 Roberta Zuntini1†
Roberta Zuntini1† Chiara Cattani1†
Chiara Cattani1† Lucia Pedace2
Lucia Pedace2 Evelina Miele2Stefano Giuseppe Caraffi1Stefano Gardini3Elena Ficarelli3
Evelina Miele2Stefano Giuseppe Caraffi1Stefano Gardini3Elena Ficarelli3 Simone Pizzi4
Simone Pizzi4 Francesca Clementina Radio4
Francesca Clementina Radio4 Angelica Barone5
Angelica Barone5 Simonetta Piana6Patrizia Bertolini5
Simonetta Piana6Patrizia Bertolini5 Domenico Corradi7Maria Marinelli1Caterina Longo8,9
Domenico Corradi7Maria Marinelli1Caterina Longo8,9 Alberico Motolese3Orsetta Zuffardi10*
Alberico Motolese3Orsetta Zuffardi10* Marco Tartaglia4*
Marco Tartaglia4* Livia Garavelli1*
Livia Garavelli1*- 1Medical Genetics Unit, Azienda USL, IRCCS, Arcispedale Santa Maria Nuova, Reggio Emilia, Italy
- 2Department of Pediatric Hematology, Oncology and Cellular and Gene Therapy, Ospedale Pediatrico Bambino Gesù, IRCCS, Rome, Italy
- 3Dermatology Unit, Azienda USL, IRCCS, Arcispedale Santa Maria Nuova, Reggio Emilia, Italy
- 4Molecular Genetics and Functional Genomics Research Unit, Ospedale Pediatrico Bambino Gesù, IRCCS, Rome, Italy
- 5Paediatric Hematology Oncology Unit, University Hospital of Parma, Parma, Italy
- 6Department of Oncology and Advanced Technologies, Pathology Unit, Azienda USL, IRCCS, Arcispedale S Maria Nuova, Reggio Emilia, Italy
- 7Department of Medicine and Surgery, Unit of Pathology, University of Parma, Parma, Italy
- 8Department of Dermatology, University of Modena and Reggio Emilia, Modena, Italy
- 9Department of Oncology and Advanced Technologies, Unit of Dermatology, Azienda USL, IRCCS, Arcispedale S Maria Nuova, Reggio Emilia, Italy
- 10Department of Molecular Medicine, University of Pavia, Pavia, Italy
We report a 7-year-old boy born with epidermal nevi (EN) arranged according to Blaschko’s lines involving the face and head, right upper limb, chest, and left lower limb, who developed a left paratesticular embryonal rhabdomyosarcoma at 18 months of age. Parallel sequencing identified a gain-of-function variant (c.37G>C, p.Gly13Arg) of HRAS in both epidermal nevus and tumor but not in leukocytes or buccal mucosal epithelial cells, indicating its postzygotic origin. The variant accounted for 33% and 92% of the total reads in the nevus and tumor DNA specimens, respectively, supporting additional somatic hits in the latter. DNA methylation (DNAm) profiling of the tumor documented a signature consistent with embryonal rhabdomyosarcoma and CNV array analysis inferred from the DNAm arrays and subsequent MLPA analysis demonstrated copy number gains of the entire paternal chromosome 11 carrying the mutated HRAS allele, likely as the result of paternal unidisomy followed by subsequent gain(s) of the paternal chromosome in the tumor. Other structural rearrangements were observed in the tumours, while no additional pathogenic variants affecting genes with role in the RAS-MAPK and PI3K-AKT-MTOR pathways were identified. Our findings provide further evidence of the contribution of “gene dosage” to the multistep process driving cell transformation associated with hyperactive HRAS function.
Introduction
The first description of a patient with a “pigmented patch” forming a linear pattern running exactly down the midline of the body, from the forehead to the groin, dates back to 1945 (Zlotnikoff, 1945). Epidermal nevi (EN) are developmental abnormalities in epidermal proliferation and are typically noted at birth or within the first year; they are related but distinct from sebaceous organoid nevi, although the two entities frequently coexist. The linear pattern often follows Blaschko’s lines, which are believed to represent dorsoventral migration patterns of neuroectodermal precursor cells during embryogenesis (Aslam et al., 2014). EN are estimated to occur in 1-3:1,000 live births, with males and females being equally affected (Garcia-Vargas et al., 2008). Although EN may occur as isolated findings, extensive evidence indicates that they are commonly associated with heterogeneous clinical features (Groesser et al., 2013), representing mosaic forms of genetically inherited conditions, as a result of postzygotic activating variants affecting genes coding for signal transducers positively controlling the RAS-MAPK and PI3K-AKT-MTOR signalling cascades (Hafner et al., 2006; Hafner et al., 2007; Hafner et al., 2012; Groesser et al., 2013). Both pathways play a key role in the control of multiple cellular processes, including proliferation, survival and energetic metabolism (Gadea et al., 2020), and their dysregulation represents a common event in cancer. Pediatric patients with widespread distribution of EN developing tumours (e.g., rhabdomyosarcomas, urothelial carcinomas, and squamous cell carcinomas) at young ages have been reported (Bourdeaut et al., 2010; Brandling-Bennett and Morel, 2010; Hafner et al., 2011). Among these, the onset of rhabdomyosarcoma in patients with EN harbouring somatic mutations in KRAS has been described (Om et al., 2017; Gadea et al., 2020; Chang et al., 2021).
Here, we report on a pediatric patient showing a diffuse EN at birth associated with a postzygotic activating missense variant in HRAS (p.Gly13Arg), who developed a left paratesticular embryonic rhabdomyosarcoma. Loss of the maternal chromosome and gains of the entire paternal chromosome 11 carrying the mutated HRAS allele, was detected in the tumour sample. This finding provides further evidence of the gene dosage effect contributing to HRAS-driven oncogenesis.
Materials and methods
Parallel sequencing analysis
Genomic DNA was extracted from the skin biopsy, peripheral blood, buccal brush and rhabdomyosarcoma tissue of the patient, and parallel sequencing was performed on them using a panel comprising 250 genes associated with tumor predisposition and progression, looking for mutations.
A panel including 250 genes associated with tumor predisposition and progression was scanned for mutations. Target enrichment and library preparation were performed using a custom-designed SeqCap EZ Hyper Cap Library assay (ROCHE, Pleasanton, CA, United States), and sequencing was performed on an Illumina NextSeq550 platform (Illumina, San Diego, CA, United States). A previously described in-house bioinformatics pipeline was used for data analysis (Lin et al., 2021; Radio et al., 2021). Reads alignment against the UCSC hg19 reference genome and variant calling (HaplotypeCaller v3.7) were attained following the GATK best practices (Van der Auwera et al., 2013). Variant annotation included occurrence in public (gnomAD) and in-house population databases, predicted functional relevance [e.g., CADD v1.4 (Kircher et al., 2014), M-CAP v1.3 (Jagadeesh et al., 2016)], and classification according to the American College of Medical Genetics and Genomics criteria (InterVar v2.0.1). Mean target coverage (>400x) was set to allow the identification of both germline and somatic events, and trio genotyping made it possible to infer allele transmission of variants of interest.
DNA methylation profiling microarray analysis
DNA methylation (DNAm) profiling was performed using the Illumina Infinium Human Methylation EPIC Bead Chip (EPIC) arrays (Illumina, San Diego, CA, United States) according to the manufacturer’s instructions, as previously reported (Miele et al., 2020). Tumour areas with the highest tumour cell content (≥70%) were selected from the formalin-fixed paraffin embedded (FFPE) tissue slides for DNA extraction. 250 ng genomic DNA were used as input material. The generated DNAm data were compared with the Heidelberg sarcoma classifier v12.2 (Koelsche et al., 2021) to assign a subgroup score for the tumour against the identified 62 different sarcoma methylation classes. The same array data were used to identify copy number alterations as previously described (Capper et al., 2018). Integrative Genomic Viewer (IGV) was used for graphical visualization of structural rearrangements and mapping genes onto regions of interest.
