS, and EZH2 were presented more frequently in PMF-AP/BP than those in overt-PMF. They have also noticed that the smaller clone size of the ASXL1 mutation had adverse risk factors in overt-PMF and PMF-AP/BP patients (Yan et al., 2022). Another study on 113 patients with overt-PMF and pre-fibrotic PMF has shown similar results. The most frequent additional mutations were observed in ASXL1, TET2, EZH2, DNMT3A, SRSF2, SF3B1, U2AF1, TP53, and IDH1/2. No significant differences were observed between overt-PMF and pre-fibrotic-PMF patients. Those patients with a high-risk mutation (ASXL1, EZH2, and SRSF2) have been associated with poorer prognosis and disease progression. In these groups of patients, leukemic transformation was detected only in patients with high-risk mutations (Morishita et al., 2021). In another study, patients with PMF were compared to patients with PV and ET. The frequencies of high-risk molecular mutations were significantly higher in patients with PMF than those in PV and ET patients. In addition, allele burdens of TET2 and DNMT3A mutations were higher in patients with PV and PMF than those in ET patients. This may suggest that TET2 and DNMT3A mutations may escalate due to the acquisition of HMR mutations in PMF and thus lead to a worse prognosis (Morishita et al., 2023). Laque Paz et al., as in other studies, observed that the most frequently mutated genes in myelofibrosis were ASXL1, TET2, SRSF2, U2AF1, and EZH2. In addition, they made very interesting observations dividing patients into four groups, namely, TP53-mutated patients, patients with ≥1 mutation in EZH2; CBL, U2AF1, SRSF2, IDH1, IDH2, NRAS, or KRAS - high-risk groups; ASXL1-only mutation; and “other” patients mainly mutated in the NFE2, DNMT3A, TET2, and SF3B1 genes. The worst prognostic effects on OS, leukemic transformation, and leukemia-free survival were in the TP53 and high-risk groups compared to those in the ASXL1-only and “other patient” groups. Moreover, the TP53 mutation and high-risk groups were correlated with a higher risk of non-AML death. Interestingly, they noted that the ASXL1-only mutation group did not have an adverse prognosis on OS and only a moderate effect on leukemic transformation. They suggested that ASXL1 mutations themselves had no negative effects but appeared to confer an adverse risk factor when correlated with high-risk mutations (Luque Paz et al., 2021). Guglielmelli et al. also investigated the prognostic role of ASXL1 but with a distinction between PMF and secondary MF (SMF). They also divided the patients into four categories: TP53 group, high-risk group, ASXL1 only, and “others.” Patients in the TP53 and ASXL1-only groups were more frequently diagnosed with SMF. Patients with TP53 and the high-risk group had the worst OS. Among patients with PMF, a negative prognostic effect of ASXL1-only was noted in comparison with the “others” category. In PMF, ASXL1 mutations among patients from the high-risk group mutation were associated with shorter OS. In PMF, VAF for ASXL1 in the TP53 and high-risk group was greater than those for ASXL1-only. Unlike SMF, VAF was lower in those groups, which may suggest that ASXL1 mutations are early driving events in PMF, but in SMF, these mutations might be acquired later. In SMF, the worst OS had the TP53 group. They noted that there were no differences between the OS of the ASXL1-only group and “other” or high-risk groups. In SMF, ASXL1 mutations in the high-risk group had no impact on OS. These recent studies have emphasized the concept that PMF and SMF are different biological entities; furthermore, they also confirmed the adverse effect of the ASXL1 mutation on PMF not in SMF (Guglielmelli et al., 2022).
Notably, RAS/CBL mutations in PMF were associated with poor response to ruxolitinib treatment and reduced survival. The authors suggested that it is important to identify RAS mutations in patients with MF (Coltro et al., 2020; Santos et al., 2020). In the existing prognostic risk assessment models for patients with PMF, apart from driver mutations, additional mutations and the karyotype were taken into account. MIPSS70 (mutation-enhanced international prognostic scoring system for transplant age patients) uses mutations and clinical data, and MIPSSv2 (the karyotype-enhanced MIPSS70) uses mutations, karyotype, and clinical data; GIPSS (the genetically-inspired prognostic scoring system) is based solely on mutations and karyotype. The use of the NGS technique to assess the risk of patients with PMF is very helpful, especially in a model that is based on genetic changes [(Guglielmelli et al., 2018; Tefferi et al., 2018b; Tefferi et al., 2018c)].
