
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Genet., 04 September 2023
Sec. Cancer Genetics and Oncogenomics
Volume 14 - 2023 | https://doi.org/10.3389/fgene.2023.1231536
This article is part of the Research TopicThe Evolution of Cancer Genetics and Oncogenomics: 2023View all 5 articles
 Fidelis Charles Bugoye1,2*
Fidelis Charles Bugoye1,2* Rispah Torrorey-Sawe2*
Rispah Torrorey-Sawe2* Richard Biegon2Nazima Dharsee3Fidelice M. S. Mafumiko1
Richard Biegon2Nazima Dharsee3Fidelice M. S. Mafumiko1 Kirtika Patel2
Kirtika Patel2 Simeon K. Mining2
Simeon K. Mining2Over the past few years, a number of studies have revealed that a significant number of men with prostate cancer had genetic defects in the DNA damage repair gene response and mismatch repair genes. Certain of these modifications, notably gene alterations known as homologous recombination (HRR) genes; PALB2, CHEK2 BRCA1, BRCA2, ATM, and genes for DNA mismatch repair (MMR); MLH1, MSH2, MSH6, and PMS2 are connected to a higher risk of prostate cancer and more severe types of the disease. The DNA damage repair (DDR) is essential for constructing and diversifying the antigen receptor genes required for T and B cell development. But this DDR imbalance results in stress on DNA replication and transcription, accumulation of mutations, and even cell death, which compromises tissue homeostasis. Due to these impacts of DDR anomalies, tumor immunity may be impacted, which may encourage the growth of tumors, the release of inflammatory cytokines, and aberrant immune reactions. In a similar vein, people who have altered MMR gene may benefit greatly from immunotherapy. Therefore, for these treatments, mutational genetic testing is indicated. Mismatch repair gene (MMR) defects are also more prevalent than previously thought, especially in patients with metastatic disease, high Gleason scores, and diverse histologies. This review summarizes the current information on the mutation spectrum and clinical significance of DDR mechanisms, such as HRR and MMR abnormalities in prostate cancer, and explains how patient management is evolving as a result of this understanding.
Prostate cancer (PCa) is the most common cancer among men worldwide and a significant public health burden. The disease is diagnosed more frequently than any other type of cancer (Sanjose et al., 2011; Lozano et al., 2012; Sung et al., 2021). Previous studies (Siegel et al., 2011; El Noor and El Noor, 2015; Bugoye et al., 2019), have shown that PCa is the most frequent solid tumor in males and one of the main causes of cancer-related deaths in men worldwide. The incidence and mortality of PCa are alarming, 2,293,818 new cases are anticipated to be diagnosed between now and 2040, and a 1.05% rise in mortality is expected (Rawla, 2019). Incidence and death of PCa are both associated with age at the time of diagnosis. African-American men have higher incidence rates than white men. Previous researchers reported 158.3 new cases per 100,000 African-American men and twice the mortality rate compared to reported data in white men, and the disparity is caused by a variety of social, environmental, and genetic factors (Panigrahi et al., 2019). PCa progression is typically gradual and symptom-free, and treatment may not even be necessary, but most frequent complaints are difficulty urinating, increased urination frequency, and nocturia, though prostatic enlargement may induce similar symptoms. The advanced stages of the disease may appear with back discomfort and urinary incontinence because the axial skeleton is the most usual site of bone metastatic sickness.
In 2018, 1,276,106 new cases of PCa were reported worldwide, making up about 7.1% of all male malignancies (Bray et al., 2018; Rawla, 2019). Prostate cancer incidence rates vary by geographic area, and these variations have been connected to a range of modifiable and non-modifiable factors (Rawla, 2019; Sung et al., 2021). Smoking, food, obesity, and exposure to chemicals are all aspects of lifestyle that can be modified to lower the chance of developing prostate cancer. These risk factors can be changed or managed to reduce a person’s chance of developing the illness. Age, race/ethnicity, family history, and some genetic alterations such as mutations in the Homologous recombination-related DNA Damage Repair genes PALB2, CHEK2, BRCA1, BRCA2, ATM, and MLH1, MSH2, MSH6, PMS2 Mismatch Repair (MMR) genes are risk factors that cannot be modified to reduce risk of developing disease (Lang et al., 2019; Sedhom and Antonarakis, 2019; van Wilpe et al., 2021). In this review, the Mutation spectrum of HRR genes, and DNA MMR genes involved in DNA damage repair mechanisms and their therapeutic applications are discussed.
The peer-reviewed articles in this review spanned the time between January 2000 and June 2023 and were taken from PubMed and Google Scholar. The search terms “prostate cancer OR genetic alteration OR mutation variant OR gene OR DNA Damage repair OR individual OR Mismatch repair OR Homologous recombination OR immunotherapy OR prevalence OR frequency OR Genetic OR Spectrum OR Landscape” were used. 1,222 publications from Google Scholar and 12,773,760 articles from Pubmed made up 12,774,982 total results for our search phrase. Since broad key terms were employed, the search string’s filtering mechanism also supplied irrelevant results. The following inclusion criteria were used to manually screen retrieved articles: 1) Articles describing a phenotypic or genetic condition associated with prostate cancer 2) Articles that detail genetic variations 3) Articles describing gene changes related to DNA damage repair 4) Articles outlining genes involved in homologous recombination repair (HRR) 5) Articles discussing DNA damage repair modifications and the clinical or therapeutic use of the Mismatch Repair (MMR) gene. To retrieve and validate the reported Mutations, Either ClinVar, European Nucleotide Archive (ENA), DNA Databank of Japan (DDBJ), or Gene Bank Genetic Variants databases were used. Also, with the exception of two articles published in 1999 and 1998, all other selected articles published in 2000 and above were considered. However, articles with irrelevant information for this review and those with redundant information and duplicate information were not selected. Thus, in this review, 174 articles were used.
Hereditary factors are known to play a role in the higher mortality and incidence rates of PCa in African American males compared to European American men (DeSantis et al., 2016). Greater genomic mutation, which results in a more aggressive phenotype, is one of the characteristics of the ethnic disparities in PCa biology. Knowing how to target genetic anomalies like DDR gene mutations offers the way to efficient treatments that can improve clinical outcomes (Kohaar et al., 2022; White et al., 2022). Compared to white males, men with African heritage and men from South America continue to have higher incidence and mortality rates of PCa (Belkahla et al., 2022). Men from the Middle East, North Africa, and Asia often have the lowest incidence of PCa in comparison to other ethnic groups, such as non-Hispanic White males, who have a chance of PCa diagnosis of 1 in 8 men over their lifetime, data from the National Cancer Institute suggest that men of African descent often have the greatest rate (1 in 6 men) (Akaza et al., 2011; Powell and Bollig-Fischer, 2013). A number of factors, including the downregulation of DNA repair genes in African men, contribute to the increased incidence and mortality rate of prostate cancer in African men. When compared with Non-Hispanic white, African American men were found with upregulation genes governing inflammatory pathways including CCL4, IFNG, CD3, IL33, and ICOSLG despite the downregulation of DNA damage response mechanisms (DeSantis et al., 2016; Nair et al., 2022). However, it should be highlighted that race is a social rather than a biological construct; in PCa, there have been conflicting results and challenges to demonstrate the prevalence and differences in DNA repair gene mutation between races, and results ought to be handled with caution (Reizine et al., 2023).
The biological heterogeneity of PCa is the root of many of the difficulties in treating the condition. Due to the clinical tumor presentation, genetic or phenotypic variability, and therapy outcomes, PCa has been described as a heterogeneous illness (Tolkach and Kristiansen, 2018; Haffner et al., 2021). Because of these heterogeneities, there is no one-size-fits-all approach to treating PCa. This has led to controversies over how to treat patients (Shore et al., 2022). For instance, the issue of whether routine PCa screening is more advantageous than the risk of over-detection and over-treatment is still up for debate and necessitates individualized treatment approaches (Force, 2018; Shore et al., 2022). Despite the promise of precision therapy in the treatment of PCa has increased due to advances in genetic testing, the use of genetic data by practitioners in developing countries is hampered by issues with insufficient capacity, the number of patients requiring genetic testing, and accessibility (Szymaniak et al., 2020). Similar to this, worries about overtreatment have changed recommendations for active monitoring, and physicians’ approaches to managing PCa vary widely (Shore et al., 2022). According to research, more than 50% of respondents disagree with management concerns relating to PCa which is advancing (Saad et al., 2019). These findings, along with others of a similar nature, have highlighted the necessity of improved knowledge of the genetic mutation spectrum in prostate cancer (Finch et al., 2022) and other concerted efforts to improve the illness management (Gillessen et al., 2015; Gillessen et al., 2018; Saad et al., 2019; Gillessen et al., 2020). Genetic Mutation in Prostate Cancer.
Genetic and epigenetic changes normally occur at different levels, and these genetic modifications have provided potential applications as biomarkers in cancers. The genetic origins of prostate cancer have been the subject of research for decades. This is because genes involved in the DNA damage repair pathways can increase the risk of developing PCa (Robinson et al., 2015a). In Cells, there are sophisticated networks of non-redundant mechanisms, including base alterations, strand breaks, and interstrand crosslinks, these mechanisms detect and repair DNA damage. Direct repair, base excision repair, nucleotide excision repair, mismatch repair (MMR), homologous recombination repair (HRR), non-homologous end joining (NHEJ), and the Fanconi anemia repair pathways are important DNA damage repair mechanisms. According to research, this PCa is substantially more likely to be developed in carriers of HRR and MMR gene mutations (Robinson et al., 2015a).
A post-replication technique called DNA mismatch repair (MMR) is used to correct base mismatches and replication-related insertion or deletion mistakes. Genetic instability is brought on by a loss of MMR gene function resulting in a buildup of errors that typically occur during DNA replication (Chen et al., 2001). The risk of PCa in men with MMR gene mutations has been demonstrated to be significantly enhanced (Rantapero et al., 2020). Individuals that have MMR mutations are typically predicted to account for 2%–5% of PCa cases (Ritch et al., 2020; Ye et al., 2020), and they are generally identified by their Gleason score 8 and de novo metastases (Dominguez-Valentin et al., 2016; Ye et al., 2020; Jiang et al., 2021; Graham and Schweizer, 2022). The genetic mutations in MMR genes in the prostate may aid in understanding PCa carcinogenesis, which may have additional repercussions for ICBs and other types of treatment. This review effort focuses on the MSH2, MSH6, MLH1, and PMS2 genes (Table 1), which are more frequently found in PCa and have recently been described by Pecina et al., though previous authors have reported on other MMR genes which are also connected to high microsatellite instability and high tumor mutational load involved in PCa progression (Chen et al., 2011; Sedhom and Antonarakis, 2019; Pećina-Šlaus et al., 2020).
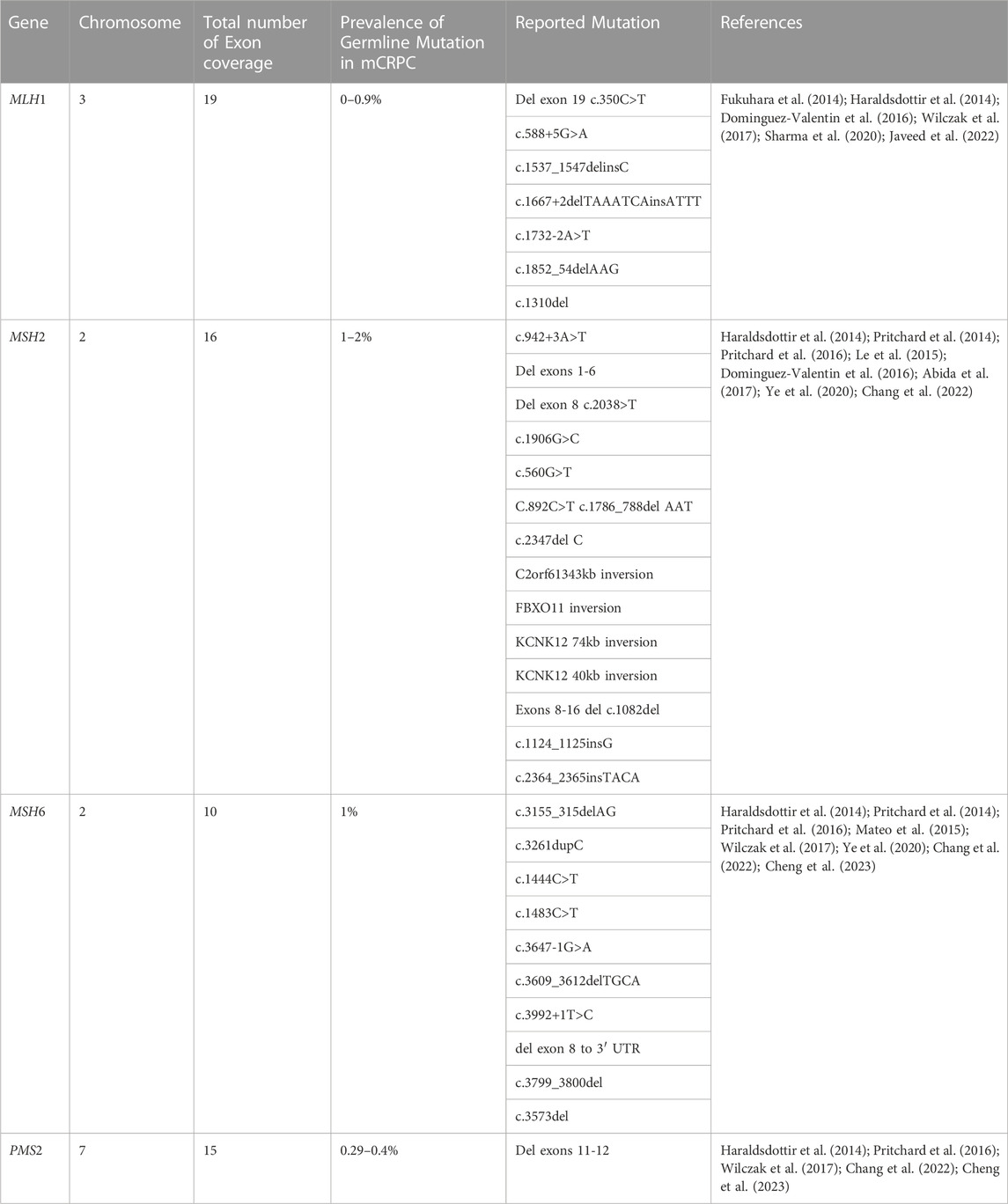
TABLE 1. Summary of mismatch repair genes related to PCa.