MS-MLPA and microsatellite analysis
Complementary approaches were adopted to characterize the structural rearrangements involving chromosome 11 and the HRAS gene using genomic DNA obtained from patient’s peripheral blood leukocytes, nevus and tumour samples. First, methylation-specific multiplex ligation-dependent probe amplification (MS-MLPA) was carried out using the MS-MLPA probe (ME030 C3-0121, MRC-Holland, Amsterdam, Netherlands) to confirm the copy number changes and analyze the CpG island methylation status of the 11p15 region. The assay was performed according to the manufacturer’s instructions. The products were run on an ABI 3500 Dx genetic analyzer (Applied Biosystems) and then analyzed using Coffalyser.net software (v.210,604.1451).
The evaluation of the parental origin of the mutated HRAS allele and duplicated chromosome 11 was evaluated on DNA extracted from patient’s peripheral blood leukocytes, nevus and tumour samples and his parents (peripheral blood leukocytes) through the analysis of fourteen microsatellites (D11S4046, D11S1338, D11S902, D11S904, D11S935, D11S905, D11S4191, D11S1314, D11S901, D11S4175, D11S925, D11S4151, D11S1320, and D11S968; panel 13 Thermo Fisher Scientific, Waltham, Massachusetts, United States) by automated sequencing.
Results
Clinical phenotype
The boy was the first child of healthy, non-consanguineous parents. The pregnancy was complicated by hypothyroidism in the mother, who was not exposed to any known teratogens. Fetal movements were described as normal. Prenatal ultrasound scans were normal. The proband was born at 40 weeks of gestation, delivered by Caesarean section due to dynamic dystocia. At birth, weight was 3,610 g (25th percentile), length was 49 cm (85th percentile), and head circumference was 36.5 cm (97th percentile). Apgar scores were 9 at 1 min and 10 at 5 min, respectively. He was admitted to the neonatal department due to diffuse proliferation of EN on the face and head, right upper limb, thorax, and left lower limb (Figures 1A–C).
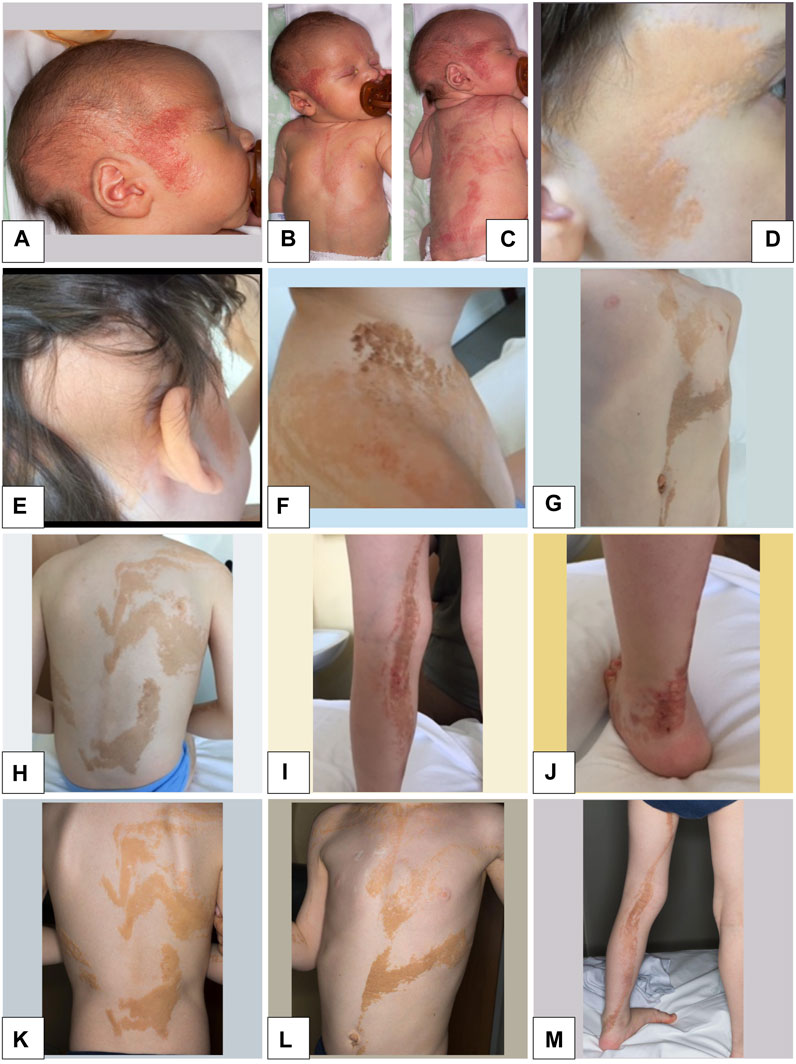
FIGURE 1. Epidermal nevi in the proband. (A–C): At Birth: diffuse epidermal nevi (EN) on the face and head, right upper limb, thorax, left lower limb, arranged according to Blaschko’s lines. (D–M): Age 7 years 2 months: diffuse EN of the face and head, right upper limb, thorax, left lower limb with rather irregular margins, partially confluent and with linear disposition to the limbs according to Blaschko’s lines of pink-orange color.
Psychomotor development was normal, with head control at 2 months, sitting without support at 6 months, autonomous walking at 18 months, first words at 18 months. At present, he attends primary school with good grades.
At the age of 18 months he developed a left paratesticular embryonic rhabdomyosarcoma, for which he was operated and underwent standard chemotherapy treatment of vincristine and actinomycin-D (Arndt et al., 2009; Oberlin et al., 2012).
At last clinical examination (7 years 2 months), height was 115.5 cm (10th percentile), weight was 17.5 Kg (<third percentile), and head circumference was 51 cm (25th-50th percentile). Pubertal stages (Tanner) were A0 P1 B1, and volume of the right testicle was 1–2 mL. He had high forehead in absence of facial dysmorphisms. He showed diffuse EN of the face and head, right upper limb, thorax, and left lower limb, with rather irregular margins, partially confluent and with linear disposition to the limbs according to Blaschko’s lines of pink-orange colour (Figures 1D–M). Some lesions were more prominent and browner in the neck and ankle, where they appeared thickened, particularly at the sites of microtrauma (Figures 1F, J).
Transfontanellar ultrasound, abdominal ultrasound, audiometric evaluation including ABR, brain MRI, abdominal MRI, EEG and ophthalmological evaluation were all normal. Echocardiography revealed mild patent foramen ovale.
The histological examination of the paratesticular lesion revealed a malignant ectomesenchymoma consisting of embryonic spindle cell-variant rhabdomyosarcoma with neuroectodermal differentiation component of ganglioneuroma type (Figures 2A–D). RT-PCR analysis was performed to assess MyoD1 expression, which was positive, and PAX3-FOX01, PAX7-FOX01, and PAX3-NCOA1 rearrangements, which were negative.