Recent studies have demonstrated the utility of NGS in refining the characterization of TN-MPNs by establishing clonality and detecting different mutations in driver mutations. Information from sequencing data analysis aids in clinical decision-making. The implementation of the NGS technique helped redefine the diagnosis of TN-MPN. Some studies highlighted the problem of the existence of non-canonical mutations that are not detected by routine methods. Studies have shown the need to search for a clonal marker to confirm the diagnosis. Another problem is that the routine techniques that we use for establishing a diagnosis of MPN have limited sensitivity. NGS helps improve sensitivity, and with this technique, we can even discover mutations in genes that have not been previously associated with MPN (Costa Melo Svidnicki et al., 2019; Geay et al., 2020; Michail et al., 2021; Maddali et al., 2022; Wu et al., 2022).
In the age of molecular medicine, new advanced technologies are needed to improve laboratory diagnostics. Detection of the driver mutation was a breakthrough discovery in the diagnosis of myeloproliferative neoplasms. This helped determine the pathogenesis of the disease. The introduction of the NGS technique has significantly changed the perception and approach to diagnosis, risk assessment, and treatment of MPN. Thanks to the ability to search for mutations in many genes simultaneously, this technique allows for a more accurate diagnosis based on the patient’s genetic profile. NGS enables the identification of a group of patients with a poor prognosis, patients with high genetic instability, and an increased risk of disease progression or transformation into AML. The identification of high-risk mutations (HMR) and the possibility of incorporating them into prognostic scoring systems highlight the importance of genetic testing in hematooncology.
AM-B designed the study and wrote the draft of the manuscript. TW contributed to writing—review and editing. All authors contributed to the article and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Al Assaf, C., Van Obbergh, F., Billiet, J., Lierman, E., Devos, T., Graux, C., et al. (2015). Analysis of phenotype and outcome in essential thrombocythemia with CALR or JAK2 mutations. Haematologica 100 (7), 893–897. doi:10.3324/haematol.2014.118299
Alvarez-Larrán, A., Díaz-González, A., Such, E., Mora, E., Andrade-Campos, M., García-Hernández, C., et al. (2021). Genomic characterization of patients with polycythemia vera developing resistance to hydroxyurea. Leukemia 35 (2), 623–627. doi:10.1038/s41375-020-0849-2
Arber, D. A., Orazi, A., Hasserjian, R. P., Borowitz, M. J., Calvo, K. R., Kvasnicka, H. M., et al. (2022). International Consensus classification of myeloid neoplasms and acute leukemias: integrating morphologic, clinical, and genomic data. Blood 140 (11), 1200–1228. doi:10.1182/blood.2022015850
Arunachalam, A. K., Suresh, H., Mathews, V., and Balasubramanian, P. (2018). Allele specific PCR: A cost effective screening method for MPL mutations in myeloproliferative neoplasms. Indian J. Hematol. Blood Transfus. 34 (4), 765–767. doi:10.1007/s12288-018-0982-5
Asp, J., Andréasson, B., Hansson, U., Wasslavik, C., Abelsson, J., Johansson, P., et al. (2016). Mutation status of essential thrombocythemia and primary myelofibrosis defines clinical outcome. Haematologica 101 (4), e129–e132. doi:10.3324/haematol.2015.138958
Barbui, T., Finazzi, G., Carobbio, A., Thiele, J., Passamonti, F., Rumi, E., et al. (2012). Development and validation of an international prognostic score of thrombosis in world Health organization-essential thrombocythemia (IPSET-thrombosis). Blood 120 (26), 5128–5133. doi:10.1182/blood-2012-07-444067