The MutL homolog 1 (MLH1) protein, a member of the MutL family of DNA mismatch repair proteins. MLH1 consists of 56kilobases is located in chromosome 3 and consists of 11 exons for coding MLH1 Protein which recognizes incorrect nucleotides, causing them to be expelled and replaced in a strand-directed manner (Fukuhara et al., 2014; Zhen et al., 2018). Several PCa cohorts have been examined for the breadth of MLH1 gene mutations using sequencing data (Pande et al., 2012; Haraldsdottir et al., 2014; Dominguez-Valentin et al., 2016). MLH1 mutations were found in 0.7% of the 150 mCRPC patient (Robinson et al., 2015b). Additionally, PCa with the MLH1 gene mutation has been connected to an elevated acute form of the illness, a higher Gleason score, a lack of differentiation, and a higher rate of distant metastasis (Shenderov et al., 2019; Antonarakis et al., 2020). Frameshift, deletion, and missense mutations are additionally the most prevalent MLH1 mutation types in PCa (McVety et al., 2005). The MLH1 mutation previously identified in PCa patients comprises the following variants associated with a higher risk of PCa; c.350C>T, c.588+5G>A, c.1537 1547delInsC, c.1667+2delTAAATCAinsATTT, c.1667+2delTAAATCAinsATTT, and c.1732-2A>T (Raymond et al., 2013; Dominguez-Valentin et al., 2016).
The MSH2 gene is responsible for the production of proteins involved in the MMR mechanism. MSH2 gene is located in the short arm of chromosome 2 (2p21) and encodes proteins that aid in DNA mismatch repair (Salo-Mullen et al., 2018; Zhen et al., 2018). In collaboration with the MutL homolog (MLH1) protein, MSH2 detects and corrects mismatched DNA base pairs that occur during DNA replication (Chakraborty and Alani, 2016; Furman et al., 2021). The MSH2 gene is commonly changed in PCa by deletion, missense, and frameshift mutations that also cause the buildup of neoantigens, raise the burden of tumor mutations, and increase the density of lymphocytes that infiltrate tumors (Haraldsdottir et al., 2014; Dominguez-Valentin et al., 2016).
The MSH6 gene is found on the short arm of chromosome 2 (2p16.3) (Salo-Mullen et al., 2018; Zhen et al., 2018). The MSH6 gene participates in MMR pathways and mutation of this gene has been associated with an increase in the likelihood of developing PCa (Pritchard et al., 2014) According to recent research, structural rearrangements such as insertions and deletions are what make MSH6 more susceptible to mutations (Dominguez-Valentin et al., 2016).
The PMS2 gene with approximately 38,000 base pairs is located on chromosome 7. The gene consists of 15 exons that code for the 862 amino acids that make up the PMS2 protein (Fukuhara et al., 2015). The produced protein is essential to the mismatch repair mechanism, which corrects small insertions and deletions as well as DNA mismatches that may occur during homologous recombination and DNA replication. As previously reported, Genomic integrity is protected by DNA mismatch repair, which corrects mismatches brought on by DNA replication and recombination (Wu et al., 2003). A multitude of essential phases in mismatch repair are orchestrated by the human MutL-alpha heterodimer. As has been previously noted, the nuclear import of MutL-alpha may be the initial regulatory step in initiating the mismatch repair mechanism (Leong et al., 2009). When this protein forms heterodimers with the MutL homolog 1 (MLH1) gene product, the resulting structure is known as the MutL-alpha heterodimer. The MutL-alpha heterodimer’s endonucleolytic activity is essential for the removal of the mismatched DNA, and the MutS-alpha and MutS-beta heterodimers identify mismatches and insertion/deletion loops (Reyes et al., 2015). Lynch Syndrome (LS), a multi-organ cancer syndrome, is caused by genetic abnormalities in the four MMR genes (MLH1, MSH2, MSH6, and PMS2). The most recent MMR gene to be connected to Lynch Syndrome is PMS2 (Hendriks et al., 2006). It has been hypothesized that PMS2 mutations, in contrast to MLH1 and MSH2 mutations, may be linked to a later stage of cancer development (Hendriks et al., 2006). In addition to the several MMR genes, the PMS2 gene like many other genes associated with PCa, has also been identified as having polymorphisms and mutations (Fukuhara et al., 2015). Earlier studies have shown that males with PMS2 mutations are more likely to develop PCa (Haraldsdottir et al., 2014).
Genomic instability and eventually cancer are brought on by the accumulation of mutations brought on by faults in the DNA damage response pathways (Bartek et al., 2001; Bartek and Lukas, 2003; Antoni et al., 2007), and the path that is frequently blocked is the homologous recombination pathway (van Wilpe et al., 2021). Since some of the proteins in this system are frequently altered in human cancers and a number of heritable conditions that are predisposed to developing cancer, disruption of this route has been demonstrated to play a significant part in the genesis of cancer (Khanna and Jackson, 2001). In the face of various DNA-damaging events, the homologous recombination mechanism is essential for maintaining genomic integrity, Men who carry inherited mutations in essential homologous recombination pathways have a significantly elevated lifetime risk of developing PCa compared to noncarriers (van Wilpe et al., 2021). The ability to test for carriers of those mutations using DNA next-generation sequencing technology has enabled the creation of prospective risk-reduction strategies and the justification for the therapeutic strategy for these individuals. There are currently promising new prospects for treating PCa in people who have mutations in homologous recombination repair genes. The use of immune-based therapies, chemotherapy with platinum, and PARP inhibitors are all viable treatment alternatives (Antonarakis et al., 2019). Castrate-resistant PCa (CRPC) patients have not responded well to immuno checkpoint blocking (ICB) or inhibitor therapy. Only 3%–5% of CRPC patients benefit from anti-PD-1 therapy (Antonarakis et al., 2019). Additionally, research has shown that using ICBs in conjunction with other treatments can improve response rates. For instance, the response rate of a combination of ipilimumab and nivolumab is predicted to be between 10%–26% (Boudadi et al., 2018). When homologous recombination repair genes BRCA1/2, ATM, PALB2, and CHEK2 are inactive, a variety of error-prone and non-conservative DNA repair pathways are used, increasing the incidence of cancer and worsening its prognosis while also causing genomic instability (Castro and Eeles, 2012; Amsi et al., 2020). The details of homologous recombination genes involved in DNA damage repair are summarized in Table 2 below.
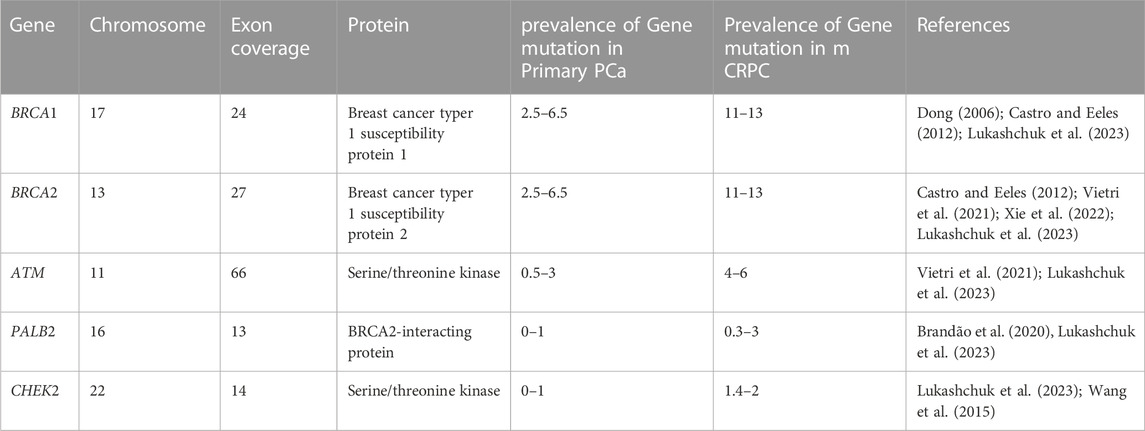
TABLE 2. Summary details of homologous recombination genes related to PCa.
Epidemiological research has connected Breast cancer gene 1(BRCA1) and Breast Cancer gene2 (BRCA2) mutations to the risk of PCa; nevertheless, 5%–15% of cases of prostate cancer are caused by high-risk hereditary variables (Ferrís-i-Tortajada et al., 2011). Pathogenic mutations, which account for less than 1% of BRCA1, and about 2% in BRCA2 of incident PCa cases, are likely to be the cause of the disease (Kote-Jarai et al., 2011; Leongamornlert et al., 2012) Click or tap here to enter text in mutation carriers. Male BRCA2 carriers have a higher lifetime chance of acquiring PCa than BRCA1 carriers (Roy et al., 2012). The DNA damage repair (DDR) process uses non-conservative and potentially mutagenic methods when these BRCA1 and/or BRCA2 genes are absent. There is evidence that this genomic instability contributes to the cancer risk associated with harmful BRCA gene mutations (Salmi et al., 2021). BRCA1 and BRCA2 pathogenic mutation carriers have a relative risk of PCa that is enhanced by 1.8–3.8 and 2.5 to 8.6 times, respectively, by the time they are 65 years old (Thompson et al., 2003; Agalliu et al., 2007; Leongamornlert et al., 2012). BRCA1 and BRCA2 gene mutation testing, as well as their correlation with higher Gleason scores (8>), and therapeutic use in PCa management, have drawn the attention of experts to familial PCa in particular (Amsi et al., 2020). Numerous authors have discussed the mutation spectrum in relation to PCa risk, but Ashkenazi Jewish and Icelandic populations have been found to have frameshift deletion 185delAG and a frameshift insertion 5382insC for BRCA1 and BRCA2 frameshift deletion, as well as 999del5 founder mutations linked to poor survival in young people (Wilkens et al., 1999; Tryggvadóttir et al., 2007). In addition, PCa patients in the United Kingdom with significantly younger ages (<56 years) were found to have two base pair deletions 5531delTT and four base pair deletions 6710del ACAA frameshift mutation in the BRCA2 gene (Gayther et al., 2000). Additionally, different studies reported various types and different mutation frequencies (Wilkens et al., 1999; Vazina et al., 2000; Ikonen et al., 2003; Gallagher et al., 2010; Castro et al., 2013; Maia et al., 2016; Shenoy et al., 2016). Most of the reported mutations are frame-shift mutations. The differences in the prevalence of BRCA1/2 mutations amongst populations may be due to differences in study sample size, inclusion criteria, patient ethnic origins, and other factors. In this review, some of the previously reported BRCA1 and BRCA2 pathogenic mutations associated with PCa in different populations are summarized in Table 3.