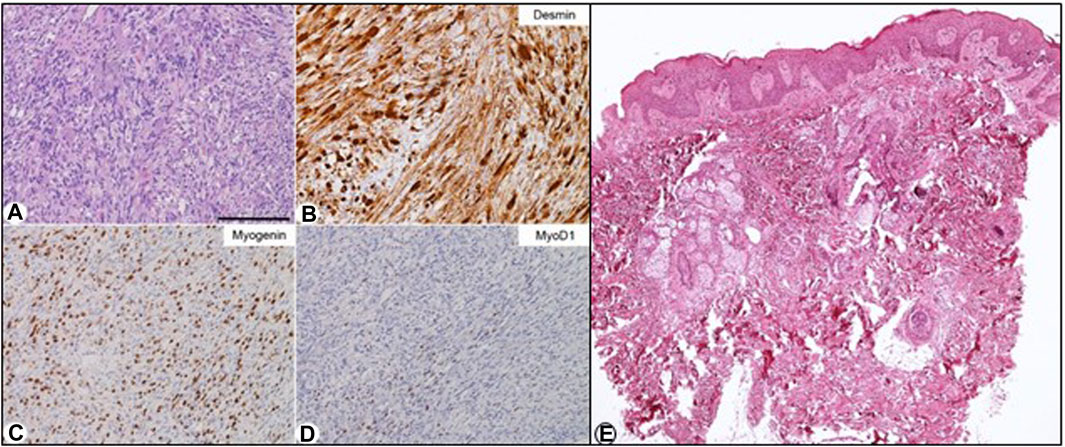
FIGURE 2. Biopsy and histological examination of the left paratesticular embryonic rhabdomyosarcoma. (A–D): Histopathology. (A) Medium-power histopathological view of this tumour, which consisted of a number of atypical spindled cells with eosinophilic cytoplasms. In this setting, occasional ganglion-like cells were present in some places. The tumour cells were immunohistochemically decorated by an anti-desmin antibody (B), anti-myogenin [(C); about 60% of the cells], and anti-MyoD1 [(D); about 10%–20% of the cells] antibodies. Stainings: (A) hematoxylin-eosin; (B–D): Immunohistochemical reactions revealed by 3,3′-Diaminobenzidine (DAB) and mildly counterstained by Harris hematoxylin. (E) Skin biopsy: histological examination of the skin of the back compatible with epidermal nevus: Mild irregular hyperplasia and focal hyperkeratosis of the epidermis and few inflammatory cells in the superficial dermis. Original magnification: (A–D): ×20 (bar is 250 μm), (E) EEx40.
The histopathological examination of a skin biopsy from the patient’s back documented irregular hyperplasia of the epidermis, acanthosis, and hyperkeratosis, consistent with EN histology (Figure 2E).
Molecular analyses
Parallel sequencing directed to scan a selected panel of genes using genomic DNA obtained from the skin lesions, blood, buccal brush and rhabdomyosarcoma tissue samples was performed, allowing the identification of a somatic pathogenic missense variant in the HRAS gene (c.37G>C, p.Gly13Arg; NM_005343.4) in both the epidermal nevus and tumour tissues. Variant reads accounted for 33% and 92% of total reads in the nevus and tumour, respectively, supporting the loss of the wild-type allele in the latter (Figure 3A). The variant was not detected in the DNA samples extracted from blood and buccal brush, indicating its postzygotic origin.
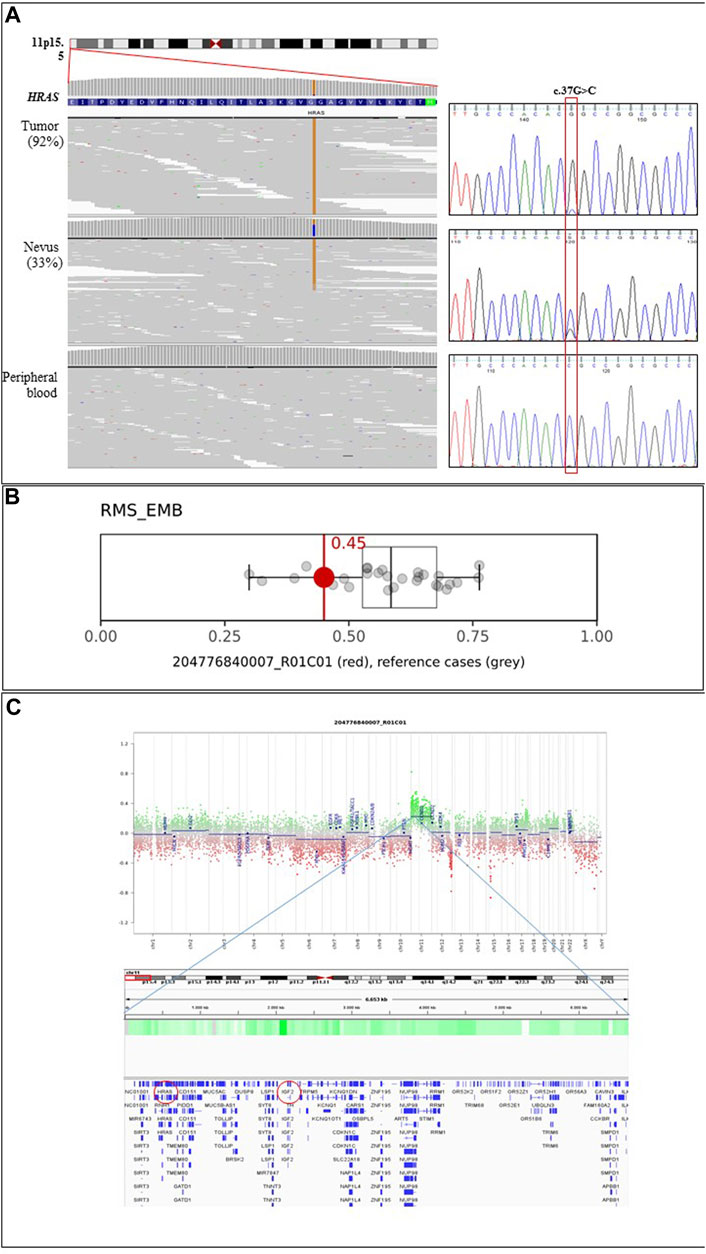
FIGURE 3. Molecular analyses. (A) The c.37G>C missense variant (p.Gly13Arg) in HRAS is found at the homozygous state in the tumour specimen of the patient, while it is observed as a heterozygous change in the epidermal nevus of the same subject. Parallel sequencing output of the portion of the first coding exon of the HRAS gene (ENST00000311189.7) is shown on the left. The variant is not observed in leukocytes. The panels show the reads alignments of three tested tissues. The variant accounted for 33% and 92% of total reads in the EN and tumour, respectively. Chromatograms from Sanger sequencing validation are reported on the right. Absence of the variant in peripheral blood documents the somatic origin of the variant. (B) Box-and-whisker plots depicting the maximum raw classification scores (0.45) of the tumour sample in the methylation class “Rhabdomyosarcoma, Embryonal (RMS_EMB)” according to Sarcoma Classifier v12.2 (https://www.molecularneuropathology.org/mnp/classifier/9). Grey dots represent the reference cases in the methylation class. (C) ERMS Copy Number Variation plots (upper panel) inferred from DNAm data and IGV snapshot (lower panel) detailing the genes mapping in the indicated duplicated regions. Red circles highlight HRAS and IGF2 genes.
DNAm microarray analysis of the proband’s tumour sample provided a profile that was consistent with the episignature characterizing embryonic rhabdomyosarcomas, showing a calibrated score of 0.99 (Figure 3B and Supplementary Table S1), in line with the pathological findings. Copy number variation analysis documented a gain of the entire chromosome 11, indicating duplication events involving the chromosome carrying the mutated HRAS allele (Figure 3C). Two genetic losses were also observed, involving chromosomes 12 (q24.21-q24.33; chr12:120,182,403_133,841,900) and 15 (q12-q13.2; chr15:28,418,871_30,574,776) (Supplementary Figure S1). The deletion on chromosome 12 involves 184 annotated genes, whose germline pathogenic variants have been associated with neurodevelopmental disorders; to date, this region has not been reported to substantially contribute to oncogenesis. The loss on chromosome 15 contains 36 genes, and considering both germline and somatic events, no information is available on its involvement in the tumourigenesis.