Barbui, T., Thiele, J., Gisslinger, H., Kvasnicka, H. M., Vannucchi, A. M., Guglielmelli, P., et al. (2018). The 2016 WHO classification and diagnostic criteria for myeloproliferative neoplasms: document summary and in-depth discussion. Blood Cancer J. 8 (2), 15. doi:10.1038/s41408-018-0054-y
Barosi, G., Massa, M., Campanelli, R., Fois, G., Catarsi, P., Viarengo, G., et al. (2017). Primary myelofibrosis: older age and high JAK2V617F allele burden are associated with elevated plasma high-sensitivity C-reactive protein levels and a phenotype of progressive disease. Leukemia Res. 60, 18–23. ISSN 0145-2126. doi:10.1016/j.leukres.2017.06.004
Baxter, E. J., Scott, L. M., Campbell, P. J., East, C., Fourouclas, N., Swanton, S., et al. (2005). Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet 365 (9464), 1054–1061. doi:10.1016/S0140-6736(05)71142-9
Cabagnols, X., Defour, J. P., Ugo, V., Ianotto, J. C., Mossuz, P., Mondet, J., et al. (2015). Differential association of calreticulin type 1 and type 2 mutations with myelofibrosis and essential thrombocytemia: relevance for disease evolution. Leukemia 29 (1), 249–252. doi:10.1038/leu.2014.270
Cattaneo, D., Croci, G. A., Bucelli, C., Tabano, S., Cannone, M. G., Gaudioso, G., et al. (2021). Triple-negative essential thrombocythemia: clinical-pathological and molecular features. A single-center cohort study. Front. Oncol. 11, 637116. doi:10.3389/fonc.2021.637116
Cerquozzi, S., Barraco, D., Lasho, T., Finke, C., Hanson, C. A., Ketterling, R. P., et al. (2017). Risk factors for arterial versus venous thrombosis in polycythemia vera: a single center experience in 587 patients. Blood Cancer J. 7 (12), 662. doi:10.1038/s41408-017-0035-6
Chachoua, I., Pecquet, C., El-Khoury, M., Nivarthi, H., Albu, R. I., Marty, C., et al. (2016). Thrombopoietin receptor activation by myeloproliferative neoplasm associated calreticulin mutants. Blood 127 (10), 1325–1335. Epub 2015 Dec 14. doi:10.1182/blood-2015-11-681932
Coltro, G., Rotunno, G., Mannelli, L., Mannarelli, C., Fiaccabrino, S., Romagnoli, S., et al. (2020). RAS/CBL mutations predict resistance to JAK inhibitors in myelofibrosis and are associated with poor prognostic features. Blood Adv. 4 (15), 3677–3687. doi:10.1182/bloodadvances.2020002175
Constantinescu, S. N., and Pecquet, C. (2020). Educational updates in hematology book: 25th congress of the European Hematology Association, Virtual Edition 2020. HemaSphere 4 (S2), e444. doi:10.1097/HS9.0000000000000444
Costa Melo Svidnicki, M., De Melo Campos, P., Alves Ferreira Filho, M., Leme Fujiura, C., Yoshizato, T., Makishima, H., et al. (2019). Mutations in triple-negative patients with myeloproliferative neoplasms. Blood 134 (1), 5395. doi:10.1182/blood-2019-128764
Defour, J. P., Chachoua, I., Pecquet, C., and Constantinescu, S. N. (2016). Oncogenic activation of MPL/thrombopoietin receptor by 17 mutations at W515: implications for myeloproliferative neoplasms. Leukemia 30 (5), 1214–1216. doi:10.1038/leu.2015.271
Furtado, L. V., Weigelin, H. C., Elenitoba-Johnson, K. S., and Betz, B. L. (2013). Detection of MPL mutations by a novel allele-specific PCR-based strategy. J. Mol. Diagn 15 (6), 810–818. doi:10.1016/j.jmoldx.2013.07.006
Geay, A., Aral, B., Bourgeois, V., Martin, P., Airaud, F., Garrec, C., et al. (2020). Diagnosis of exon 12-positive polycythemia vera rescued by NGS. Clin. Case Rep. 8 (5), 790–792. doi:10.1002/ccr3.2720
Giannopoulos, A., Rougkala, N., Loupis, T., Mantzourani, M., Viniou, N. A., Variami, E., et al. (2019). Detection of CALR mutations using high resolution melting curve analysis (HRM-A); application on a large cohort of Greek ET and MF patients. Mediterr. J. Hematol. Infect. Dis. 11 (1), e2019009. doi:10.4084/MJHID.2019.009
Grinfeld, J., Nangalia, J., Baxter, E. J., Wedge, D. C., Angelopoulos, N., Cantrill, R., et al. (2018). Classification and personalized prognosis in myeloproliferative neoplasms. N. Engl. J. Med. 379 (15), 1416–1430. doi:10.1056/NEJMoa1716614