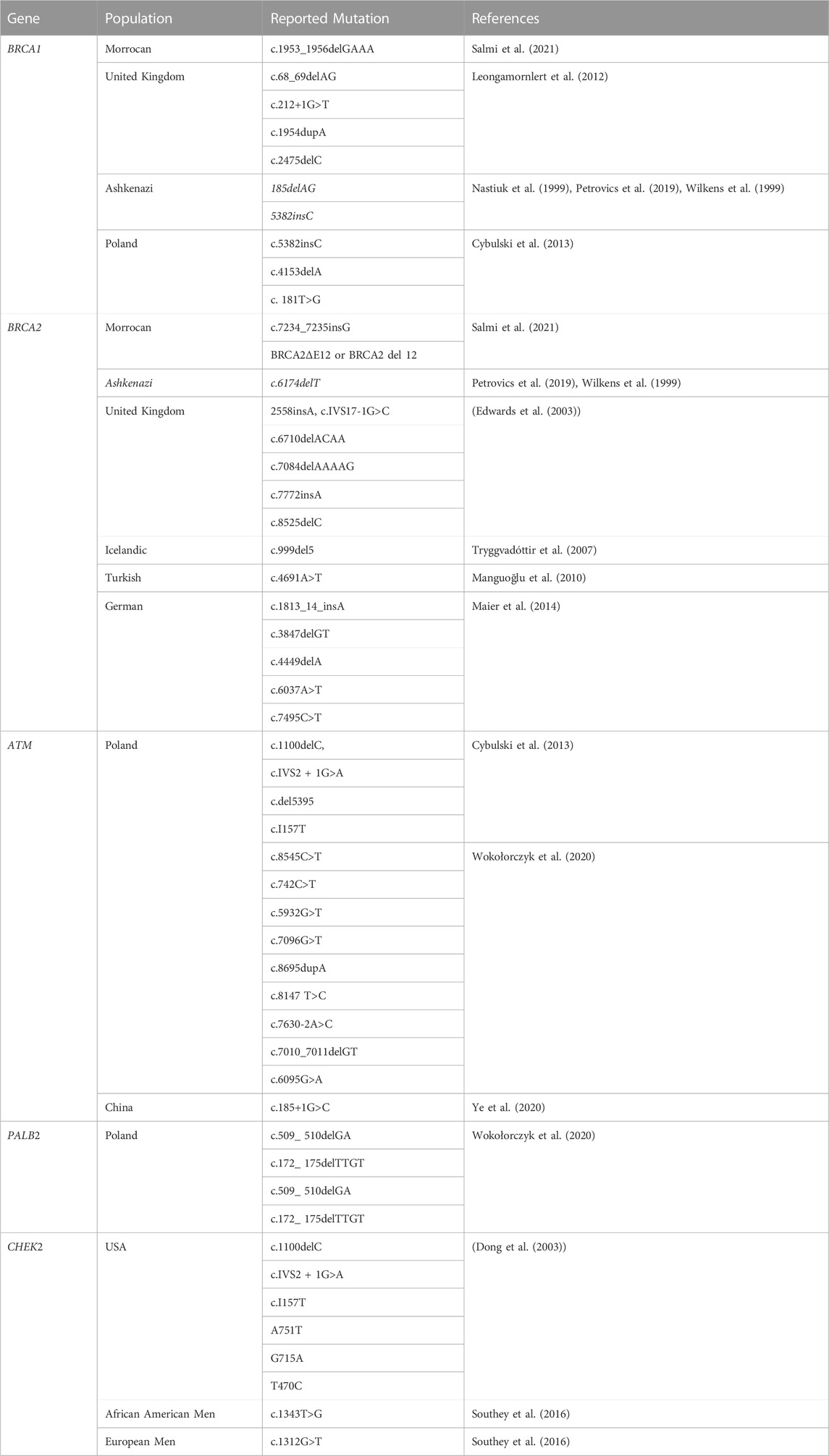
TABLE 3. Mutation spectrum of homologous recombination genes related to PCa.
The Ataxia Telangiectasia Mutated (ATM) gene is in charge of producing the serine/threonine kinase that is connected to PI3K and is thought to be involved in maintaining genomic integrity. On chromosomes 11q22–23, ATM is located and has 66 exons and a coding sequence of 9,168 base pairs (Choi et al., 2016). In the double-strand break (DSB) repair process, ATM functions as a signal transducer and is essential for the detection of DNA damage and the cellular response to it (Virtanen et al., 2019). The rearrangement of antibody genes during B-cell maturation or meiotic recombination, as well as ionizing radiation, chemotherapeutic drugs, or oxidative stress, can all result in DSB, according to other findings (Lieber, 2011; Bednarski and Sleckman, 2012; Choi et al., 2016). RAG DSBs trigger traditional DNA damage responses by activating ATM and DNA-PKcs, both of which are serine-threonine kinases that are a member of the phosphatidylinositol-3-kinase (PI3K)-like family (Callén et al., 2009). This is similar to other DSBs created in the G1 phase of the cell cycle (Bednarski and Sleckman, 2012). Both ATM and DNA-PKcs function as transducers in the DNA damage response, phosphorylating a range of downstream effectors (Callén et al., 2009).
Due to the different and redundant functions that ATM and DNA-PKcs play in the response to RAG-mediated DSBs, immune system deficiencies and errors in DNA end repair occur in mice and individuals who lack either of these kinases (Bednarski and Sleckman, 2012). According to the information that is currently available, castration-resistant PCa have an enrichment of approximately two times the frequency of localized PCa in terms of tumoral germline or somatic ATM mutations, which vary from 5% to 8% of the tumors, also Men with pathogenic ATM mutations have an increased risk of developing PCa, which may potentially cause the condition to appear earlier (Giri and Beebe-Dimmer, 2016; Thalgott et al., 2018; Wokołorczyk et al., 2020). There is limited information about populations in Africa, despite some populations being known to carry these mutations and being discussed in this review. In the research conducted on 390 Polish people, eight genes were mutated and 76 males (19.5%) had a mutation in one of the BRCA1, BRCA2, ATM, CHEK2, MSH2, or MSH6 genes. A total of 11 mutations (2.8%) associated with ATM were among the reported gene variations. A few one-stop gains in the ATM gene have been documented, including c.8545C>T, c.742C>T, c.5932G>T, and c.7096G>T. A frameshift mutation, c.7010_7011del, two splice acceptor variations, c.7630-2A>C, and two missense mutations, c.6095G>A and c.8147 T>C, are all found in the ATM gene, but all of those alterations were not reported in the control groups (Wokołorczyk et al., 2020).
The BRCA1-PALB2-BRCA2 protein, which is encoded by the partner and localizer of BRCA2 (PALB2), is found on chromosome 16 and forms the PALB2 complex. The molecular scaffold, PALB2, interacts with BRCA2. It is essential for homologous recombination and DNA double-strand break (DSB) repair (Sy et al., 2009). Fanconi anemia is also brought on by germline homozygous loss of function (LoF) mutations of PALB2, like BRCA2 (Reid et al., 2007; Xia et al., 2007), although heterozygous LoF mutations have been associated with hereditary breast cancer and pancreatic cancer (Jones et al., 2009; Antoniou et al., 2014). Despite new correlations between pathogenic PALB2 mutations and an elevated risk of different cancer, research on the PALB2 gene’s role in PCa has produced conflicting results. The authors of this study are aware of new results linking PALB2 to a statistically significant increase in the chance of developing PCa (Yang et al., 2020). While it has long been known that these polymorphisms increase the risk of breast cancer, data on PALB2’s role in PCa have also been decisively proven (Horak et al., 2019; Wokołorczyk et al., 2020; Bouras et al., 2022).
The authors also acknowledge that there has not been much research on PALB2 pathogenic mutations in PCa patients, but research conducted in Poland in 2021 by Wokolorczyk and others found that aggressive cancers with high Gleason scores of 8–10 were more frequently diagnosed with the two founder mutations of PALB2 c.509_510delGA and c.172_ 175delTTGT, which together account for 80% of all PALB2 mutations. Furthermore, the 5′ ends of the PALB2 gene were the location of these founder mutations, as described by Wokolorczyk (Wokołorczyk et al., 2021). Additionally, these c.509_510delGA and c.172_ 175delTTGT variants result in a translational frameshift with a predicted alternate stop codon and are predicted to cause PALB2 function to be lost through premature protein truncation or nonsense-mediated messenger RNA decay. These research findings are strengthening the case that the two Polish founder mutations of PALB2 are also pathogenic for breast cancer (Wokołorczyk et al., 2021).
Cell cycle checkpoint kinase 2 (CHEK2) is a protein that participates in DNA damage response in many distinct cell types. Ataxia Telangiectasia Mutated (ATM) protein activates CHEK2 to prevent the buildup of mutations and reduce the risk of cancer when DNA is damaged (Bartek et al., 2001; Bartek and Lukas, 2003; Antoni et al., 2007). By activating CHEK2 in response to DNA damage, the checkpoint control pathways’ downstream targets, such as p53, Cdc25A, Cdc25C, BRCA1, E2F1, Pml1, Plk3, and other substrates, are signaled. These substrates altered biological activities resulting in cell cycle blockage, enhanced DNA repair, or apoptosis (Bartek et al., 2001; Bartek and Lukas, 2003; Antoni et al., 2007). Cell cycle regulator CHEK2 controls the homologous recombinant DNA repair process, suppresses tumors, and genetic changes in it render cancers more susceptible to more advanced targeted therapies. CHEK2 is phosphorylated and activated in an ATM-dependent way in response to certain DNA-damaging substances (Matsuoka et al., 1998).
In the cell-cycle checkpoint control, active CHEK2 and other DNA damage-triggered protein kinases stabilize TP53 or speed up Cdc25A degradation through the coordination of DNA repair, cell-cycle progression, and death (Matsuoka et al., 1998; Hirao et al., 2000; Falck et al., 2001; Zannini et al., 2014) The tumor-suppressing function of the cell cycle regulator CHEK2 is mediated via homologous recombinant DNA repair, and genetic changes in it render malignancies susceptible to more advanced targeted therapies. CHEK2 is phosphorylated and activated in an ATM-dependent way in response to certain DNA-damaging agents (Matsuoka et al., 1998). Activated CHEK2 and other DNA damage protein kinases stabilize TP53 or hasten Cdc25A degradation in the cell-cycle checkpoint regulation via coordinating DNA repair, cell-cycle progression, and apoptosis (Matsuoka et al., 1998; Hirao et al., 2000; Bartek et al., 2001; Falck et al., 2001; Zannini et al., 2014) The frameshift 1100delC mutation in the CHEK2 gene, which causes the translation of a truncated CHEK2 protein product lacking kinase function, is the gene variation that has received the most attention. A substitution G>A in the exon 2 splice site has also been known as the IVS2 + 1G>A (or 444 + 1G>A) mutation. Because of this substitution, an aberrant splicing event occurs which also causes the insertion of a 4-base pair fragment, and finally, a frameshift that terminates at codon 154 in exon 3 (Dong et al. (2003)). Men with the CHEK2 1100delC, IVS2 + 1G>A, and I157T missense mutations have a higher chance of developing PCa, while there is no evidence that these CHEK2 1100delC, variants cause the disease in all cases of familial PCa. A previous study using 178 patients identified 13 CHECK2 mutations, of which 9 (10.7%) were found in the prostate cancer of 84 unselected patients and 4 (4.3%) were found in the early-onset cancer of 94 patients (Dong et al. (2003)). Men from both Europe and Africa have shown similar evidence of a CHEK2 risk mutation for PCa. African men had a CHEK2 c.1343T>G OR of 3.03 (95% CI 1.53 to 6.03, p = 0.0006), whereas European men had a CHEK2 c.1312G>T OR of 2.21 (95% CI 1.06 to 4.63, p = 0.030) (Southey et al., 2016).
The tumor is more likely to be recognized by the immune system when it contains more neoantigens. The accumulation of these neo-antigens is triggered by mutations in the DNA repair process. The prevalence of mutations, which might increase the number of neoantigens, would increase if specific DNA repair mechanisms, including MMR and HRR are altered (Jiricny, 2013; De Mattos-Arruda et al., 2020; Golan et al., 2021; Ma et al., 2022; Amodio et al., 2023). Neoantigen-reactive T cells have been demonstrated in studies to be one of the critical components of immunotherapy efficacy, particularly in tumors with a high tumor mutational burden (TMB) (Ye et al., 2020). When ICBs like CTLA4 and PD1 are used to treat some forms of cancer, tumors with high TMB have superior clinical results (Lu and Robbins, 2016; Ye et al., 2020; Graf et al., 2022; Shi et al., 2022) Cancer-associated antigens, including neoantigens derived from genetic mutations, are presented to CD8+ T cells through the major histocompatibility complex (MHC) on dendritic cells (DCs), and professional antigen-presenting cells (APCs). However, the majority of neoantigens are usually not recognized by the immune system, thus identification of highly tumor-specific antigens is important for the development of personalized immunotherapy (Kiyotani et al., 2018; De Mattos-Arruda et al., 2020) In patients with advanced and other types of cancers, greater neoantigen load is a predictive factor for a better result when utilizing ICBs (Hopfner and Hornung, 2020). Further studies are needed to fill this knowledge gap because there is a lack of enough evidence in the literature to relate specific DDR gene mutations to TMB, resistance to DNA-damaging treatments, or radiation therapy.