Chromosome 11 contains two clusters of imprinted genes located in the 11p15.5 region regulated by separate Imprinting Control regions (IC1 and IC2). To assess the parental origin of the somatic mutation we first performed the MS-MLPA analysis, which showed an altered methylation index of 0.93 on IC1 and a methylation index of 0.12 on IC2 (Figure 4A), which was consistent with an amplification of the paternal chromosome as reported by Jiang et al. (2021). This finding was confirmed by microsatellite analysis (Figure 4B). Overall, the molecular analyses support a complex series of somatic events involving the HRAS allele of the paternal chromosome 11 consisting of an intragenic activating mutation in the EN, a duplication of the mutated allele with loss of the wild-type allele possibly due to a parental unidisomy event, and subsequent gain(s) of the duplicated chromosome.
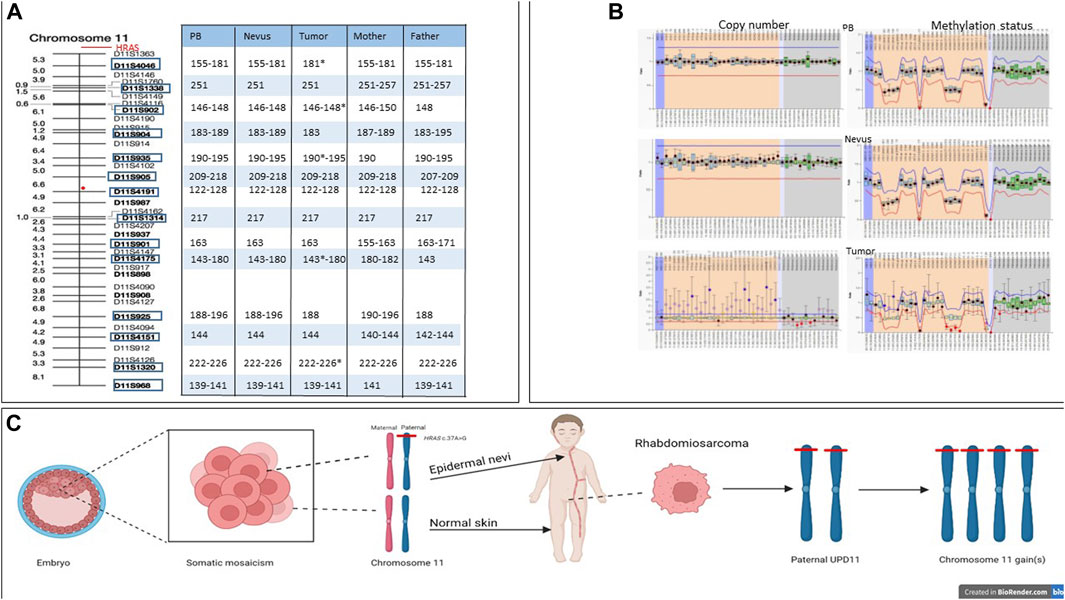
FIGURE 4. Microsatellite and MLPA analyses and multi-hit model in HRAS-associated neoplastic transformation. (A) Microsatellite analysis of patient’s peripheral blood normal and tumor tissues and peripheral blood of his parents, showing length of each marker for each tissues. The red circle indicates the centromere position. Stars indicate alleles with increased copy number. (B) MLPA analysis of normal tissue (upper plot), nevus (middle plot) and tumor tissue (lower plot). (C) The c.37G>C missense variant (p.Gly13Arg) arose postzygotically in the paternal HRAS allele, resulting in diffuse EN along Blaschko’s line. Later, in the left paratestis, paternal unidisomy of chromosome 11 and subsequent gain(s) of the same chromosome carrying the mutated HRAS allele is hypothesized.
Review of the literature
A systematic literature search was performed using PubMed (https://www.ncbi.nlm.nih.gov/pubmed; data freeze 28 March 2023) to identify reports on patients presenting with molecularly characterized EN by using the following search equations: (“epidermal nevi”) AND (“somatic mutation”); (“epidermal nevi”) AND (“HRAS”). One hundred and thirty-four papers, published between 2002 and 2023, were retrieved. Thirty-seven articles reported somatic variants in the FGFR3, FGFR2, PIK3CA, KRAS, and HRAS genes from 90 patients presenting with EN. Among these, 61 (68%) cases presented alterations in RAS genes. The gene variants, analyzed tissues and clinical features of the patients are summarized in Supplementary Table S2. Thirty-two patients (35%) had developed tumours, which were benign in 19 subjects (59%) and malignant neoplasms in 13 (41%). Among the 19 patients who developed benign tumours, only one (6%) carried somatic variant in KRAS, 16 (84%) showed a mutated HRAS allele, and two (10%) had mutations in PIK3CA genes; the 13 patients with cancer harboured somatic variants exclusively in the KRAS (31%) and HRAS (69%) genes.
Discussion
We describe the second case of a somatic variant in HRAS (c.37G>C, p.Gly13Arg) causing EN and contributing to rhabdomyosarcoma, the latter process involving multiple gains of the entire paternal chromosome 11 carrying the mutated HRAS allele.
Rhabdomyosarcoma is the most common soft-tissue sarcoma of childhood (Ognjanovic et al., 2009). It is classified by histology into two main subtypes, alveolar (ARMS) and embryonal (ERMS), having distinct molecular and clinical profiles. Somatic mutations in RAS genes are frequently found in EN and are also involved in the onset of ERMS, being observed in approximately 25% of the cases (Agaram et al., 2022). To date, five patients carrying somatic mutation in codon 35 of KRAS and codon 13 of HRAS with extensive EN and rhabdomyosarcoma have been reported (Bourdeaut et al., 2010; Om et al., 2017; Chang et al., 2021; Luo et al., 2021; Davies et al., 2022). Two additional patients have been described without molecular characterization (Rongioletti and Rebora, 1991; Vidaurri-de la Cruz et al., 2004). RAS proteins are molecular switches controlling the activation of several signal transduction pathways, most of which supporting cell proliferation and survival, and are commonly mutated in cancer (Bos, 1989). Germline mutations in these genes also underlie a well-defined group of developmental disorders collectively known as “RASopathies”, which are characterized by variable cancer predisposition (Tartaglia and Gelb, 2010). Among the RASopathies, a greater risk of developing malignancy characterizes patients with Costello Syndrome (CS), with a 15% cumulative risk of cancer at age 20, generally represented by RMS, neuroblastoma and bladder cancer (Zampino et al., 2007; Dunnett-Kane et al., 2020). More than 90% of the mutations in CS patients are clustered in codons 12 and 13 (p.Gly12Ala/Ser/Val/Cys/Asp/Glu and p.G13Cys/Asp), constituting a mutational hotspot (Giannoulatou et al., 2013). Similarly, 98% of the RAS gene mutations implicated in cancer involve Gly12, Gly13 and Gln61 (COSMIC database, https://cancer.sanger.ac.uk/cosmic) (COSMIC, 2023), which are key residues for the intrinsic and GAP-mediated RAS GTPase activity, and when mutated result in constitutively active proteins. The somatic p.Gly13Arg substitution in HRAS is frequently found in cancer and EN, while it has not been described in CS patients, suggesting that it may be tolerated only in a mosaic state, resulting lethal as a germline event (Hafner et al., 2012). Of note, while patients with variants involving the PI3K-AKT pathway have been associated with benign neoplasms (e.g., lipomas and trichoblastomas), malignancies (e.g., rhabdomyosarcomas and bladder cancer) have exclusively been reported in patients with EN associated with somatic variants of RAS genes (Supplementary Table S2).