Guglielmelli, P., and Calabresi, L. (2021). The MPL mutation. Int. Rev. Cell Mol. Biol. 365, 163–178. doi:10.1016/bs.ircmb.2021.09.003
Guglielmelli, P., Coltro, G., Mannelli, F., Rotunno, G., Loscocco, G. G., Mannarelli, C., et al. (2022). ASXL1 mutations are prognostically significant in PMF, but not MF following essential thrombocythemia or polycythemia vera. Blood Adv. 6 (9), 2927–2931. doi:10.1182/bloodadvances.2021006350
Guglielmelli, P., Gangat, N., Coltro, G., Lasho, T. L., Loscocco, G. G., Finke, C. M., et al. (2021b). Mutations and thrombosis in essential thrombocythemia. Blood Cancer J. 11, 77. doi:10.1038/s41408-021-00470-y
Guglielmelli, P., Lasho, T. L., Rotunno, G., Mudireddy, M., Mannarelli, C., Nicolosi, M., et al. (2018). MIPSS70: mutation-enhanced international prognostic score system for transplantation-age patients with primary myelofibrosis. J. Clin. Oncol. 36 (4), 310–318. doi:10.1200/JCO.2017.76.4886
Guglielmelli, P., Lasho, T. L., Rotunno, G., Score, J., Mannarelli, C., Pancrazzi, A., et al. (2014). The number of prognostically detrimental mutations and prognosis in primary myelofibrosis: an international study of 797 patients. Leukemia 28 (9), 1804–1810. doi:10.1038/leu.2014.76
Guglielmelli, P., Loscocco, G. G., Mannarelli, C., Rossi, E., Mannelli, F., Ramundo, F., et al. (2021a). JAK2V617F variant allele frequency >50% identifies patients with polycythemia vera at high risk for venous thrombosis. Blood Cancer J. 11 (12), 199. doi:10.1038/s41408-021-00581-6
Guglielmelli, P., Maccari, C., Sordi, B., Balliu, M., Atanasio, A., Mannarelli, C., et al. (2023). Phenotypic correlations of CALR mutation variant allele frequency in patients with myelofibrosis. Blood Cancer J. 13, 21. doi:10.1038/s41408-023-00786-x
Guglielmelli, P., Pacilli, A., Rotunno, G., Rumi, E., Rosti, V., Delaini, F., et al. (2017). Presentation and outcome of patients with 2016 WHO diagnosis of prefibrotic and overt primary myelofibrosis. Blood 129, 3227–3236. doi:10.1182/blood-2017-01-761999
Haslam, K., and Langabeer, S. E. (2016). Monitoring minimal residual disease in the myeloproliferative neoplasms: current applications and emerging approaches. Biomed. Res. Int. 2016, 7241591. Epub. doi:10.1155/2016/7241591
How, J., Hobbs, G., and Mullally, A. (2019). Mutant calreticulin in myeloproliferative neoplasms. Blood 134 (25), 2242–2248. doi:10.1182/blood.2019000622
James, C., Ugo, V., Le Couédic, J. P., Staerk, J., Delhommeau, F., Lacout, C., et al. (2005). A unique clonal JAK2 mutation leading toconstitutive signalling causes polycythaemia vera. Nature 434 (7037), 1144–1148. doi:10.1038/nature03546
Jones, A. V., Ward, D., Lyon, M., Leung, W., Callaway, A., Chase, A., et al. (2015). Evaluation of methods to detect CALR mutations in myeloproliferative neoplasms. Leuk. Res. 39 (1), 82–87. doi:10.1016/j.leukres.2014.11.019
Khoury, J. D., Solary, E., Abla, O., Akkari, Y., Alaggio, R., Apperley, J. F., et al. (2022). The 5th edition of the world Health organization classification of haematolymphoid Tumours: myeloid and histiocytic/dendritic neoplasms. Leukemia 36, 1703–1719. doi:10.1038/s41375-022-01613-1
Kim, H-Y., Han, Y., Jang, J. H., Jung, C. W., Kim, S-H., and Kim, H-J. (2022). Effects of CALR-mutant type and burden on the phenotype of myeloproliferative neoplasms. Diagnostics 12 (11), 2570. doi:10.3390/diagnostics12112570
Klampfl, T., Gisslinger, H., Harutyunyan, A. S., Nivarthi, H., Rumi, E., Milosevic, J. D., et al. (2013). Somatic mutations of calreticulin in myeloproliferative neoplasms. N. Engl. J. Med. 369, 2379–2390. doi:10.1056/NEJMoa1311347
Knudsen, T. A., Skov, V., Stevenson, K., Werner, L., Duke, W., Laurore, C., et al. (2022). Genomic profiling of a randomized trial of interferon-α vs hydroxyurea in MPN reveals mutation-specific responses. Blood Adv. 6 (7), 2107–2119. doi:10.1182/bloodadvances.2021004856