In order to detect PCa and increase survival rates without over diagnosing and overtreating patients, screening techniques have been developed (Force, 2018). Oncologists can use risk stratification in therapeutic decision-making, and the stratification groups are routinely used to establish the inclusion or exclusion criteria for patients to take part in studies looking at drugs that are specifically targeted for a given risk group (Rodrigues et al., 2012). There are a number of prostate risk stratification methods in place, including the Gleason score, PSA levels, clinical staging, and histological staging, among others; however, none of these are enough to predict outcomes (S.-Y. Wang et al., 2014). As a result, this drove the need for additional research until recently, when 333 primary prostate tumors were examined for the first time by the Cancer Genome Atlas Research Network, which discovered that 19% of them contained DNA repair gene abnormalities (“Cancer Genome Atlas Research Network,” 2015). New classification schemes based on molecular traits have been described, which will improve PCa precision medicine. Recent studies have demonstrated that the DDR gene mutation raises the risk of PCa (Castro et al., 2019; Chung et al., 2020).
Pathogenic mutation in the DDR genes BRCA1/2 and ATM status differs between risk for aggressive and indolent PCa and is linked to a younger death age and a shorter survival period (Na et al., 2017). According to research, while some people have aggressive cancers that metastasize and result in disease-related death, many with indolent tumors can be successfully treated with early therapy or monitored (Sakellakis et al., 2022). Furthermore, studies conducted in various contexts with various populations have demonstrated that homologous recombination repair (HRR) and mismatch repair genes, which are mutated in a high proportion of patients, are involved in aggressive PCa biology (Lukashchuk et al., 2023). When managing familial cancer risk, knowledge of the therapeutic sensitivity to novel targeted medications imparted by these mutations in DNA damage response could be life-saving (Herberts et al., 2023; Lukashchuk et al., 2023; Sorrentino and Di Carlo, 2023).
Patients with homologous recombination repair (HRR) and mismatch repair (MMR) gene mutations are more likely to have poor clinical outcomes, intraductal and cribriform structure, and higher Gleason scores than patients without these mutations (Risbridger et al., 2015; Schweizer et al., 2019). According to studies, localized PCa with mutations in the HRR and MMR genes displays a biological profile that is very aggressive and resembles a treatment-resistant metastatic disease. The dysregulation of MED12L/MED12 in localized PCa with HRR and MMR gene alterations is an illustration of this. A combination of genomic dysregulation of the Wnt-pathway mediator complexes and enhanced genomic instability can account for this. This imbalance is a typical castration-resistant PCa molecular feature (Taylor et al., 2017). Genomic instability, which is a characteristic of cancer cells that is not present in normal cells due to DDR defects, is a well-known characteristic of these cells. Men with genomic instability seem to have a worse prognosis and are more likely to be diagnosed with PCa, especially in somatic cells with shorter telomere lengths (Heaphy et al., 2013). According to the most recent study that thoroughly investigated the function of DDR mutation in PCa survival (Zhang et al., 2022), the majority of DDR pathway mutations have been reported to be linked to a poor prognosis (Heaphy et al., 2013). Castro and others highlighted the function of the DDR gene mutation as a predictive biomarker after finding a substantial relationship between germline BRCA1/2 gene mutation and PCa aggressiveness. In hereditary BRCA1/2 mutation carriers, Castro found that the risk of stage T3/T4 cancer, nodal involvement, a Gleason score of 8 or higher, and metastases at the time of initial diagnosis was increased and that the survival time was also decreased (Castro et al., 2013). In another study, it was discovered that patients with lethal PCa had a significantly higher combined rate of germline BRCA/ATM alterations than those with localized PCa, and those with local disease or a diagnosis of metastases had a shorter prostate-cancer-specific survival time than non-carriers (Na et al., 2017). These findings were recently supported by research conducted by Lukashchuk, Zhang, Silvestri, and others (Silvestri et al., 2020; Zhang et al., 2022; Lukashchuk et al., 2023) thus, in addition to other reported findings elsewhere, the current data suggest that DDR gene mutation would be a helpful marker for disease screening. Additionally, among reported DDR genes, the CHEK2 mutation provides reliable evidence of PCa risk in African men (Southey et al., 2016).
The antigen recognition and immune cell activation in the adaptive immune system depends on the interaction between molecules of MHC on the surface of cells that present antigen (APCs) and costimulatory molecules found on the surface of naive T cells (Jiang et al., 2021). According to previous research, antigen recognition by MHC molecules of Intratumoral T cell lymphocytes depends on the activation and amount of T cells (Alexandrov et al., 2013; Jiang et al., 2021). As reported by Alexandrov and others (Alexandrov et al., 2013), the number of mutations that cause alterations in the sequence of amino acids of the protein in a tumor has a positive correlation with the quantity of tumor neoantigen. Prostate and various forms of cancer develop at various rates in terms of intra-tumoral T-cell activation and the number of cells involved (Alexandrov et al., 2013). Additionally, when the non-synonymous mutation occurs in DDR genes, the newly produced peptide is identified as foreign by immune cells and elicits an adaptive immune response (Riaz et al., 2016). These non-synonymous mutation rates could rise as a result of DDR deficits and they have also been linked to the synthesis of peptides that affect the proinflammatory responses, particularly in MMR genes (Jiricny, 2013).
Immune Checkpoint Blockades (ICB)are therapeutic drugs that specifically target negative inhibition receptors on host T lymphocytes, such as cytotoxic T lymphocyte antigen 4 (CTLA-4) and programmed death 1 (PD1), which are frequently taken over tumors to hinder an efficient antitumor immune response. ICB acts on the immune system to strengthen antitumoral immunity, in contrast to conventional cancer therapies, which directly target cancer cells (Lei et al., 2021). The therapeutic efficacy of ICB using antibodies against inhibitory compounds produced on tumor and immune cells has been shown for a wide range of tumor types. Recent studies have demonstrated the efficacy and approval of anti-CTLA-4, anti-PD1, anti-PDL1, and their combinations with other medications. Compared to chemotherapy alone, the administration of these ICB has considerably reduced the incidence of cancer (Wei et al., 2018; Akinleye and Rasool, 2019). The findings indicate that not all PCa patients who are not actively selected benefit from ICB because of the diversity of the disease and therapy response. Unfortunately, the exact reason why different people respond to ICB therapy in different ways is still unknown. Additionally, while employing ICB, increased cancer treatment costs and unnecessary immune-related side effects should be considered (Lei et al., 2021). However, significant research is needed to address inherent and acquired problems that limit the number of individuals who benefit from ICB. Similarly, for efficient utilization of ICBs, it will be necessary to identify potential candidates for therapies. This helps to determine whether a patient will be a long-term responder, a short-term responder, or a non-responder (Lei et al., 2021). Through the use of these biomarkers, we would be able to treat responders to the fullest extent possible without unnecessarily harming non-responders (Lei et al., 2021).
Checkpoint inhibitors have also been demonstrated to be beneficial for patients with advanced PCa (Teply and Antonarakis, 2017; Boudadi et al., 2018; Hansen et al., 2018; Antonarakis et al., 2020). In men with BRCA1/2 mutations, subset analysis of larger trials revealed a greater effect for single-agent PD-1 inhibitors. For instance, in a clinical trial on pembrolizumab monotherapy for PCa, Antonarakis, and others found that men with BRCA1/2 or ATM mutations had an objective response rate of 12%, which was much greater compared to the 4% objective response rate seen in men with no those mutations (Antonarakis et al., 2020). Three of the five men (60%) who had tumors with altered HRR genes (BRCA1/2) demonstrated objective responses in previous clinical trials in 78 men with mCRPC treated with the combination of ipilimumab and nivolumab (Sharma et al., 2020). Similar results were also reported from a study carried out by Boudadi at John Hopkins (Boudadi et al., 2018).
The amount of DNA damage brought on by internal and external sources would be fatal in the absence of the DNA damage repair (DDR). In order to prevent damage from being duplicated during the S-phase or transferred to the daughter cells during mitosis, the DDR evolved as a structured network of pathways that repair DNA and stop cell division. Hanahan and Weinberg assert that DDR gene alterations can cause genomic instability and mutations that may result in cancer (Hanahan and Weinberg, 2011). Additionally, active oncogenes enhance replication stress by driving cells into the S-phase before they are ready. This leads to DNA sequence alterations and aberrant DNA structures that need to be resolved by the DDR (Saxena and Zou, 2022). According to Curtin, a mutation in one DDR gene that is associated with the DDR pathway may result in enhanced activity of compensatory pathways, leading to resistance to DNA-damaging radiation therapy and chemotherapy (Curtin, 2023). In order to treat PCa, the development of drugs that target the DDR was initially justified by the need to get around these mechanisms of resistance. However, the approach used by chemo- or radiotherapies to stop the DDR also causes more harm to healthy cells while maintaining increased antitumor effects (Curtin, 2012). Previous researchers suggested the inhibition of PARP, which interferes with DNA damage repair and results in tumor cell death, is connected to the anticancer mechanism of medicines targeting DDR. While PARP inhibition is tolerated in normal cells, it has a significant impact on tumor cells with HRR mutations (Keung et al., 2019; Lang et al., 2019; Rose et al., 2020).
Single-strand breaks (SSB) which occur as a result of PARP inhibition facilitate the formation of damage in DSB which is mainly repaired by HRR (Yi et al., 2019). The coincident loss of PARP function in cancer cells with changed HRR proteins engaged in HRR inadequate repair leads to the formation of Double strand breaks (DSBs) and consequently cell death (Yi et al., 2019). Authors have recently reported a breakthrough in PCa treatment that uses the DDR gene mutation to target the tumor only while protecting healthy cells with intact DDR pathways. This discovery has led to numerous clinical trials that take advantage of these defects, and numerous PARP inhibitors, such as olaparib, rucaparib, niraparib, talazoparib, and veliparib, have been successfully tested in patients with a variety of DDR gene mutations, including BRCA1, BRCA2, ATR, ATM, CHEK1/2, and PALB2 (Catalano et al., 2023).
In recent years, the PCa treatment PARPi—poly (ADP-ribose) polymerase inhibitors—has been approved by the FDA (Catalano et al., 2023). Patients with mutations in the DDR response genes BRCA2, ATM, and BRCA1 reported significantly greater response rates to olaparib than noncarriers in one of the earliest trials of PARPi in patients with advanced PCa (Gurley and Kemp, 2001). Similar to this, rucaparib’s phase II TRITON2 research results by Abida and others hastened its approval after demonstrating a 51% radiographic response rate for docetaxel-resistant patients with BRCA1/2 mutation in mCRPC (Abida et al., 2020). Researchers have recently demonstrated that next-generation hormonal drug combined with PARPi therapy for mCRPC provides the strongest PARPi benefit (Agarwal et al., 2022; Chi et al., 2022; Clarke et al., 2022; Herberts et al., 2023).
Despite the difficulties and variations in PCa diagnosis and treatment that have been mentioned, the most current study is showing gaps in the recognized therapy guidelines. To better diagnose and treat PCa patients, a multidisciplinary approach involving molecular biologists, oncologists, and immunologists is necessary. Research on effective and targeted treatments for prostate cancer is therefore critically needed, as well as investigations on the variety of genetic abnormalities prevalent in prostate cancer. Future studies should focus on generating genetic data that could be used to improve the health of PCa patients through immunologically based targeted therapies or combination therapeutic options, particularly in African settings where knowledge of the genetic spectrum of genes associated with the disease is limited.
FB developed the concept, planning, execution, critical review, summary, in-introduction, abstract, drafting, and editing of the manuscript. RT-S, supervision oversight and leadership of the review, conclusion, and referencing. RB, review, and editing of the manuscript. KP, funding acquisition, project administration, review, and editing of the manuscript. SKM, funding acquisition, project administration, review, and editing of the manuscript. ND; critical review, alternative approaches, and editing of the manuscript. FMMS; administration, review, and editing of the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by the Intra-Africa Academic Mobility Scholarship scheme through P4HPTII project-Moi University, Kenya, and Government Chemist Laboratory Authority (GCLA), Tanzania to facilitate the preparation of this manuscript.
We thank the Staff at the Department of Pathology and Immunology Section at Moi University, Ocean Road Cancer Institute, and Government Chemist Laboratory Authority for providing support in reviewing and for valuable contributions.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
ADT, Androgen deprivation therapy; PCa, Prostate cancer; BER, BER base excision repair; BRCA1, Breast cancer gene 1; BRCA2, Breast cancer gene 2; CRPC, Castration-resistant prostate cancer; DDR, DDR DNA damage repair; HRR, Homologous recombination repair; mCRPC, Metastatic castration-resistant prostate cancer; MMR, Mismatch repair; NCCN, National Comprehensive Cancer Network; PARP, Poly (ADP-ribose) polymerase; PARPi, Poly (ADP-ribose) polymerase inhibitor; PSA, Prostate-specific antigen; DSBs, Double strand breaks; SSBs, SSBs single-strand breaks.