We detected a variant allele frequency (VAF) of 33% in the EN. The same variant was not detected in blood and buccal brush samples, supporting its post-zygotic origin, in line with previous reports (Supplementary Table S2). The same variant showed an increased VAF (92%) in the tumor, which is in line with previous findings reported in Costello Syndrome indicating a second somatic hit generally associated with uniparental disomy (UPD) (Kratz et al., 2007). CNV and MLPA analyses performed on the tumor tissue, however, indicated the occurrence of additional entire chromosome 11 gain(s) (Figure 3C and Figure 4B), which had not previously been observed (Hafner et al., 2011; Parham and Barr, 2013; Avitan-Hersh et al., 2014; Li et al., 2014; Farschtschi et al., 2015; Om et al., 2017; Gadea et al., 2020), further indicating a role of a dosage effect for the mutated HRAS allele in tumorigenesis.
Gain of chromosome 8 represents the most common cytogenetic alteration described in ERMS (70%). Additional recurrent structural rearrangements involve chromosomes 2, 7, 11, 12, 13, 20 (gains, 25%–50%) and chromosomes 9 and 10 (losses, 20%–30%) (Bourdeaut et al., 2010; Parham and Barr, 2013; Clay et al., 2021). However, a major hallmark of ERMS is the occurrence of UPD at 11p15.5, a region that contains HRAS and a cluster of imprinted genes (Kratz et al., 2007; Robbins et al., 2016), including IGF2 (Figure 3C), whose increased RNA expression is a characteristic of these tumors. The molecular mechanisms underlying IGF2 overexpression can be due to either loss of heterozygosity (LOH) or loss of imprinting (LOI) at the 11p15.5 locus (Martins et al., 2011). In our patient, we demonstrated that the activating HRAS mutation arose on the paternal allele and the consequent gain(s) of that chromosome was possibly the result of an UPD with loss of the maternal allele followed by subsequent gain(s) of the paternal chromosome, the latter representing an additional hit contributing to neoplastic transformation (Figure 4C). Unfortunately, no information on the parental origin of the HRAS alleles carrying the pathogenetic variants in the previously reported cases is available.
As reported by Hobbs et al. (2016) and in the COSMIC database (COSMIC, 2023), different mutations in RAS genes are associated with predisposition to specific tumors, therefore molecular analysis is recommended in newborns presenting diffuse EN to detect pathogenic variants. The risk of cancer in patients harboring somatic mutations depends on the stage of embryonic development in which the variant occurs and therefore on the number and type of tissues involved. Although the incidence and the individual risk of tumors in patients with EN is not known, it is conceivable that patients with EN might have an increased risk of non-cutaneous malignancies of various organs during life; therefore, it would be advisable to follow these patients periodically in order to limit the risk of carcinogenesis from early childhood. In line with these considerations, the present findings outline the importance of a careful follow-up of these patients requiring periodic oncohematological and dermatological evaluations, urine tests, and kidney, urinary tract and thyroid ultrasound. Furthermore, given the high incidence of cardiac anomalies (60%–90%) associated with germline RASopathies (Calcagni et al., 2020), cardiac evaluation with echocardiogram at least at the time of diagnosis might be also indicated in these patients. We appreciate that, while the development of therapies targeting RAS proteins has been challenging (Moore et al., 2020; Conroy et al., 2021), new opportunities resulting from the development of inhibitors that are able to target RAS proteins in their active or inactive state are emerging, and therapeutic strategies for targeting oncogenic RAS are currently explored in clinical trials (Moore et al., 2020; Punekar et al., 2022), making even more relevant the identification of the driver somatic variants for a more effective surveillance and care.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/snp/, rs104894228.
Ethics statement
The studies involving humans were approved by Ethical Committee of the Ospedale Pediatrico Bambino Gesù (ref. 1702_OPBG_2018). The studies were conducted in accordance with the local legislation and institutional requirements. The human samples used in this study were acquired from primarily isolated as part of your previous study for which ethical approval was obtained. Written informed consent for participation was not required from the participants or the participants’ legal guardians/next of kin in accordance with the national legislation and institutional requirements. Written informed consent was obtained from the minor(s)’ legal guardian/next of kin for the publication of any potentially identifiable images or data included in this article. Written informed consent was obtained from the participant/patient(s) for the publication of this case report.
Author contributions
Conceptualization: LG, OZ, and MT. Methodology: RZ, SGC, and LP. Investigation: LP, EM, SPiz, SPia, DC, and FCR. Data collection: CC, CL, SG, EF, AB, PB, MM, and AM. Data curation: RZ, SGC, CC, and LG. Writing–original draft preparation: RZ, CC, and SGC. Writing–review and editing: MT, LG, OZ, and PB. Supervision: LG, OZ, and MT. All authors contributed to the article and approved the submitted version.
Funding
This work was supported, in part, by funding from Fondazione Bambino Gesù (Vite Coraggiose), Italian Ministry of Health (CCR-2017-23669081, 5x1000_2019), and AIRC (IG 21614) to MT, and partially supported by Italian Ministry of Health–Ricerca Corrente Annual Program 2024 to LG
Acknowledgments
This work has been generated within the European Reference Network on Rare Congenital Malformations and Rare Intellectual Disability (ERN-ITHACA) [EU Framework Partnership Agreement ID: 3HP-HP-FPA ERN-01-2016/739516]. We are very grateful to the parents for their cooperation in providing the medical data and photographs necessary for this publication and for their willingness to share information about their child and family for the purpose of advancing science and providing clinical care to similarly affected families. The authors also wish to thank the photographers Marco Bonazzi and Luca Valcavi and Jonathan MacKenzie for reading the manuscript and improving the English. The authors are grateful for the contribution made by the Fondazione Cassa di Risparmio Manodori of Reggio Emilia.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2023.1231434/full#supplementary-material