Kralovics, R., Passamonti, F., Buser, A. S., Teo, S. S., Tiedt, R., Passweg, J. R., et al. (2005). A gain-of-function mutation of JAK2 in myeloproliferative disorders. N. Engl. J. Med. 352 (17), 1779–1790. doi:10.1056/NEJMoa051113
Lasho, T. L., Mudireddy, M., Finke, C. M., Hanson, C. A., Ketterling, R. P., Szuber, N., et al. (2018). Targeted next-generation sequencing in blast phase myeloproliferative neoplasms. Blood Adv. 2 (4), 370–380. doi:10.1182/bloodadvances.2018015875
Lee, A. J., Kim, S. G., Nam, J. Y., Yun, J., Ryoo, H. M., and Bae, S. H. (2021). Clinical features and outcomes of JAK2 V617F-positive polycythemia vera and essential thrombocythemia according to the JAK2 V617F allele burden. Blood Res. 56 (4), 259–265. doi:10.5045/br.2021.2021089
Levine, R. L., Wadleigh, M., Cools, J., Ebert, B. L., Wernig, G., Huntly, B. J., et al. (2005). Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasiawith myelofibrosis. Cancer Cell 7 (4), 387–397. doi:10.1016/j.ccr.2005.03.023
Li, M. M., Datto, M., Duncavage, E. J., Kulkarni, S., Lindeman, N. I., Roy, S., et al. (2017). Standards and guidelines for the interpretation and reporting of sequence variants in cancer: A joint Consensus recommendation of the association for molecular Pathology, American society of clinical Oncology, and College of American Pathologists. J. Mol. Diagn 19 (1), 4–23. doi:10.1016/j.jmoldx.2016.10.002
Link-Lenczowska, D., Pallisgaard, N., Cordua, S., Zawada, M., Czekalska, S., Krochmalczyk, D., et al. (2018). A comparison of qPCR and ddPCR used for quantification of the JAK2 V617F allele burden in Ph negative MPNs. Ann. Hematol. 97 (12), 2299–2308. Epub. doi:10.1007/s00277-018-3451-1
Loomila, L., Naseem, S., Rastogi, P., Jain, A., and Malhotra, P. (2022). Study of JAK2 V617F allele burden by droplet digital PCR in BCR-ABL1 negative myeloproliferative neoplasms and its implications on disease phenotype. Blood 140 (1), 12270. Page 12270, ISSN 0006-4971. doi:10.1182/blood-2022-168835
Loscocco, G. G., Coltro, G., Guglielmelli, P., and Vannucchi, A. M. (2021a). Integration of molecular information in risk assessment of patients with myeloproliferative neoplasms. Cells 10 (8), 1962. doi:10.3390/cells10081962
Loscocco, G. G., Guglielmelli, P., Gangat, N., Rossi, E., Mannarelli, C., Betti, S., et al. (2021b). Clinical and molecular predictors of fibrotic progression in essential thrombocythemia: A multicenter study involving 1607 patients. Am. J. Hematol. 96 (11), 1472–1480. doi:10.1002/ajh.26332
Loscocco, G. G., Guglielmelli, P., and Vannucchi, A. M. (2020). Impact of mutational profile on the management of myeloproliferative neoplasms: A short review of the emerging data. Onco Targets Ther. 13, 12367–12382. PMID: 33293830; PMCID: PMC7718985. doi:10.2147/OTT.S287944
Luque Paz, D., Chauveau, A., Boyer, F., Buors, C., Samaison, L., Cottin, L., et al. (2017). Sequential analysis of 18 genes in polycythemia vera and essential thrombocythemia reveals an association between mutational status and clinical outcome. Genes Chromosom. Cancer 56 (5), 354–362. doi:10.1002/gcc.22437
Luque Paz, D., Jouanneau-Courville, R., Riou, J., Ianotto, J. C., Boyer, F., Chauveau, A., et al. (2020). Leukemic evolution of polycythemia vera and essential thrombocythemia: genomic profiles predict time to transformation. Blood Adv. 4 (19), 4887–4897. doi:10.1182/bloodadvances.2020002271
Luque Paz, D., Kralovics, R., and Skoda, R. C. (2023). Genetic basis and molecular profiling in myeloproliferative neoplasms. Blood 141 (16), 1909–1921. doi:10.1182/blood.2022017578
Luque Paz, D., Riou, J., Verger, E., Cassinat, B., Chauveau, A., Ianotto, J. C., et al. (2021). Genomic analysis of primary and secondary myelofibrosis redefines the prognostic impact of ASXL1 mutations: a FIM study. Blood Adv. 5 (5), 1442–1451. doi:10.1182/bloodadvances.2020003444