Abida, W., Armenia, J., Gopalan, A., Brennan, R., Walsh, M., Barron, D., et al. (2017). Prospective genomic profiling of prostate cancer across disease States reveals germline and somatic alterations that may affect clinical decision making. JCO Precis. Oncol. 2017, 1–16. doi:10.1200/PO.17.00029
Abida, W., Patnaik, A., Campbell, D., Shapiro, J., Bryce, A. H., McDermott, R., et al. (2020). Rucaparib in men with metastatic castration-resistant prostate cancer harboring a BRCA1 or BRCA2 gene alteration. J. Clin. Oncol. 38 (32), 3763–3772. doi:10.1200/JCO.20.01035
Agalliu, I., Karlins, E., Kwon, E. M., Iwasaki, L. M., Diamond, A., Ostrander, E. A., et al. (2007). Rare germline mutations in the BRCA2 gene are associated with early-onset prostate cancer. Br. J. Cancer 97 (6), 826–831. doi:10.1038/sj.bjc.6603929
Agarwal, N., Azad, A., Shore, N. D., Carles, J., Fay, A. P., Dunshee, C., et al. (2022). Talazoparib plus enzalutamide in metastatic castration-resistant prostate cancer: TALAPRO-2 phase III study design. Future Oncol. Lond. Engl. 18 (4), 425–436. doi:10.2217/fon-2021-0811
Akaza, H., Kanetake, H., Tsukamoto, T., Miyanaga, N., Sakai, H., Masumori, N., et al. (2011). Efficacy and safety of dutasteride on prostate cancer risk reduction in asian men: the results from the REDUCE study. Jpn. J. Clin. Oncol. 41 (3), 417–423. doi:10.1093/jjco/hyq221
Akinleye, A., and Rasool, Z. (2019). Immune checkpoint inhibitors of PD-L1 as cancer therapeutics. J. Hematol. Oncol. 12 (1), 92. doi:10.1186/s13045-019-0779-5
Alexandrov, L. B., Nik-Zainal, S., Wedge, D. C., Aparicio, S. A. J. R., Behjati, S., Biankin, A. V., et al. (2013). Signatures of mutational processes in human cancer. Nature 500 (7463), 415–421. doi:10.1038/nature12477
Amodio, V., Lamba, S., Chilà, R., Cattaneo, C. M., Mussolin, B., Corti, G., et al. (2023). Genetic and pharmacological modulation of DNA mismatch repair heterogeneous tumors promotes immune surveillance. Cancer Cell 41 (1), 196–209.e5. doi:10.1016/j.ccell.2022.12.003
Amsi, P. T., Yahaya, J. J., Kalungi, S., and Odida, M. (2020). Immunohistochemical expression of BRCA1 and BRCA2 in a cohort of Ugandan men with prostate cancer: an analytical cross - sectional study. Afr. J. Urology 2020, 71. doi:10.1186/s12301-020-00079-w
Antonarakis, E. S., Piulats, J. M., Gross-Goupil, M., Goh, J., Ojamaa, K., Hoimes, C. J., et al. (2020). Pembrolizumab for treatment-refractory metastatic castration-resistant prostate cancer: multicohort, open-label phase II KEYNOTE-199 study. J. Clin. 38 (5), 395–405. doi:10.1200/JCO.19.01638
Antonarakis, E. S., Shaukat, F., Isaacsson Velho, P., Kaur, H., Shenderov, E., Pardoll, D. M., et al. (2019). Clinical features and therapeutic outcomes in men with advanced prostate cancer and DNA mismatch repair gene mutations. Eur. Urol. 75 (3), 378–382. doi:10.1016/j.eururo.2018.10.009
Antoni, L., Sodha, N., Collins, I., and Garrett, M. D. (2007). CHK2 kinase: cancer susceptibility and cancer therapy – two sides of the same coin? Nat. Rev. Cancer 7 (12), 925–936. doi:10.1038/nrc2251
Antoniou, A. C., Casadei, S., Heikkinen, T., Barrowdale, D., Pylkäs, K., Roberts, J., et al. (2014). Breast-cancer risk in families with mutations in PALB2. N. Engl. J. Med. 371 (6), 497–506. doi:10.1056/nejmoa1400382
Bartek, J., Falck, J., and Lukas, J. (2001). CHK2 kinase-a busy messenger. Nat. Rev. Mol. Cell Biol. 2 (12), 877–886. doi:10.1038/35103059
Bartek, J., and Lukas, J. (2003). Chk1 and Chk2 kinases in checkpoint control and cancer. Cancer Cell 3 (5), 421–429. doi:10.1016/S1535-6108(03)00110-7
Bednarski, J. J., and Sleckman, B. P. (2012). Chapter six - lymphocyte development: Integration of DNA damage response signaling (F. W. Alt (Ed.); Vol. 116, pp. 175–204). Cambridge, Massachusetts, United States: Academic Press. doi:10.1016/B978-0-12-394300-2.00006-5
Belkahla, S., Nahvi, I., Biswas, S., Nahvi, I., and Ben Amor, N. (2022). Advances and development of prostate cancer, treatment, and strategies: A systemic review. Front. Cell Dev. Biol. 10, 991330. doi:10.3389/fcell.2022.991330
Boudadi, K., Suzman, D. L., Anagnostou, V., Fu, W., Luber, B., Niknafs, N., et al. (2018). Ipilimumab plus nivolumab and DNA-repair defects in AR-V7- expressing metastatic prostate cancer. Oncotarget 9 (47), 28561–28571. doi:10.18632/oncotarget.25564
Boudadi, K., Suzman, D. L., Anagnostou, V., Fu, W., Luber, B., Wang, H., et al. (2018). Ipilimumab plus nivolumab and DNA-repair defects in AR-V7-expressing metastatic prostate cancer. Oncotarget 9 (47), 28561–28571. doi:10.18632/oncotarget.25564
Bouras, A., Lafaye, C., Leone, M., Kherraf, Z. E., Martin-Denavit, T., Fert-Ferrer, S., et al. (2022). Identification and characterization of an exonic duplication in PALB2 in a man with synchronous breast and prostate cancer. Int. J. Mol. Sci. 23 (2), 667. doi:10.3390/ijms23020667
Brandão, A., Paulo, P., and Teixeira, M. R. (2020). Hereditary predisposition to prostate cancer: from genetics to clinical implications. Int. J. Mol. Sci. 21 (14), 5036–5123. doi:10.3390/ijms21145036
Bray, F., Ferlay, J., Soerjomataram, I., Siegel, R. L., Torre, L. A., and Jemal, A. (2018). Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA A Cancer J. Clin. 68 (6), 394–424. doi:10.3322/caac.21492
Bugoye, F. C., Leyna, G. H., Moen, K., and Mmbaga, E. J. (2019). Knowledge, perceived risk and utilization of prostate cancer screening services among men in dar Es salaam, Tanzania. Prostate Cancer 2019, 2463048. doi:10.1155/2019/2463048
Callén, E., Jankovic, M., Wong, N., Zha, S., Chen, H., Virgilio, M. D., et al. (2009). Essential role for DNA-PKcs in DNA double-strand break repair and apoptosis in ATM-deficient lymphocytes. NIH Public Access 34 (3), 285–297. doi:10.1016/j.molcel.2009.04.025
Cancer Genome Atlas Research Network (2015). The molecular Taxonomy of primary prostate cancer. Cell 163 (4), 1011–1025. doi:10.1016/j.cell.2015.10.025
Castro, E., and Eeles, R. (2012). The role of BRCA1 and BRCA2 in prostate cancer. Asian J. Androl. 14 (3), 409–414. doi:10.1038/aja.2011.150
Castro, E., Goh, C., Olmos, D., Saunders, E., Leongamornlert, D., Tymrakiewicz, M., et al. (2013). Germline BRCA mutations are associated with higher risk of nodal involvement, distant metastasis, and poor survival outcomes in prostate cancer. J. Clin. Oncol. 31 (14), 1748–1757. doi:10.1200/JCO.2012.43.1882
Castro, E., Romero-Laorden, N., Del Pozo, A., Lozano, R., Medina, A., Puente, J., et al. (2019). PROREPAIR-B: A prospective cohort study of the impact of germline DNA repair mutations on the outcomes of patients with metastatic castration-resistant prostate cancer. J. Clin. Oncol. 37 (6), 490–503. doi:10.1200/JCO.18.00358
Catalano, M., Generali, D., Gatti, M., Riboli, B., Paganini, L., Nesi, G., et al. (2023). DNA repair de fi ciency as circulating biomarker in prostate cancer. January 13, 1115241–1115312. doi:10.3389/fonc.2023.1115241
Chakraborty, U., and Alani, E. (2016). Understanding how mismatch repair proteins participate in the repair/anti-recombination decision. FEMS Yeast Res. 16 (6), fow071. doi:10.1093/femsyr/fow071
Chang, H.-H., Lee, C.-H., Chen, Y.-T., Huang, C.-Y., Yu, C.-C., Lin, V. C., et al. (2022). Genetic analysis reveals the prognostic significance of the DNA mismatch repair gene MSH2 in advanced prostate cancer. Cancers 14 (1), 223. doi:10.3390/cancers14010223
Chen, X., Reiter, P. L., and McRee, A.-L. (2011). 乳鼠心肌提取 HHS public access. Physiology Behav. 176 (10), 139–148. doi:10.1053/j.semdp.2015.02.018
Chen, Y., Wang, J., Fraig, M. M., Metcalf, J., Turner, W. R., Bissada, N. K., et al. (2001). Defects of DNA mismatch repair in human prostate cancer. Cancer Res. 61 (10), 4112–4121.
Cheng, H. H., Sokolova, A. O., Gulati, R., Bowen, D., Knerr, S. A., Klemfuss, N., et al. (2023). Internet-based germline genetic testing for men with metastatic prostate cancer. JCO Precis. Oncol. 7, e2200104. doi:10.1200/PO.22.00104
Chi, K. N., Rathkopf, D. E., Smith, M. R., Efstathiou, E., Attard, G., Olmos, D., et al. (2022). Phase 3 MAGNITUDE study: first results of niraparib (NIRA) with abiraterone acetate and prednisone (AAP) as first-line therapy in patients (pts) with metastatic castration-resistant prostate cancer (mCRPC) with and without homologous recombination repair (HRR) gene alterations. J. Clin. Oncol. 40 (6), 12. doi:10.1200/JCO.2022.40.6_suppl.012
Choi, M., Kipps, T., and Kurzrock, R. (2016). ATM mutations in cancer: therapeutic implications. Mol. Cancer Ther. 15 (8), 1781–1791. doi:10.1158/1535-7163.MCT-15-0945
Chung, J.-S., Morgan, T. M., and Hong, S. K. (2020). Clinical implications of genomic evaluations for prostate cancer risk stratification, screening, and treatment: A narrative review. Prostate Int. 8 (3), 99–106. doi:10.1016/j.prnil.2020.09.001
Clarke, N. W., Armstrong, A. J., Thiery-Vuillemin, A., Oya, M., Shore, N., Loredo, E., et al. (2022). Abiraterone and olaparib for metastatic castration-resistant prostate cancer. NEJM Evid. 1 (9), 2200043. doi:10.1056/EVIDoa2200043
Curtin, N. J. (2012). DNA repair dysregulation from cancer driver to therapeutic target. Nat. Rev. Cancer 12 (12), 801–817. doi:10.1038/nrc3399
Curtin, N. J. (2023). Targeting the DNA damage response for cancer therapy. Biochem. Soc. Trans. 51 (1), 207–221. doi:10.1042/BST20220681
Cybulski, C., Wokołorczyk, D., Kluźniak, W., Jakubowska, A., Górski, B., Gronwald, J., et al. (2013). An inherited NBN mutation is associated with poor prognosis prostate cancer. Br. J. Cancer 108, 461–468. doi:10.1038/bjc.2012.486
De Mattos-Arruda, L., Vazquez, M., Finotello, F., Lepore, R., Porta, E., Hundal, J., et al. (2020). Neoantigen prediction and computational perspectives towards clinical benefit: recommendations from the ESMO precision medicine working group. Ann. Oncol. 31 (8), 978–990. doi:10.1016/j.annonc.2020.05.008
DeSantis, C. E., Siegel, R. L., Sauer, A. G., Miller, K. D., Fedewa, S. A., Alcaraz, K. I., et al. (2016). Cancer statistics for african Americans, 2016: progress and opportunities in reducing racial disparities. CA A Cancer J. Clin. 66 (4), 290–308. doi:10.3322/caac.21340
Dominguez-Valentin, M., Joost, P., Therkildsen, C., Jonsson, M., Rambech, E., and Nilbert, M. (2016). Frequent mismatch-repair defects link prostate cancer to Lynch syndrome. BMC Urol. 16 (1), 15. doi:10.1186/s12894-016-0130-1
Dong, X., Wang, L., Taniguchi, K., Wang, X., Cunningham, J. M., McDonnell, S. K., et al. (2003). Mutations in CHEK2 associated with prostate cancer risk. Am. J. Hum. Genet. 72, 270–280. doi:10.1086/346094
Dong, J.-T. (2006). Prevalent mutations in prostate cancer. J. Cell. Biochem. 97 (3), 433–447. doi:10.1002/jcb.20696
Edwards, S. M., Kote-Jarai, Z., Meitz, J., Hamoudi, R., Hope, Q., Osin, P., et al. (2003). Two percent of men with early-onset prostate cancer harbor germline mutations in the BRCA2 gene. Am. J. Hum. Genet. 72, 1–12. doi:10.1086/345310
El Noor, N. I. A., and El Noor, M. K. A. (2015). Quality of life in prostate cancer survivors in developing countries: the case of the gaza strip, Palestine. Nurs. Pract. Today 1 (2), 53–62.