References
Agaram, N. P., Huang, S-C., Tap, W. D., Wexler, L. H., and Antonescu, C. R. (2022). Clinicopathologic and survival correlates of embryonal rhabdomyosarcoma driven by RAS/RAF mutations. Genes. Chromosom. Cancer 61, 131–137. doi:10.1002/gcc.23010
Arndt, C. A., Stoner, J. A., Hawkins, D. S., Rodeberg, D. A., Hayes-Jordan, A. A., Paidas, C. N., et al. (2009). Vincristine, actinomycin, and cyclophosphamide compared with vincristine, actinomycin, and cyclophosphamide alternating with vincristine, topotecan, and cyclophosphamide for intermediate-risk rhabdomyosarcoma: children's oncology group study D9803. J. Clin. Oncol. 27, 5182–5188. doi:10.1200/JCO.2009.22.3768
Aslam, A., Salam, A., Griffiths, C. E., and McGrath, J. A. (2014). Naevus sebaceus: a mosaic RASopathy. Clin. Exp. Dermatol 39, 1–6. doi:10.1111/ced.12209
Avitan-Hersh, E., Tatur, S., Indelman, M., Gepstein, V., Shreter, R., Hershkovitz, D., et al. (2014). Postzygotic HRAS mutation causing both keratinocytic epidermal nevus and thymoma and associated with bone dysplasia and hypophosphatemia due to elevated FGF23. J. Clin. Endocrinol. Metab. 99, E132–E136. doi:10.1210/jc.2013-2813
Bessis, D., Plaisancié, J., Gaston, V., and Bieth, E. (2017). Fibroblast growth factor receptor 3 epidermal naevus syndrome with urothelial mosaicism for the activating p.Ser249Cys FGFR3 mutation. Acta Derm. Venereol. 97, 402–403. doi:10.2340/00015555-2554
Bourdeaut, F., Hérault, A., Gentien, D., Pierron, G., Ballet, S., Reynaud, S., et al. (2010). Mosaicism for oncogenic G12D KRAS mutation associated with epidermal nevus, polycystic kidneys and rhabdomyosarcoma. J. Med. Genet. 47, 859–862. doi:10.1136/jmg.2009.075374
Brandling-Bennett, H. A., and Morel, K. D. (2010). Epidermal nevi. Pediatr. Clin. North Am. 57, 1177–1198. doi:10.1016/j.pcl.2010.07.004
Bygum, A., Fagerberg, C. R., Clemmensen, O. J., Fiebig, B., and Hafner, C. (2011). Systemic epidermal nevus with involvement of the oral mucosa due to FGFR3 mutation. BMC Med. Genet. 12, 79. doi:10.1186/1471-2350-12-79
Calcagni, G., Gagliostro, G., Limongelli, G., Unolt, M., De Luca, E., Digilio, M. C., et al. (2020). Atypical cardiac defects in patients with RASopathies: updated data on CARNET study. Birth Defects Res. 112, 725–731. doi:10.1002/bdr2.1670
Capper, D., Jones, D., Sill, M., Hovestadt, V., Schrimpf, D., Sturm, D., et al. (2018). DNA methylation-based classification of central nervous system tumours. Nature 555, 469–474. doi:10.1038/nature26000
Chang, C. A., Perrier, R., Kurek, K. C., Estrada-Veras, J., Lehman, A., Yip, S., et al. (2021). Novel findings and expansion of phenotype in a mosaic RASopathy caused by somatic KRAS variants. Am. J. Med. Genet. Part A 185A, 2829–2845. doi:10.1002/ajmg.a.62356
Chantorn, R., and Shwayder, T. (2011). Phacomatosis pigmentokeratotica: a further case without extracutaneous anomalies and review of the condition. Pediatr. Dermatol. 28, 715–719. doi:10.1111/j.1525-1470.2011.01655.x
Clay, M. R., Patel, A., Tran, Q., Hedges, D. J., Chang, T. C., Stewart, E., et al. (2021). Methylation profiling reveals novel molecular classes of rhabdomyosarcoma. Sci. Rep. 11, 22213. doi:10.1038/s41598-021-01649-w
Collin, B., Taylor, I. B., Wilkie, A. O., and Moss, C. (2007). Fibroblast growth factor receptor 3 (FGFR3) mutation in a verrucous epidermal naevus associated with mild facial dysmorphism. Br. J. Dermatol 156, 1353–1356. doi:10.1111/j.1365-2133.2007.07869.x
Conroy, M., Cowzer, D., Kolch, W., and Duffy, A. G. (2021). Emerging RAS-directed therapies for cancer. Cancer Drug Resist 4, 543–558. doi:10.20517/cdr.2021.07
COSMIC (2023). Cosmic. https://cancer.sanger.ac.uk/cosmic.
Davies, O. M. T., Bruckner, A. L., McCalmont, T., Mascarenhas, L., Oza, V., Williams, M. L., et al. (2022). Cutaneous mosaic RASopathies associated with rhabdomyosarcoma. Pediatr. Blood Cancer 69, e29639. doi:10.1002/pbc.29639
Denorme, P., Morren, M. A., Hollants, S., Spaepen, M., Suaer, K., Zutterman, N., et al. (2018). Phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit alpha (PIK3CA)-related overgrowth spectrum: a brief report. Pediatr. Dermatol 35, e186–e188. doi:10.1111/pde.13441
Dunnett-Kane, V., Burkitt-Wright, E., Blackhall, F. H., Malliri, A., Evans, D. G., and Lindsay, C. R. (2020). Germline and sporadic cancers driven by the RAS pathway: parallels and contrasts. Ann. Oncol. 31, 873–883. doi:10.1016/j.annonc.2020.03.291
Farschtschi, S., Mautner, V. F., Hollants, S., Hagel, C., Spaepen, M., Schulte, C., et al. (2015). Keratinocytic epidermal nevus syndrome with Schwann cell proliferation, lipomatous tumour and mosaic KRAS mutation. BMC Med. Genet. 16, 6. doi:10.1186/s12881-015-0146-5
Gadea, A., Hernández-Muñoz, I., Vicente, A., Andrades, E., García-Calvente, M., Camacho, L., et al. (2020). Molecular characterisation of oncogenic urothelial mosaic mutations in patients withextensive keratinocytic epidermal naevi. J. Med. Genet. 57, 601–604. doi:10.1136/jmedgenet-2019-106296
Garcia-Vargas, A., Hafner, C., Perez-Rodriguez, A. G., Rodrı ´guez-Rojas, L. X., Gonza´lez-Esqueda, P., Stoehr, R., et al. (2008). An epidermal nevus syndrome with cerebral involvement caused by a mosaic FGFR3 mutation. Am. J. Med. Genet. A 146A, 2275–2279. doi:10.1002/ajmg.a.32429
Giannoulatou, E., McVean, G., Taylor, I. B., McGowan, S. J., Maher, G. J., Iqbal, Z., et al. (2013). Contributions of intrinsic mutation rate and selfish selection to levels of de novo HRAS mutations in the paternal germline. Proc. Natl. Acad. Sci. U. S. A. 110, 20152–20157. doi:10.1073/pnas.1311381110
Groesser, L., Herschberger, E., Sagrera, A., Shwayder, T., Flux, K., Ehmann, L., et al. (2013). Phacomatosis pigmentokeratotica is caused by a postzygotic HRAS mutation in a multipotent progenitor cell. J. Invest. Dermatol 133, 1998–2003. doi:10.1038/jid.2013.24
Hafner, C., van Oers, J. M., Vogt, T., Landthaler, M., Stoehr, R., Blaszyk, H., et al. (2006). Mosaicism of activating FGFR3 mutations in human skin causes epidermal nevi. J. Clin. Invest. 116, 2201–2207. doi:10.1172/JCI28163
Hafner, C., López-Knowles, E., Luis, N. M., Toll, A., Baselga, E., Fernández-Casado, A., et al. (2007). Oncogenic PIK3CA mutations occur in epidermal nevi and seborrheic keratoses with a characteristic mutation pattern. Proc. Natl. Acad. Sci. U. S. A. 104, 13450–13454. doi:10.1073/pnas.0705218104