Maddali, M., Venkatraman, A., Kulkarni, U. P., Mani, S., Raj, S., Sigamani, E., et al. (2022). Molecular characterization of triple-negative myeloproliferative neoplasms by next-generation sequencing. Ann. Hematol. 101 (9), 1987–2000. doi:10.1007/s00277-022-04920-w
Michail, O., McCallion, P., McGimpsey, J., Hindley, A., Greenfield, G., McAllister, R., et al. (2021). Mutational profiling in suspected triple-negative essential thrombocythaemia using targeted next-generation sequencing in a real-world cohort. J. Clin. Pathol. 74 (12), 808–811. doi:10.1136/jclinpath-2020-206570
Morishita, S., Hashimoto, Y., Furuya, C., Edahiro, Y., Ochiai, T., Shirane, S., et al. (2023). Non-driver gene mutation analysis in a large cohort of polycythemia vera and essential thrombocythemia. Eur. J. Haematol. 110 (2), 131–136. doi:10.1111/ejh.13882
Morishita, S., Ochiai, T., Misawa, K., Osaga, S., Inano, T., Fukuda, Y., et al. (2021). Clinical impacts of the mutational spectrum in Japanese patients with primary myelofibrosis. Int. J. Hematol. 113 (4), 500–507. doi:10.1007/s12185-020-03054-x
Nangalia, J., Massie, C. E., Baxter, E. J., Nice, F. L., Gundem, G., Wedge, D. C., et al. (2013). Somatic CALR mutations in myeloproliferative neoplasms with nonmutated JAK2. N. Engl. J. Med. 369, 2391–2405. doi:10.1056/NEJMoa1312542
Nivarthi, H., Chen, D., Cleary, C., Kubesova, B., Jäger, R., Bogner, E., et al. (2016). Thrombopoietin receptor is required for the oncogenic function of CALR mutants. Leukemia 30, 1759–1763. doi:10.1038/leu.2016.32
Park, S. H., Chi, H. S., Cho, Y. U., Jang, S., and Park, C. J. (2013). The allele burden of JAK2 V617F can aid in differential diagnosis of Philadelphia Chromosome-Negative Myeloproliferative Neoplasm. Blood Res. 48 (2), 128–132. Epub. doi:10.5045/br.2013.48.2.128
Pasca, S., Chifotides, H. T., Verstovsek, S., and Bose, P. (2022). Mutational landscape of blast phase myeloproliferative neoplasms (MPN-BP) and antecedent MPN. Int. Rev. Cell Mol. Biol. 366, 83–124. Cellular and molecular aspects of MPNs - part B, Volume N, Galuzzi L, Serial. Editor N Bartalucci. 1st edition (Academic Press). doi:10.1016/bs.ircmb.2021.02.008
Pei, Y. Q., Wu, Y., Wang, F., and Cui, W. (2016). Prognostic value of CALR vs. JAK2V617F mutations on splenomegaly, leukemic transformation, thrombosis, and overall survival in patients with primary fibrosis: a meta-analysis. Ann. Hematol. 95 (9), 1391–1398. Epub. doi:10.1007/s00277-016-2712-0
Pietra, D., Rumi, E., Ferretti, V. V., Di Buduo, C. A., Milanesi, C., Cavalloni, C., et al. (2016). Differential clinical effects of different mutation subtypes in CALR-mutant myeloproliferative neoplasms. Leukemia 30 (2), 431–438. doi:10.1038/leu.2015.277
Pikman, Y., Lee, B. H., Mercher, T., McDowell, E., Ebert, B. L., Gozo, M., et al. (2006). MPLW515L is a novel somatic activating mutation in myelofibrosis with myeloid metaplasia. PLoS Med. 3 (7), e270. doi:10.1371/journal.pmed.0030270
Quintás-Cardama, A., Abdel-Wahab, O., Manshouri, T., Kilpivaara, O., Cortes, J., Roupie, A. L., et al. (2013). Molecular analysis of patients with polycythemia vera or essential thrombocythemia receiving pegylated interferon α-2a. Blood 122 (6), 893–901. Epub 2013 Jun 19. doi:10.1182/blood-2012-07-442012
Regimbeau, M., Mary, R., Hermetet, F., and Girodon, F. (2022). Genetic background of polycythemia vera. Genes (Basel). 13 (4), 637. doi:10.3390/genes13040637
Rotunno, G., Mannarelli, C., Guglielmelli, P., Pacilli, A., Pancrazzi, A., Pieri, L., et al. (2014). Impact of calreticulin mutations on clinical and hematological phenotype and outcome in essential thrombocythemia. Blood 123 (10), 1552–1555. Epub. doi:10.1182/blood-2013-11-538983
Rumi, E., Pietra, D., Ferretti, V., Klampfl, T., Harutyunyan, A. S., Milosevic, J. D., et al. (2014). JAK2 or CALR mutation status defines subtypes of essential thrombocythemia with substantially different clinical course and outcomes. Blood 123 (10), 1544–1551. doi:10.1182/blood-2013-11-539098