Falck, J., Mailand, N., Syljuåsen, R. G., Bartek, J., and Lukas, J. (2001). The ATM–Chk2–Cdc25A checkpoint pathway guards against radioresistant DNA synthesis. Nature 410 (6830), 842–847. doi:10.1038/35071124
Ferrís-i-Tortajada, J., García-i-Castell, J., Berbel-Tornero, O., and Ortega-García, J. A. (2011). Constitutional risk factors in prostate cancer. Actas Urológicas Españolas 35 (5), 282–288. doi:10.1016/j.acuro.2010.12.009
Finch, A., Clark, R., Vesprini, D., Lorentz, J., Kim, R. H., Thain, E., et al. (2022). An appraisal of genetic testing for prostate cancer susceptibility. Npj Precis. Oncol. 6 (1), 43. doi:10.1038/s41698-022-00282-8
Force, U. S. P. S. T., Grossman, D. C., Curry, S. J., Owens, D. K., Bibbins-Domingo, K., Caughey, A. B., et al. (2018). Screening for prostate cancer: US preventive services task force recommendation statement. JAMA 319 (18), 1901–1913. doi:10.1001/jama.2018.3710
Fukuhara, S., Chang, I., Mitsui, Y., Chiyomaru, T., Yamamura, S., Majid, S., et al. (2015). Functional role of DNA mismatch repair gene PMS2 in prostate cancer cells. Oncotarget 6 (18), 16341–16351. doi:10.18632/oncotarget.3854
Fukuhara, S., Chang, I., Mitsui, Y., Chiyomaru, T., Yamamura, S., Majid, S., et al. (2014). DNA mismatch repair gene MLH1 induces apoptosis in prostate cancer cells. Oncotarget 5 (22), 11297–11307. doi:10.18632/oncotarget.2315
Furman, C. M., Elbashir, R., and Alani, E. (2021). Expanded roles for the MutL family of DNA mismatch repair proteins. Yeast (Chichester, Engl. 38 (1), 39–53. doi:10.1002/yea.3512
Gallagher, D. J., Gaudet, M. M., Pal, P., Kirchhoff, T., Balistreri, L., Vora, K., et al. (2010). Germline BRCA mutations denote a clinicopathologic subset of prostate cancer. Clin. Cancer Res. 16 (7), 2115–2121. doi:10.1158/1078-0432.CCR-09-2871
Gayther, S. A., de Foy, K. A., Harrington, P., Pharoah, P., Dunsmuir, W. D., Edwards, S. M., et al. (2000). The frequency of germ-line mutations in the breast cancer predisposition genes BRCA1 and BRCA2 in familial prostate cancer. The Cancer Research Campaign/British Prostate Group United Kingdom Familial Prostate Cancer Study Collaborators. Cancer Res. 60 (16), 4513–4518.
Gillessen, S., Attard, G., Beer, T. M., Beltran, H., Bjartell, A., Bossi, A., et al. (2020). Management of patients with advanced prostate cancer: report of the advanced prostate cancer consensus conference 2019. Eur. Urol. 77 (4), 508–547. doi:10.1016/j.eururo.2020.01.012
Gillessen, S., Attard, G., Beer, T. M., Beltran, H., Bossi, A., Bristow, R., et al. (2018). Management of patients with advanced prostate cancer: the report of the advanced prostate cancer consensus conference APCCC 2017. Eur. Urol. 73 (2), 178–211. doi:10.1016/j.eururo.2017.06.002
Gillessen, S., Omlin, A., Attard, G., Bono, J. S. De, Efstathiou, E., Fizazi, K., et al. (2015). Management of patients with advanced prostate cancer: recommendations of the st gallen advanced prostate cancer consensus conference (APCCC) 2015. Ann. Oncol. 26, 1589–1604. doi:10.1093/annonc/mdv257
Giri, V. N., and Beebe-Dimmer, J. L. (2016). Familial prostate cancer. Seminars Oncol. 43 (5), 560–565. doi:10.1053/j.seminoncol.2016.08.001
Golan, T., O’Kane, G. M., Denroche, R. E., Raitses-Gurevich, M., Grant, R. C., Holter, S., et al. (2021). Genomic features and classification of homologous recombination deficient pancreatic ductal adenocarcinoma. Gastroenterology 160 (6), 2119–2132.e9. doi:10.1053/j.gastro.2021.01.220
Graf, R. P., Fisher, V., Weberpals, J., Gjoerup, O., Tierno, M. B., Huang, R. S. P., et al. (2022). Comparative effectiveness of immune checkpoint inhibitors vs chemotherapy by tumor mutational burden in metastatic castration-resistant prostate cancer. JAMA Netw. Open 5 (3), e225394. doi:10.1001/jamanetworkopen.2022.5394
Graham, L. S., and Schweizer, M. T. (2022). Mismatch repair deficiency and clinical implications in prostate cancer. Prostate 82 (1), S37–S44. doi:10.1002/pros.24343
Gurley, K. E., and Kemp, C. J. (2001). Synthetic lethality between mutation in Atm and DNA-PK(cs) during murine embryogenesis. Curr. Biol. CB 11 (3), 191–194. doi:10.1016/s0960-9822(01)00048-3
Haffner, M. C., Zwart, W., Roudier, M. P., True, L. D., Nelson, W. G., Epstein, J. I., et al. (2021). Genomic and phenotypic heterogeneity in prostate cancer. Nat. Rev. Urol. 18 (2), 79–92. doi:10.1038/s41585-020-00400-w
Hanahan, D., and Weinberg, R. A. (2011). Hallmarks of cancer: the next generation. Cell 144 (5), 646–674. doi:10.1016/j.cell.2011.02.013
Hansen, A. R., Massard, C., Ott, P. A., Haas, N. B., Lopez, J. S., Ejadi, S., et al. (2018). Pembrolizumab for advanced prostate adenocarcinoma: findings of the KEYNOTE-028 study. Ann. Oncol. Official J. Eur. Soc. Med. Oncol. 29 (8), 1807–1813. doi:10.1093/annonc/mdy232
Haraldsdottir, S., Hampel, H., Wei, L., Wu, C., Frankel, W., Bekaii-Saab, T., et al. (2014). Prostate cancer incidence in males with Lynch syndrome. Genet. Med. 16 (7), 553–557. doi:10.1038/gim.2013.193
Heaphy, C. M., Yoon, G. S., Peskoe, S. B., Joshu, C. E., Lee, T. K., Giovannucci, E., et al. (2013). Prostate cancer cell telomere length variability and stromal cell telomere length as prognostic markers for metastasis and death. Cancer Discov. 3 (10), 1130–1141. doi:10.1158/2159-8290.CD-13-0135
Hendriks, Y. M. C., Jagmohan-Changur, S., van der Klift, H. M., Morreau, H., van Puijenbroek, M., Tops, C., et al. (2006). Heterozygous mutations in PMS2 cause hereditary nonpolyposis colorectal carcinoma (Lynch syndrome). Gastroenterology 130 (2), 312–322. doi:10.1053/j.gastro.2005.10.052
Herberts, C., Wyatt, A. W., Nguyen, P. L., and Cheng, H. H. (2023). Genetic and genomic testing for prostate cancer: beyond DNA repair. Am. Soc. Clin. Oncol. Educ. Book 43, e390384. doi:10.1200/EDBK_390384
Hirao, A., Kong, Y.-Y., Matsuoka, S., Wakeham, A., Ruland, J., Yoshida, H., et al. (2000). DNA damage-induced activation of p53 by the checkpoint kinase Chk2. Science 287 (5459), 1824–1827. doi:10.1126/science.287.5459.1824
Hopfner, K.-P., and Hornung, V. (2020). Molecular mechanisms and cellular functions of cGAS–STING signalling. Nat. Rev. Mol. Cell Biol. 21 (9), 501–521. doi:10.1038/s41580-020-0244-x
Horak, P., Weischenfeldt, J., von Amsberg, G., Beyer, B., Schütte, A., Uhrig, S., et al. (2019). Response to olaparib in a PALB2 germline mutated prostate cancer and genetic events associated with resistance. Cold Spring Harb. Mol. Case Stud. 5 (2), a003657. doi:10.1101/mcs.a003657
Ikonen, T., Matikainen, M. P., Syrjäkoski, K., Mononen, N., Koivisto, P. A., Rökman, A., et al. (2003). BRCA1 and BRCA2 mutations have no major role in predisposition to prostate cancer in Finland. J. Med. Genet. 40 (8), e98. doi:10.1136/jmg.40.8.e98
Javeed, S., Chughtai, A., Zafar, G., Khalid, F., Batool, A., and Chughtai, A. S. (2022). An evaluation of the immunohistochemical expression of mismatch repair proteins (MSH2, MSH6, MLH1, and PMS2) in prostate adenocarcinoma. Cureus 14 (7), e27448. doi:10.7759/cureus.27448
Jiang, M., Jia, K., Wang, L., Li, W., Chen, B., Liu, Y., et al. (2021). Alterations of DNA damage response pathway: biomarker and therapeutic strategy for cancer immunotherapy. Acta Pharm. Sin. B 11 (10), 2983–2994. doi:10.1016/j.apsb.2021.01.003
Jiang, X., Hu, X., Gu, Y., Li, Y., Jin, M., Zhao, H., et al. (2021). Homologous recombination repair gene mutations in Chinese localized and locally advanced prostate cancer patients. Pathology Res. Pract. 224, 153507. doi:10.1016/j.prp.2021.153507
Jiricny, J. (2013). Postreplicative mismatch repair. Cold Spring Harb. Perspect. Biol. 5 (4), a012633. doi:10.1101/cshperspect.a012633
Jones, S., Hruban, R. H., Kamiyama, M., Borges, M., Zhang, X., Parsons, D. W., et al. (2009). Exomic sequencing identifies PALB2 as a pancreatic cancer susceptibility gene. Science 324 (5924), 217. doi:10.1126/science.1171202
Keung, M. Y. T., Wu, Y., and Vadgama, J. V. (2019). PARP inhibitors as a therapeutic agent for homologous recombination deficiency in breast cancers. J. Clin. Med. 8 (4), 435. doi:10.3390/jcm8040435
Khanna, K. K., and Jackson, S. P. (2001). DNA double-strand breaks: signaling, repair and the cancer connection. Nat. Genet. 27 (3), 247–254. doi:10.1038/85798
Kiyotani, K., Chan, H. T., and Nakamura, Y. (2018). Immunopharmacogenomics towards personalized cancer immunotherapy targeting neoantigens. Cancer Sci. 109 (3), 542–549. doi:10.1111/cas.13498
Kohaar, I., Zhang, X., Tan, S.-H., Nousome, D., Babcock, K., Ravindranath, L., et al. (2022). Germline mutation landscape of DNA damage repair genes in African Americans with prostate cancer highlights potentially targetable RAD genes. Nat. Commun. 13, 1361. doi:10.1038/s41467-022-28945-x
Kote-Jarai, Z., Leongamornlert, D., Saunders, E., Tymrakiewicz, M., Castro, E., Mahmud, N., et al. (2011). BRCA2 is a moderate penetrance gene contributing to young-onset prostate cancer: implications for genetic testing in prostate cancer patients. Br. J. Cancer 105 (8), 1230–1234. doi:10.1038/bjc.2011.383
Lang, S. H., Swift, S. L., White, H., Misso, K., Kleijnen, J., and Quek, R. G. W. (2019). A systematic review of the prevalence of DNA damage response gene mutations in prostate cancer. Int. J. Oncol. 55 (3), 597–616. doi:10.3892/ijo.2019.4842
Le, D. T., Uram, J. N., Wang, H., Bartlett, B. R., Kemberling, H., Eyring, A. D., et al. (2015). PD-1 blockade in tumors with mismatch-repair deficiency. N. Engl. J. Med. 372 (26), 2509–2520. doi:10.1056/NEJMoa1500596
Lei, Y., Li, X., Huang, Q., Zheng, X., and Liu, M. (2021). Progress and challenges of predictive biomarkers for immune checkpoint blockade. Front. Oncol. 11, 617335. doi:10.3389/fonc.2021.617335
Leong, V., Lorenowicz, J., Kozij, N., and Guarné, A. (2009). Nuclear import of human MLH1, PMS2, and MutLalpha: redundancy is the key. Mol. Carcinog. 48, 742–750. doi:10.1002/mc.20514
Leongamornlert, D., Mahmud, N., Tymrakiewicz, M., Saunders, E., Dadaev, T., Castro, E., et al. (2012). Germline BRCA1 mutations increase prostate cancer risk. Br. J. Cancer 106 (10), 1697–1701. doi:10.1038/bjc.2012.146