Hafner, C., Toll, A., and Real, F. X. (2011). HRAS mutation mosaicism causing urothelial cancer and epidermal nevus. N. Engl. J. Med. 365, 1940–1942. doi:10.1056/NEJMc1109381
Hafner, C., Toll, A., Gantner, S., Mauerer, A., Lurkin, I., Acquadro, F., et al. (2012). Keratinocytic epidermal nevi are associated with mosaic RAS mutations. J. Med. Genet. 49, 249–253. doi:10.1136/jmedgenet-2011-100637
Hobbs, G. A., Der, C. J., and Rossman, K. L. (2016). RAS isoforms and mutations in cancer at a glance. J. Cell. Sci. 129, 1287–1292. doi:10.1242/jcs.182873
Honda, A., Umegaki-Arao, N., Sasaki, T., Nakabayashi, K., Hata, K., Matsubara, Y., et al. (2017). Somatic HRAS p.G12S mosaic mutation causes unilaterally distributed epidermal nevi, woolly hair and palmoplantar keratosis. J. Dermatol 44, e109–e110. doi:10.1111/1346-8138.13801
Huang, L., Zhang, J., Chen, F., Zhang, X., Cao, Y., Cheng, R., et al. (2022). The first case of Chinesephacomatosis pigmentokeratotica diagnosed by a missenseHRAS mosaicism. J. Dermatol 00, 921–924. doi:10.1111/1346-8138.16434
Igawa, S., Honma, M., Minami-Hori, M., Tsuchida, E., Iizuka, H., and Ishida-Yamamoto, A. (2016). Novel postzygotic KRAS mutation in a Japanese case of epidermal nevus syndrome presenting with two distinct clinical features, keratinocytic epidermal nevi and sebaceous nevi. J. Dermatol 43, 103–104. doi:10.1111/1346-8138.13153
Jagadeesh, K., Wenger, A., Berger, M., Guturu, H., Stenson, P., Cooper, D., et al. (2016). M-CAP eliminates a majority of variants of uncertain significance in clinical exomes at high sensitivity. Nat. Genet. 48 (12), 1581–1586. doi:10.1038/ng.3703
Jiang, H., Ping, Z., Wang, J., Liu, X., Jin, Y., Li, S., et al. (2021). A Beckwith-Wiedemann syndrome case with de novo 24 Mb duplication of chromosome 11p15.5p14.3. Mol. Cytogenet 14 (1), 14. doi:10.1186/s13039-021-00532-7
Keppler-Noreuil, K. M., Rios, J. J., Parker, V. E., Semple, R. K., Lindhurst, M. J., Sapp, J. C., et al. (2015). PIK3CA-related overgrowth spectrum (PROS): diagnostic and testing eligibility criteria, differential diagnosis, and evaluation. Am. J. Med. Genet. A 167A, 287–295. doi:10.1002/ajmg.a.36836
Kircher, M., Witten, D. M., Jain, P., O'Roak, B. J., Cooper, G. M., and Shendure, J. (2014). A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 46, 310–315. doi:10.1038/ng.2892
Kitamura, S., Yanagi, T., Imafuku, K., Hata, H., Fujii, K., Nishihara, H., et al. (2017). Seborrheic keratosis arising on an epidermal nevus with HRAS p.G13R mutation. Int. J. Dermatol 56, e177–e180. doi:10.1111/ijd.13573
Koelsche, C., Schrimpf, D., Stichel, D., Sill, M., Sahm, F., Reuss, D. E., et al. (2021). Sarcoma classification by DNA methylation profiling. Nat. Commun. 12, 498. doi:10.1038/s41467-020-20603-4
Kratz, C. P., Steinemann, D., Niemeyer, C. M., Schlegelberger, B., Koscielniak, E., Kontny, U., et al. (2007). Uniparental disomy at chromosome 11p15.5 followed by HRAS mutations in embryonal rhabdomyosarcoma: lessons from Costello syndrome. Hum. Mol. Genet. 16 (4), 374–379. doi:10.1093/hmg/ddl458
Levinsohn, J. L., Tian, L. C., Boyden, L. M., McNiff, J. M., Narayan, D., Loring, E. S., et al. (2013). Whole-exome sequencing reveals somatic mutations in HRAS and KRAS, which cause nevus sebaceus. J. Invest. Dermatol 133, 827–830. doi:10.1038/jid.2012.379
Levinsohn, J. L., Teng, J., Craiglow, B. G., Loring, E. C., Burrow, T. A., Mane, S. S., et al. (2014). Somatic HRAS p.G12S mutation causes woolly hair and epidermal nevi. J. Invest. Dermatol 134, 1149–1152. doi:10.1038/jid.2013.430
Li, J. Y., Berger, M. F., Marghoob, A., Bhanot, U. K., Toyohara, J. P., and Pulitzer, M. P. (2014). Combined melanocytic and sweat gland neoplasm: cell subsets harbor an identical HRAS mutation in phacomatosis pigmentokeratotica. J. Cutan. Pathol. 41, 663–671. doi:10.1111/cup.12339
Lim, Y. H., Ovejero, D., Sugarman, J. S., Deklotz, C. M., Maruri, A., Eichenfield, L. F., et al. (2014). Multilineage somatic activating mutations in HRAS and NRAS cause mosaic cutaneous and skeletal lesions, elevated FGF23 and hypophosphatemia. Hum. Mol. Genet. 23, 397–407. doi:10.1093/hmg/ddt429
Lim, Y. H., Ovejero, D., and Derrick, K. M.Yale Center for Mendelian Genomics (2016). Cutaneous skeletal hypophosphatemia syndrome (CSHS) is a multilineage somatic mosaic RASopathy. J. Am. Acad. Dermatol 75, 420–427. doi:10.1016/j.jaad.2015.11.012
Lin, Y. C., Niceta, M., Muto, V., Vona, B., Pagnamenta, A. T., Maroofian, R., et al. (2021). SCUBE3 loss-of-function causes a recognizable recessive developmental disorder due to defective bone morphogenetic protein signaling. Am. J. Hum. Genet. 108, 115–133. doi:10.1016/j.ajhg.2020.11.015
Luo, Q., Zhang, Q., Shen, J., Guan, W., Li, M., Zhang, J., et al. (2021). Expanding mutational spectrum of HRAS by a patient withSchimmelpenning–Feuerstein–Mimssyndrome. J. Dermatol 48, 1273–1276. doi:10.1111/1346-8138.15922
Martin, R. J., Arefi, M., Splitt, M., Redford, L., Moss, C., and Rajan, N. (2018). Phacomatosis pigmentokeratotica and precocious puberty associated with HRAS mutation. Br. J. Dermatol 178, 289–291. doi:10.1111/bjd.15643
Martins, A. S., Olmos, D., Missiaglia, E., and Shipley, J. (2011). Targeting the insulin-like growth factor pathway in rhabdomyosarcomas: rationale and future perspectives. Sarcoma 2011, 209736. doi:10.1155/2011/209736
Mestach, L., Polubothu, S., Calder, A., Denayer, E., Gholam, K., Legius, E., et al. (2020). Keratinocytic epidermal nevi associated with localized fibro-osseous lesions without hypophosphatemia. Pediatr. Dermatol 37, 890–895. doi:10.1111/pde.14254
Miele, E., De Vito, R., Ciolfi, A., Pedace, L., Russo, I., De Pasquale, M. D., et al. (2020). DNA methylation profiling for diagnosing undifferentiated sarcoma with capicua transcriptional receptor (CIC) alterations. Int. J. Mol. Sci. 21, 1818. doi:10.3390/ijms21051818
Moore, A. R., Rosenberg, S. C., McCormick, F., and Malek, S. (2020). RAS-Targeted therapies: is the undruggable drugged? Nat. Rev. Drug Discov. 19, 533–552. doi:10.1038/s41573-020-0068-6
Narahira, A., Kitamura, S., Maeda, T., Nishihara, H., and Yanagi, T. (2021). Multiple squamous cell carcinomas arising on an epidermal nevus harboring HRAS p.G13R mutation. J. Dermatol 48, e92–e93. doi:10.1111/1346-8138.15682