Rumi, E., Trotti, C., Vanni, D., Casetti, I. C., Pietra, D., and Sant’Antonio, E. (2020). The genetic basis of primary myelofibrosis and its clinical relevance. nternational J. Mol. Sci. 21 (23), 8885. doi:10.3390/ijms21238885
Santos, F. P. S., Getta, B., Masarova, L., Famulare, C., Schulman, J., Datoguia, T. S., et al. (2020). Prognostic impact of RAS-pathway mutations in patients with myelofibrosis. Leukemia 34 (3), 799–810. doi:10.1038/s41375-019-0603-9
Sazawal, S., Singh, K., Chhikara, S., Chaubey, R., Mahapatra, M., and Saxena, R. (2019). Influence of JAK2V617F allele burden on clinical phenotype of polycythemia vera patients: A study from India. South Asian J. Cancer 8 (2), 127–129. doi:10.4103/sajc.sajc_161_18
Scott, L. M. (2011). The JAK2 exon 12 mutations: a comprehensive review. Am. J. Hematol. 86 (8), 668–676. Epub 2011 Jun 14. doi:10.1002/ajh.22063
Scott, L. M., Tong, W., Levine, R. L., Scott, M. A., Beer, P. A., Stratton, M. R., et al. (2007). JAK2 exon 12 mutations in polycythemia vera and idiopathic erythrocytosis. N. Engl. J. Med. 356 (5), 459–468. doi:10.1056/NEJMoa065202
Segura-Díaz, A., Stuckey, R., Florido, Y., González-Martín, J. M., López-Rodríguez, J. F., Sánchez-Sosa, S., et al. (2020). Thrombotic risk detection in patients with polycythemia vera: the predictive role of dnmt3a/TET2/ASXL1 mutations. Cancers (Basel) 12 (4), 934. doi:10.3390/cancers12040934
Senín, A., Fernández-Rodríguez, C., Bellosillo, B., Camacho, L., Longarón, R., Angona, A., et al. (2018). Non-driver mutations in patients with JAK2V617F-mutated polycythemia vera or essential thrombocythemia with long-term molecular follow-up. Ann. Hematol. 97 (3), 443–451. doi:10.1007/s00277-017-3193-5
Sørensen, A. L., Mikkelsen, S. U., Knudsen, T. A., Bjørn, M. E., Andersen, C. L., Bjerrum, O. W., et al. (2020). Ruxolitinib and interferon-α2 combination therapy for patients with polycythemia vera or myelofibrosis: a phase II study. Haematologica 105 (9), 2262–2272. doi:10.3324/haematol.2019.235648
Stegelmann, F., Teichmann, L. L., Heidel, F. H., Crodel, C. C., Ernst, T., Kreil, S., et al. (2023). Clinicohematologic and molecular response of essential thrombocythemia patients treated with pegylated interferon-α: a multi-center study of the German study group-myeloproliferative neoplasms (GSG-MPN). Leukemia 37, 924–928. doi:10.1038/s41375-023-01837-9
Szuber, N., and Tefferi, A. (2018). Driver mutations in primary myelofibrosis and their implications. Curr. Opin. Hematol. 25 (2), 129–135. doi:10.1097/MOH.0000000000000406
Tefferi, A., Guglielmelli, P., Nicolosi, M., Mannelli, F., Mudireddy, M., Bartalucci, N., et al. (2018c). Gipss: genetically inspired prognostic scoring system for primary myelofibrosis. Leukemia 32 (7), 1631–1642. doi:10.1038/s41375-018-0107-z
Tefferi, A., Guglielmelli, P., Larson, D. R., Finke, C., Wassie, E. A., Pieri, L., et al. (2014b). Long-term survival and blast transformation in molecularly annotated essential thrombocythemia, polycythemia vera, and myelofibrosis. Blood 124 (16), 2507–2513. doi:10.1182/blood-2014-05-579136
Tefferi, A., Guglielmelli, P., Lasho, T. L., Gangat, N., Ketterling, R. P., Pardanani, A., et al. (2018b). MIPSS70+ version 2.0: mutation and karyotype-enhanced international prognostic scoring system for primary myelofibrosis. J. Clin. Oncol. 36 (17), 1769–1770. doi:10.1200/JCO.2018.78.9867
Tefferi, A., Lasho, T. L., Finke, C. M., Elala, Y., Hanson, C. A., Ketterling, R. P., et al. (2016b). Targeted deep sequencing in primary myelofibrosis. Blood Adv. 1 (2), 105–111. doi:10.1182/bloodadvances.2016000208
Tefferi, A., Lasho, T. L., Guglielmelli, P., Finke, C. M., Rotunno, G., Elala, Y., et al. (2016a). Targeted deep sequencing in polycythemia vera and essential thrombocythemia. Blood Adv. 1 (1), 21–30. doi:10.1182/bloodadvances.2016000216