Lieber, M. R. (2011). The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annu. Rev. Biochem. 79 (3), 181–211. doi:10.1146/annurev.biochem.052308.093131
Lozano, R., Naghavi, M., Foreman, K., Lim, S., Shibuya, K., Aboyans, V., et al. (2012). Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010: A systematic analysis for the global burden of disease study 2010. Lancet 380 (9859), 2095–2128. doi:10.1016/S0140-6736(12)61728-0
Lu, Y.-C., and Robbins, P. F. (2016). Targeting neoantigens for cancer immunotherapy. Int. Immunol. 28 (7), 365–370. doi:10.1093/intimm/dxw026
Lukashchuk, N., Barnicle, A., Adelman, C. A., Armenia, J., Kang, J., Barrett, J. C., et al. (2023). Impact of DNA damage repair alterations on prostate cancer progression and metastasis. Front. Oncol. 13, 1162644–1162713. doi:10.3389/fonc.2023.1162644
Ma, X., Dong, L., Liu, X., Ou, K., and Yang, L. (2022). POLE/POLD1 mutation and tumor immunotherapy. J. Exp. Clin. Cancer Res. 41 (1), 216. doi:10.1186/s13046-022-02422-1
Maia, S., Cardoso, M., Paulo, P., Pinheiro, M., Pinto, P., Santos, C., et al. (2016). The role of germline mutations in the BRCA1/2 and mismatch repair genes in men ascertained for early-onset and/or familial prostate cancer. Fam. Cancer 15 (1), 111–121. doi:10.1007/s10689-015-9832-x
Maier, C., Herkommer, K., Luedeke, M., Rinckleb, A., Schrader, M., Vogel, W., et al. (2014). Subgroups of familial and aggressive prostate cancer with considerable frequencies of BRCA2 mutations. Prostate 74, 1444–1451. doi:10.1002/pros.22860
Manguoğlu, E., Güran, S., Yamaç, D., Colak, T., Simşek, M., Baykara, M., et al. (2010). Germline mutations of BRCA1 and BRCA2 genes in Turkish breast, ovarian, and prostate cancer patients. Cancer Genet. Cytogenet. 203 (2), 230–237. doi:10.1016/j.cancergencyto.2010.07.125
Mateo, J., Carreira, S., Sandhu, S., Miranda, S., Mossop, H., Perez-Lopez, R., et al. (2015). DNA-repair defects and olaparib in metastatic prostate cancer. N. Engl. J. Med. 373 (18), 1697–1708. doi:10.1056/NEJMoa1506859
Matsuoka, S., Huang, M., and Elledge, S. J. (1998). Linkage of ATM to cell cycle regulation by the Chk2 protein kinase. Science 282 (5395), 1893–1897. doi:10.1126/science.282.5395.1893
McVety, S., Younan, R., Li, L., Gordon, P. H., Wong, N., Foulkes, W. D., et al. (2005). Novel genomic insertion-- deletion in MLH1: Possible mechanistic role for non-homologous end-joining DNA repair. Clin. Genet. 68, 234–238. doi:10.1111/j.1399-0004.2005.00486.x
Na, R., Zheng, S. L., Han, M., Yu, H., Jiang, D., Shah, S., et al. (2017). Germline mutations in ATM and BRCA1/2 distinguish risk for lethal and indolent prostate cancer and are associated with early age at death. Eur. Urol. 71 (5), 740–747. doi:10.1016/j.eururo.2016.11.033
Nair, S. S., Chakravarty, D., Dovey, Z. S., Zhang, X., and Tewari, A. K. (2022). Why do African-American men face higher risks for lethal prostate cancer? Curr. Opin. Urology 32 (1), 96–101. doi:10.1097/MOU.0000000000000951
Nastiuk, K. L., Mansukhani, M., Terry, M. B., Kularatne, P., Rubin, M. A., Melamed, J., et al. (1999). Common mutations in BRCA1 and BRCA2 do not contribute to early prostate cancer in Jewish men. Prostate 40, 172–177. doi:10.1002/(sici)1097-0045(19990801)40:3<172::aid-pros5>3.0.co;2-r
Pande, M., Wei, C., Chen, J., Amos, C. I., Lynch, P. M., Lu, K. H., et al. (2012). Cancer spectrum in DNA mismatch repair gene mutation carriers: results from a hospital based Lynch syndrome registry. Fam. Cancer 11 (3), 441–447. doi:10.1007/s10689-012-9534-6
Panigrahi, G. K., Praharaj, P. P., Kittaka, H., Mridha, A. R., Black, O. M., Singh, R., et al. (2019). Exosome proteomic analyses identify inflammatory phenotype and novel biomarkers in African American prostate cancer patients. Cancer Med. 8 (3), 1110–1123. doi:10.1002/cam4.1885
Pećina-Šlaus, N., Kafka, A., Salamon, I., and Bukovac, A. (2020). Mismatch repair pathway, genome stability and cancer. Front. Mol. Biosci. 7, 122. doi:10.3389/fmolb.2020.00122
Petrovics, G., Price, D. K., Lou, H., Chen, Y., Garland, L., Bass, S., et al. (2019). Increased frequency of germline BRCA2 mutations associates with prostate cancer metastasis in a racially diverse patient population. Prostate Cancer Prostatic Dis. 22, 406–410. doi:10.1038/s41391-018-0114-1
Powell, I. J., and Bollig-Fischer, A. (2013). Minireview: the molecular and genomic basis for prostate cancer health disparities. Mol. Endocrinol. Baltim. Md 27 (6), 879–891. doi:10.1210/me.2013-1039
Pritchard, C. C., Mateo, J., Walsh, M. F., De Sarkar, N., Abida, W., Beltran, H., et al. (2016). Inherited DNA-repair gene mutations in men with metastatic prostate cancer. N. Engl. J. Med. 375 (5), 443–453. doi:10.1056/NEJMoa1603144
Pritchard, C. C., Morrissey, C., Kumar, A., Zhang, X., Smith, C., Coleman, I., et al. (2014). Complex MSH2 and MSH6 mutations in hypermutated microsatellite unstable advanced prostate cancer. Nat. Commun. 5, 4988–4996. doi:10.1038/ncomms5988
Rantapero, T., Wahlfors, T., Kähler, A., Hultman, C., Lindberg, J., Tammela, T. L., et al. (2020). Inherited DNA repair gene mutations in men with lethal prostate cancer. Genes 11 (3), 314. doi:10.3390/genes11030314
Rawla, P. (2019). Epidemiology of prostate cancer. World J. Oncol. 10 (2), 63–89. doi:10.14740/wjon1191
Raymond, V. M., Mukherjee, B., Wang, F., Huang, S.-C., Stoffel, E. M., Kastrinos, F., et al. (2013). Elevated risk of prostate cancer among men with Lynch syndrome. J. Clin. Oncol. 31 (14), 1713–1718. doi:10.1200/JCO.2012.44.1238
Reid, S., Schindler, D., Hanenberg, H., Barker, K., Hanks, S., Kalb, R., et al. (2007). Biallelic mutations in PALB2 cause Fanconi anemia subtype FA-N and predispose to childhood cancer. Nat. Genet. 39 (2), 162–164. doi:10.1038/ng1947
Reizine, N., Socco, S., Weng, E., Atueyi, U., Mehta, V., and Patnaik, A. (2023). Utilization of next-generation sequencing in conjunction with immunohistochemistry to predict exceptional response to combination immune checkpoint therapy in a heavily pretreated patient with castration-resistant prostate cancer. JCO Precis. Oncol. 7, e2200483. doi:10.1200/PO.22.00483
Reyes, G. X., Schmidt, T. T., Kolodner, R. D., and Hombauer, H. (2015). New insights into the mechanism of DNA mismatch repair. Chromosoma 124 (4), 443–462. doi:10.1007/s00412-015-0514-0
Riaz, N., Morris, L., Havel, J. J., Makarov, V., Desrichard, A., and Chan, T. A. (2016). The role of neoantigens in response to immune checkpoint blockade. Int. Immunol. 28 (8), 411–419. doi:10.1093/intimm/dxw019
Risbridger, G. P., Taylor, R. A., Clouston, D., Sliwinski, A., Thorne, H., Hunter, S., et al. (2015). Patient-derived xenografts reveal that intraductal carcinoma of the prostate is a prominent Pathology in BRCA2 mutation carriers with prostate cancer and correlates with poor prognosis. Eur. Urol. 67 (3), 496–503. doi:10.1016/j.eururo.2014.08.007
Ritch, E., Fu, S. Y. F., Herberts, C., Wang, G., Warner, E. W., Schonlau, E., et al. (2020). Identification of hypermutation and defective mismatch repair in ctDNA from metastatic prostate cancer. Clin. Cancer Res. 26 (5), 1114–1125. doi:10.1158/1078-0432.CCR-19-1623
Robinson, D., Van Allen, E. M., Wu, Y.-M., Schultz, N., Lonigro, R. J., Mosquera, J.-M., et al. (2015a). Integrative clinical genomics of advanced prostate cancer. Cell 161 (5), 1215–1228. doi:10.1016/j.cell.2015.05.001
Robinson, D., Van Allen, E. M., Wu, Y. M., Schultz, N., Lonigro, R. J., Mosquera, J. M., et al. (2015b). Erratum: integrative clinical genomics of advanced prostate cancer. Cell 162 (2), 454. doi:10.1016/j.cell.2015.06.053
Rodrigues, G., Warde, P., Pickles, T., Crook, J., Brundage, M., Souhami, L., et al. (2012). Pre-treatment risk stratification of prostate cancer patients: A critical review. Can. Urological Assoc. J. = J. de l’Association Des Urologues Du Can. 6 (2), 121–127. doi:10.5489/cuaj.11085
Rose, M., Burgess, J. T., O’Byrne, K., Richard, D. J., and Bolderson, E. (2020). PARP inhibitors: clinical relevance, mechanisms of action and tumor resistance. Front. Cell Dev. Biol. 8, 564601. doi:10.3389/fcell.2020.564601
Roy, R., Chun, J., and Powell, S. N. (2012). BRCA1 and BRCA2: different roles in a common pathway of genome protection. Nat. Rev. Cancer 12 (1), 68–78. doi:10.1038/nrc3181
Saad, F., Canil, C., Finelli, A., Hotte, S. J., Malone, S., Shayegan, B., et al. (2019). Controversial issues in the management of patients with advanced prostate cancer: results from a Canadian consensus forum. Can. Urological Assoc. J. 14 (4), E137–49. doi:10.5489/cuaj.6082
Sakellakis, M., Jacqueline Flores, L., and Ramachandran, S. (2022). Patterns of indolence in prostate cancer (Review). Exp. Ther. Med. 23 (5), 351. doi:10.3892/etm.2022.11278
Salmi, F., Maachi, F., Tazzite, A., Aboutaib, R., Fekkak, J., Azeddoug, H., et al. (2021). Next-generation sequencing of BRCA1 and BRCA2 genes in Moroccan prostate cancer patients with positive family history. PLoS ONE 16 (7), e0254101–e0254114. doi:10.1371/journal.pone.0254101
Salo-Mullen, E. E., Lynn, P. B., Wang, L., Walsh, M., Gopalan, A., Shia, J., et al. (2018). Contiguous gene deletion of chromosome 2p16.3-p21 as a cause of Lynch syndrome. Fam. Cancer 17 (1), 71–77. doi:10.1007/s10689-017-0006-x
Sanjose, S. De, Bruni, L., Saraiya, M., Bray, F., Arbyn, M., Castellsague, X., et al. (2011). Worldwide burden of cervical cancer in 2008. April 22, 2675–2686. doi:10.1093/annonc/mdr015
Saxena, S., and Zou, L. (2022). Hallmarks of DNA replication stress. Mol. Cell 82 (12), 2298–2314. doi:10.1016/j.molcel.2022.05.004
Schweizer, M. T., Antonarakis, E. S., Bismar, T. A., Guedes, L. B., Cheng, H. H., Tretiakova, M. S., et al. (2019). Genomic characterization of prostatic ductal adenocarcinoma identifies a high prevalence of DNA repair gene mutations. JCO Precis. Oncol. 3, 1–9. doi:10.1200/PO.18.00327
Sedhom, R., and Antonarakis, E. S. (2019). Clinical implications of mismatch repair deficiency in prostate cancer. Future Oncol. Lond. Engl. 15 (20), 2395–2411. doi:10.2217/fon-2019-0068
Sharma, M., Yang, Z., and Miyamoto, H. (2020). Loss of DNA mismatch repair proteins in prostate cancer. Medicine 99 (19), e20124. doi:10.1097/MD.0000000000020124