Nishihara, K., Tohyama, M., and Kubo, A. (2019). A case of woolly hair nevus, multiple linear pigmentation, and epidermal nevi with somatic HRAS p.G12S mutation. Pediatr. Dermatol 36, 368–371. doi:10.1111/pde.13783
Oberlin, O., Rey, A., Sanchez de Toledo, J., Martelli, H., Jenney, M. E., Scopinaro, M., et al. (2012). Randomized comparison of intensified six-drug versus standard three-drug chemotherapy for high-risk nonmetastatic rhabdomyosarcoma and other chemotherapy-sensitive childhood soft tissue sarcomas: long-term results from the international society of pediatric oncology MMT95 study. J. Clin. Oncol. 30, 2457–2465. doi:10.1200/JCO.2011.40.3287
Ognjanovic, S., Linabery, A. M., Charbonneau, B., and Ross, J. A. (2009). Trends in childhood rhabdomyosarcoma incidence and survival in the United States, 1975-2005. Cancer 115, 4218–4226. doi:10.1002/cncr.24465
Om, A., Cathey, S. S., Gathings, R. M., Hudspeth, M., Lee, J. A., Marzolf, S., et al. (2017). Phacomatosis pigmentokeratotica: a mosaic RASopathy with malignant potential. Pediatr. Dermatol 34, 352–355. doi:10.1111/pde.13119
Ousager, L. B., Bygum, A., and Hafner, C. (2012). Identification of a novel S249C FGFR3 mutation in a keratinocytic epidermal naevus syndrome. Br. J. Dermatol 167, 202–204. doi:10.1111/j.1365-2133.2012.10812.x
Parham, D. M., and Barr, F. G. (2013). Classification of rhabdomyosarcoma and its molecular basis. Adv. Anat. Pathol. 20, 387–397. doi:10.1097/PAP.0b013e3182a92d0d
Punekar, S. R., Velcheti, V., Neel, B. G., and Wong, K. K. (2022). The current state of the art and future trends in RAS-targeted cancer therapies. Nat. Rev. Clin. Oncol. 19, 637–655. PMID: 36028717. doi:10.1038/s41571-022-00671-9
Radio, F. C., Pang, K., Ciolfi, A., Levy, M. A., Hernández-García, A., Pedace, L., et al. (2021). SPEN haploinsufficiency causes a neurodevelopmental disorder overlapping proximal 1p36 deletion syndrome with an episignature of X chromosomes in females. Am. J. Hum. Genet. 108, 502–516. doi:10.1016/j.ajhg.2021.01.015
Robbins, K. M., Stabley, D. L., Holbrook, J., Sahraoui, R., Sadreameli, A., Conard, K., et al. (2016). Paternal uniparental disomy with segmental loss of heterozygosity of chromosome 11 are hallmark characteristics of syndromic and sporadic embryonal rhabdomyosarcoma. Am. J. Med. Genet. A 170 (12), 3197–3206. doi:10.1002/ajmg.a.37949
Rongioletti, F., and Rebora, A. (1991). Epidermal nevus with transitional cell carcinomas of the urinary tract. J. Am. Acad. Dermatol 25 (5), 856–858. doi:10.1016/s0190-9622(08)80987-5
Slack, J. C., Bründler, M. A., Chang, C. A., Perrier, R., Lafay-Cousin, L., and Kurek, K. C. (2021). Bilateral nephroblastic tumors and a complex renal vascular anomaly in a patient with a mosaic RASopathy: novel histopathologic features and molecular insights. Pediatr. Dev. Pathol. 24, 235–240. doi:10.1177/1093526620986502
Tanaka, R., Umegaki-Arao, N., Sasaki, T., Aoki, S., Yoshida, K., Niizeki, H., et al. (2018). Linear keratinocytic epidermal nevi on trunk skin caused by a somatic FGFR2 p.C382R mutation. J. Dermatol 45, e302–e303. doi:10.1111/1346-8138.14344
Tartaglia, M., and Gelb, B. D. (2010). Disorders of dysregulated signal traffic through the RAS-MAPK pathway: phenotypic spectrum and molecular mechanisms. Ann. N. Y. Acad. Sci. 1214, 99–121. doi:10.1111/j.1749-6632.2010.05790.x
Toll, A., Fernández, L. C., Pons, T., Groesser, L., Sagrera, A., Carrillo-de Santa Pau, E., et al. (2016). Somatic embryonic FGFR2 mutations in keratinocytic epidermal nevi. J. Invest. Dermatol 136, 1718–1721. doi:10.1016/j.jid.2016.03.040
Van der Auwera, G. A., Carneiro, M., Hartl, C., Poplin, R., del Angel, G., Levy-Moonshine, A., et al. (2013). From FastQ data to high-confidence variant calls: the genome analysis toolkit best practices pipeline. Curr. Protoc. Bioinforma. 43, 11.10.1–11. doi:10.1002/0471250953.bi1110s43
Vidaurri-de la Cruz, H., Tamayo-Sanchez, L., Duran-McKinster, C., McKinster, C., de la Luz Orozco-Covarrubias, M., and Ruiz-Maldonado, R. (2004). Epidermal nevus syndromes: clinical findings in 35 patients. Pediatr. Dermatol 21, 432–439. doi:10.1111/j.0736-8046.2004.21402.x
Whyte, M. P., Griffith, M., Trani, L., Mumm, S., Gottesman, G. S., McAlister, W. H., et al. (2017). Melorheostosis: exome sequencing of an associated dermatosis implicates postzygotic mosaicism of mutated KRAS. Bone 101, 145–155. doi:10.1016/j.bone.2017.04.010
Wiedemeyer, K., and Hartschuh, W. (2009). Trichoblastomas with merkel cell proliferation in nevi sebacei in schimmelpenning-feuerstein-mims syndrome—Histological differentiation between trichoblastomas and basal cell carcinomas. J. Dtsch. Dermatol Ges. 7, 612–615. doi:10.1111/j.1610-0387.2009.07036.x
Wollenberg, A., Butnaru, C., and Oppel, T. (2002). Phacomatosis pigmentokeratotica (Happle) in a 23-year-old man. Acta Derm. Venereol. 82, 55–57. doi:10.1080/000155502753600911
Zampino, G., Pantaleoni, F., Carta, C., Cobellis, G., Vasta, I., Neri, C., et al. (2007). Diversity, parental germline origin, and phenotypic spectrum of de novo HRAS missense changes in Costello syndrome. Hum. Mutat. 28, 265–272. doi:10.1002/humu.20431
Keywords: epidermal nevus, rhabdomyosarcoma, HRAS, postzygotic mutation, paternal UPD11
Citation: Zuntini R, Cattani C, Pedace L, Miele E, Caraffi SG, Gardini S, Ficarelli E, Pizzi S, Radio FC, Barone A, Piana S, Bertolini P, Corradi D, Marinelli M, Longo C, Motolese A, Zuffardi O, Tartaglia M and Garavelli L (2023) Case Report: Sequential postzygotic HRAS mutation and gains of the paternal chromosome 11 carrying the mutated allele in a patient with epidermal nevus and rhabdomyosarcoma: evidence of a multiple-hit mechanism involving HRAS in oncogenic transformation. Front. Genet. 14:1231434. doi: 10.3389/fgene.2023.1231434
Received: 30 May 2023; Accepted: 01 August 2023;
Published: 10 August 2023.
Edited by:
Dong Li, Children’s Hospital of Philadelphia, United StatesReviewed by:
Talita Aguiar, Columbia University Irving Medical Center, United StatesSankar Jagadeeshan, Ben-Gurion University of the Negev, Israel
Copyright © 2023 Zuntini, Cattani, Pedace, Miele, Caraffi, Gardini, Ficarelli, Pizzi, Radio, Barone, Piana, Bertolini, Corradi, Marinelli, Longo, Motolese, Zuffardi, Tartaglia and Garavelli. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Orsetta Zuffardi, b3JzZXR0YS56dWZmYXJkaUB1bmlwdi5pdA==; Marco Tartaglia, bWFyY28udGFydGFnbGlhQG9wYmcubmV0; Livia Garavelli, bGl2aWEuZ2FyYXZlbGxpQGF1c2wucmUuaXQ=
†These authors have contributed equally to this work