Tefferi, A., Nicolosi, M., Mudireddy, M., Szuber, N., Finke, C. M., Lasho, T. L., et al. (2018a). Driver mutations and prognosis in primary myelofibrosis: mayo-careggi MPN alliance study of 1,095 patients. Am. J. Hematol. 93 (3), 348–355. doi:10.1002/ajh.24978
Tefferi, A., Wassie, E. A., Guglielmelli, P., Gangat, N., Belachew, A. A., Lasho, T. L., et al. (2014a). Type 1 versus type 2 calreticulin mutations in essential thrombocythemia: a collaborative study of 1027 patients. Am. J. Hematol. 89 (8), E121–E124. Epub. doi:10.1002/ajh.23743
Ullah, N., Khan, S. N., Umair, M., Khan, A. A., Liu, X., Khattak, A. A., et al. (2022). Development of a real-time qPCR assay for detection of common MPL mutations in myeloproliferative neoplasms (MPNS). Appl. Biochem. Biotechnol. 194 (12), 5907–5917. doi:10.1007/s12010-022-04051-y
Vainchenker, W., and Kralovics, R. (2017). Genetic basis and molecular pathophysiology of classical myeloproliferative neoplasms. Blood 129, 667–679. doi:10.1182/blood-2016-10-695940
Vannucchi, A., Lasho, T., Guglielmelli, P., Biamonte, F., Pardanani, A., Pereira, A., et al. (2013). Mutations and prognosis in primary myelofibrosis. Leukemia 27, 1861–1869. doi:10.1038/leu.2013.119
Verger, E., Cassinat, B., Chauveau, A., Dosquet, C., Giraudier, S., Schlageter, M. H., et al. (2015). Clinical and molecular response to interferon-α therapy in essential thrombocythemia patients with CALR mutations. Blood 126 (24), 2585–2591. Epub 2015 Oct 20. PMID: 26486786. doi:10.1182/blood-2015-07-659060
Weir, P., Hindley, A., Catherwood, M., and McMullin, M. F. (2023). CALR type 1 mutations are associated with an increased incidence of myelofibrosis in young male patients. Ir. J. Med. Sci. 192 (2), 591–593. doi:10.1007/s11845-022-03047-1
Wu, S., Luo, P., Yu, Y., Xiong, B., Wang, Y., and Zuo, X. (2022). Next-generation sequencing redefines the diagnosis of triple-negative myeloproliferative neoplasms. Ann. Hematol. 101 (3), 705–708. doi:10.1007/s00277-021-04561-5
Yan, X., Xu, Z., Qu, S., Qin, T., Huang, G., Xiao, Z., et al. (2022). Mutational landscape, clonal evolution and prognostic effect of non-driver mutations during disease progression of primary myelofibrosis. Blood 140 (1), 3965–3967. doi:10.1182/blood-2022-165185
Zhang, Y., Zhou, Y., Wang, Y., Teng, G., Li, D., Wang, Y., et al. (2020). Thrombosis among 1537 patients with JAK2V617F -mutated myeloproliferative neoplasms: risk factors and development of a predictive model. Cancer Med. 9 (6), 2096–2105. Epub 2020 Jan 28. doi:10.1002/cam4.2886
Zhuge, J., Zhang, W., Zhang, W., Xu, M., and Hoffman, R. (2010). Sensitive detection of MPLW515L/K mutations by amplification refractory mutation system (ARMS)-PCR. Clin. Chim. Acta 411 (1-2), 122–123. doi:10.1016/j.cca.2009.10.012
Zulkeflee, R. H., Zulkafli, Z., Johan, M. F., Husin, A., Islam, M. A., and Hassan, R. (2021). Clinical and laboratory features of JAK2 V617F, CALR, and MPL mutations in Malaysian patients with classical myeloproliferative neoplasm (MPN). Int. J. Environ. Res. Public Health 18 (14), 7582. doi:10.3390/ijerph18147582
Keywords: hematooncology, myeloproliferative neoplasms, gene mutations, next-generation sequencing, polycythemia vera, essential thrombocythemia, myelofibrosis
Citation: Mroczkowska-Bękarciak A and Wróbel T (2023) BCR::ABL1-negative myeloproliferative neoplasms in the era of next-generation sequencing. Front. Genet. 14:1241912. doi: 10.3389/fgene.2023.1241912
Received: 17 June 2023; Accepted: 22 August 2023;
Published: 08 September 2023.
Edited by:
Gokce Toruner, University of Texas MD Anderson Cancer Center, United StatesReviewed by:
Rajko Kusec, Clinical Hospital Dubrava, CroatiaCopyright © 2023 Mroczkowska-Bękarciak and Wróbel. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Aleksandra Mroczkowska-Bękarciak, b21yb2N6a293c2thQGludGVyaWEucGw=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.