Sharma, P., Pachynski, R. K., Narayan, V., Fléchon, A., Gravis, G., Galsky, M. D., et al. (2020). Nivolumab plus ipilimumab for metastatic castration-resistant prostate cancer: preliminary analysis of patients in the CheckMate 650 trial. Cancer Cell 38 (4), 489–499. doi:10.1016/j.ccell.2020.08.007
Shenderov, E., Isaacsson Velho, P., Awan, A. H., Wang, H., Mirkheshti, N., Lotan, T. L., et al. (2019). Genomic and clinical characterization of pulmonary-only metastatic prostate cancer: A unique molecular subtype. Prostate 79 (13), 1572–1579. doi:10.1002/pros.23881
Shenoy, D., Packianathan, S., Chen, A. M., and Vijayakumar, S. (2016). Do African-American men need separate prostate cancer screening guidelines? BMC Urol. 16 (1), 19. doi:10.1186/s12894-016-0137-7
Shi, C., Qin, K., Lin, A., Jiang, A., Cheng, Q., Liu, Z., et al. (2022). The role of DNA damage repair (DDR) system in response to immune checkpoint inhibitor (ICI) therapy. J. Exp. Clin. Cancer Res. 41 (1), 268–327. doi:10.1186/s13046-022-02469-0
Shore, N. D., Morgans, A. K., El-Haddad, G., Srinivas, S., and Abramowitz, M. (2022). Addressing challenges and controversies in the management of prostate cancer with multidisciplinary teams. Target. Oncol. 17 (6), 709–725. doi:10.1007/s11523-022-00925-7
Siegel, R., Ward, E., Brawley, O., and Jemal, A. (2011). Cancer statistics, 2011 the impact of eliminating socioeconomic and racial disparities on premature cancer deaths. CA Cancer J. Clin. 61, 212–236. doi:10.3322/caac.20121
Silvestri, V., Leslie, G., Barnes, D. R., Agnarsson, B. A., Aittomäki, K., Alducci, E., et al. (2020). Characterization of the cancer spectrum in men with germline BRCA1 and BRCA2 pathogenic variants: results from the consortium of investigators of modifiers of BRCA1/2 (CIMBA). JAMA Oncol. 6 (8), 1218–1230. doi:10.1001/jamaoncol.2020.2134
Sorrentino, C., and Di Carlo, E. (2023). Molecular targeted therapies in metastatic prostate cancer: recent advances and future challenges. Cancers 15 (11), 2885. doi:10.3390/cancers15112885
Southey, M. C., Goldgar, D. E., Winqvist, R., Pylkäs, K., Couch, F., Tischkowitz, M., et al. (2016). PALB2, CHEK2 and ATM rare variants and cancer risk: data from COGS. J. Med. Genet. 53 (12), 800–811. doi:10.1136/jmedgenet-2016-103839
Sung, H., Ferlay, J., Siegel, R. L., Laversanne, M., Soerjomataram, I., Jemal, A., et al. (2021). Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA A Cancer J. Clin. 71 (3), 209–249. doi:10.3322/caac.21660
Sy, S. M. H., Huen, M. S. Y., and Chen, J. (2009). PALB2 is an integral component of the BRCA complex required for homologous recombination repair. Proc. Natl. Acad. Sci. U. S. A. 106 (17), 7155–7160. doi:10.1073/pnas.0811159106
Szymaniak, B. M., Facchini, L. A., Giri, V. N., Antonarakis, E. S., Beer, T. M., Carlo, M. I., et al. (2020). Practical considerations and challenges for germline genetic testing in patients with prostate cancer: Recommendations from the germline genetics working group of the PCCTC. JCO Oncol. Pract. 16, 811–819. doi:10.1200/OP.20.00431
Taylor, R. A., Fraser, M., Livingstone, J., Espiritu, S. M. G., Thorne, H., Huang, V., et al. (2017). Germline BRCA2 mutations drive prostate cancers with distinct evolutionary trajectories. Nat. Commun. 8 (1), 13671. doi:10.1038/ncomms13671
Teply, B. A., and Antonarakis, E. S. (2017). Treatment strategies for DNA repair-deficient prostate cancer. Expert Rev. Clin. Pharmacol. 10 (8), 889–898. doi:10.1080/17512433.2017.1338138
Thalgott, M., Kron, M., Brath, J. M., Ankerst, D. P., Thompson, I. M., Gschwend, J. E., et al. (2018). Men with family history of prostate cancer have a higher risk of disease recurrence after radical prostatectomy. World J. Urology 36 (2), 177–185. doi:10.1007/s00345-017-2122-5
Thompson, D., Easton, D. F., and Consortium, the B. C. L. (2003). Cancer incidence in BRCA1 mutation carriers. Obstetrical Gynecol. Surv. 58 (1), 27–28. doi:10.1097/00006254-200301000-00016
Tolkach, Y., and Kristiansen, G. (2018). The heterogeneity of prostate cancer: A practical approach. Pathobiology 85 (1–2), 108–116. doi:10.1159/000477852
Tryggvadóttir, L., Vidarsdóttir, L., Thorgeirsson, T., Jonasson, J. G., Olafsdóttir, E. J., Olafsdóttir, G. H., et al. (2007). Prostate cancer progression and survival in BRCA2 mutation carriers. J. Natl. Cancer Inst. 99 (12), 929–935. doi:10.1093/jnci/djm005
van Wilpe, S., Tolmeijer, S. H., Koornstra, R. H. T., de Vries, I. J. M., Gerritsen, W. R., Ligtenberg, M., et al. (2021). Homologous recombination repair deficiency and implications for tumor immunogenicity. Cancers 13 (9), 2249. doi:10.3390/cancers13092249
Vazina, A., Baniel, J., Yaacobi, Y., Shtriker, A., Engelstein, D., Leibovitz, I., et al. (2000). The rate of the founder Jewish mutations in BRCA1 and BRCA2 in prostate cancer patients in Israel. Br. J. Cancer 83 (4), 463–466. doi:10.1054/bjoc.2000.1249
Vietri, M. T., D’Elia, G., Caliendo, G., Resse, M., Casamassimi, A., Passariello, L., et al. (2021). Hereditary prostate cancer: genes related, target therapy and prevention. Int. J. Mol. Sci. 22 (7), 3753. doi:10.3390/ijms22073753
Virtanen, V., Paunu, K., Ahlskog, J. K., Varnai, R., Sipeky, C., and Sundvall, M. (2019). PARP inhibitors in prostate cancer—the preclinical rationale and current clinical development. Genes 10 (8), 565. doi:10.3390/genes10080565
Wang, S.-Y., Cowan, J. E., Cary, K. C., Chan, J. M., Carroll, P. R., and Cooperberg, M. R. (2014). Limited ability of existing nomograms to predict outcomes in men undergoing active surveillance for prostate cancer. BJU Int. 114 (6), E18–E24. doi:10.1111/bju.12554
Wang, Y., Dai, B., and Ye, D. (2015). CHEK2 mutation and risk of prostate cancer: A systematic review and meta-analysis. Int. J. Clin. Exp. Med. 8 (9), 15708–15715.
Wei, S. C., Duffy, C. R., and Allison, J. P. (2018). Fundamental mechanisms of immune checkpoint blockade therapy. Cancer Discov. 8 (9), 1069–1086. doi:10.1158/2159-8290.CD-18-0367
White, J. A., Kaninjing, E. T., Adeniji, K. A., Jibrin, P., Obafunwa, J. O., Ogo, C. N., et al. (2022). Whole-exome sequencing of Nigerian prostate tumors from the prostate cancer transatlantic consortium (CaPTC) reveals DNA repair genes associated with african ancestry. Cancer Res. Commun. 2 (9), 1005–1016. doi:10.1158/2767-9764.CRC-22-0136
Wilczak, W., Rashed, S., Hube-Magg, C., Kluth, M., Simon, R., Büscheck, F., et al. (2017). Up-regulation of mismatch repair genes MSH6, PMS2 and MLH1 parallels development of genetic instability and is linked to tumor aggressiveness and early PSA recurrence in prostate cancer. Carcinogenesis 38 (1), 19–27. doi:10.1093/carcin/bgw116
Wilkens, E. P., Freije, D., Xu, J., Nusskern, D. R., Suzuki, H., Isaacs, S. D., et al. (1999). No evidence for a role of BRCA1 or BRCA2 mutations in Ashkenazi Jewish families with hereditary prostate cancer. Prostate 39 (4), 280–284. doi:10.1002/(sici)1097-0045(19990601)39:4<280:aid-pros8>3.0.co;2-f
Wokołorczyk, D., Kluźniak, W., Huzarski, T., Gronwald, J., Szymiczek, A., Rusak, B., et al. (2020). Mutations in ATM, NBN and BRCA2 predispose to aggressive prostate cancer in Poland. Int. J. Cancer 147 (10), 2793–2800. doi:10.1002/ijc.33272
Wokołorczyk, D., Kluźniak, W., Stempa, K., Rusak, B., Huzarski, T., Gronwald, J., et al. (2021). PALB2 mutations and prostate cancer risk and survival. Br. J. Cancer 125 (4), 569–575. doi:10.1038/s41416-021-01410-0
Wu, X., Platt, J. L., and Cascalho, M. (2003). Dimerization of MLH1 and PMS2 limits nuclear localization of MutLalpha. Mol. Cell. Biol. 23 (9), 3320–3328. doi:10.1128/MCB.23.9.3320-3328.2003
Xia, B., Dorsman, J. C., Ameziane, N., de Vries, Y., Rooimans, M. A., Sheng, Q., et al. (2007). Fanconi anemia is associated with a defect in the BRCA2 partner PALB2. Nat. Genet. 39 (2), 159–161. doi:10.1038/ng1942
Xie, C., Luo, J., He, Y., Jiang, L., Zhong, L., and Shi, Y. (2022). BRCA2 gene mutation in cancer. Medicine 101 (45), e31705. doi:10.1097/MD.0000000000031705
Yang, X., Leslie, G., Doroszuk, A., Schneider, S., Allen, J., Decker, B., et al. (2020). Cancer risks associated with germline PALB2 pathogenic variants: an international study of 524 families. J. Clin. Oncol. 38 (7), 674–685. doi:10.1200/JCO.19.01907
Ye, S., Wang, H., He, K., Peng, M., Wang, Y., Li, Y., et al. (2020). Clinical characterization of mismatch repair gene-deficient metastatic castration-resistant prostate cancer. Front. Oncol. 10, 533282. doi:10.3389/fonc.2020.533282
Yi, T., Feng, Y., Sundaram, R., Tie, Y., Zheng, H., Qian, Y., et al. (2019). Antitumor efficacy of PARP inhibitors in homologous recombination deficient carcinomas. Int. J. Cancer 145 (5), 1209–1220. doi:10.1002/ijc.32143
Zannini, L., Delia, D., and Buscemi, G. (2014). CHK2 kinase in the DNA damage response and beyond. J. Mol. Cell Biol. 6 (6), 442–457. doi:10.1093/jmcb/mju045
Zhang, D., Xu, X., Wei, Y., Chen, X., Li, G., Lu, Z., et al. (2022). Prognostic role of DNA damage response genes mutations and their association with the sensitivity of olaparib in prostate cancer patients. Cancer Control J. Moffitt Cancer Cent. 29, 10732748221129451. doi:10.1177/10732748221129451
Keywords: mutation, DNA damage, prostate cancer, mismatch, repair-deficient, landscape
Citation: Bugoye FC, Torrorey-Sawe R, Biegon R, Dharsee N, Mafumiko FMS, Patel K and Mining SK (2023) Mutational spectrum of DNA damage and mismatch repair genes in prostate cancer. Front. Genet. 14:1231536. doi: 10.3389/fgene.2023.1231536
Received: 30 May 2023; Accepted: 16 August 2023;
Published: 04 September 2023.
Edited by:
Thierry Mp Gauthier, National Institutes of Health (NIH), United StatesReviewed by:
Liliana C. Patiño, National Institute of Dental and Craniofacial Research (NIH), United StatesCopyright © 2023 Bugoye, Torrorey-Sawe, Biegon, Dharsee, Mafumiko, Patel and Mining. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Fidelis Charles Bugoye, YnVnb3llODFAeWFob28uY28udWs=; Rispah Torrorey-Sawe, dG9ycm9yZXlAZ21haWwuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.