 Yuri Yoh1
Yuri Yoh1 Tadashi Shiohama1*Tomoko Uchida1
Tadashi Shiohama1*Tomoko Uchida1 Ryota Ebata1
Ryota Ebata1 Hironobu Kobayashi1Kentaro Okunushi1
Hironobu Kobayashi1Kentaro Okunushi1 Mitsuhiro Kato2
Mitsuhiro Kato2 Kazuki Watanabe3
Kazuki Watanabe3 Mitsuko Nakashima3
Mitsuko Nakashima3 Hirotomo Saitsu3
Hirotomo Saitsu3 Hiromichi Hamada1
Hiromichi Hamada1- 1Department of Pediatrics, Chiba University Hospital, Chiba, Japan
- 2Department of Pediatrics, Showa University School of Medicine, Tokyo, Japan
- 3Department of Biochemistry, Hamamatsu University School of Medicine, Hamamatsu, Japan
Megalencephaly-capillary malformation syndrome (MCAP, OMIM # 602501) is caused by hyperactivity of the thephosphoinositide-3-kinase (PI3K)–Vakt murine thymoma viral oncogene homolog (AKT)–mammalian target of rapamycin (mTOR) pathway, which results in megalencephaly, capillary malformations, asymmetrical overgrowth, and connective tissue dysplasia. Herein, we report the case of a 7-month-old girl with MCAP due to a PIK3CA somatic mosaic variant who presented with atrial tachycardia, finally diagnosed as pulmonary arterial hypertension (PAH). Oxygen therapy and sildenafil decreased pulmonary blood pressure and improved atrial tachycardia. Previous studies reported an association between the PI3K/AKT/mTOR pathway and abnormal pulmonary arterial smooth muscle cell proliferation, which may be associated with PAH. PAH should be considered a potentially lethal complication in MCAP patients, even when no structural cardiac abnormalities are identified in the neonatal period.
1 Introduction
Megalencephaly-capillary malformation syndrome (MCAP, OMIM # 602501) is an overgrowth disorder characterized by a highly recognizable constellation of features involving the brain and body, including megalencephaly or hemimegalencephaly with perisylvian polymicrogyria, focal or generalized somatic overgrowth, vascular malformations, particularly capillary malformations, digital anomalies, variable connective tissue dysplasia, hypotonia, and mild-to-severe intellectual disability (Mirzaa G. M. et al., 2013). A heterozygous pathogenic variant of the PIK3CA gene has been identified in MCAP, and variants in this gene activate the thephosphoinositide-3-kinase (PI3K)–Vakt murine thymoma viral oncogene homolog (AKT)–mammalian target of rapamycin (mTOR) pathway, which leads to excessive growth in affected tissues (Riviére et al., 2012).
As the symptoms and severity of MCAP vary greatly from one person to another, their life expectancy is also variable. Sudden infant death has been reported in at least 12 children with MCAP (Yano and Watanabe, 2001; Nakamura et al., 2014; reviewed by Harada et al., 2015). The causes of death include heart failure due to congenital heart disease, arrhythmia, and cerebellar tonsillar herniation; however, in many cases, the reasons remain unclear (Harada et al., 2015).
Herein, we report a patient with MCAP with a somatic heterozygous pathogenic variant of PIK3CA who presented with atrial tachycardia and progressive pulmonary arterial hypertension (PAH). Non-structural cardiovascular abnormalities in MCAP have not been reported in detail (Mirzaa G. et al., 2013; Garde et al., 2021), except for the patients with structural heart defects (e.g., atrial and ventricular septal defects) and/or abnormalities of the great vessels (Yano and Watanabe, 2001; Mirzaa G. et al., 2013; Mirzaa et al., 2016), the clinical course in our case may shed light on PAH as a potential life-threatening comorbidity.
2 Case presentation
A girl was referred at 22 weeks of pregnancy because due to large for her gestational age with an estimated weight of 1,072 g (+4.4 SD). She also showed enlarged lateral ventricles and excessive amniotic fluid. She had no family history of genetic disorders or PAH. She was born by cesarean section at 35 weeks and 5 days of age, with a birth weight of 4,391 g (+5.4 SD), height of 52 cm (+3.2 SD), and occipitofrontal circumference of 44 cm (+9.6 SD). She showed macrocephaly, neonatal hypotonia, frontal bossing, cutis marmorata (Figure 1A), hyperextensibility of the lateral abdomen, syndactyly and sandal gap toe on both feet, and polydactyly of the right hand. Brain magnetic resonance imaging on 18 days of age revealed extensive polymicrogyria (Figure 1B) and mild sagging of the central cerebellum (Figure 1C). Her physical and neuroimaging findings were compatible with MCAP.
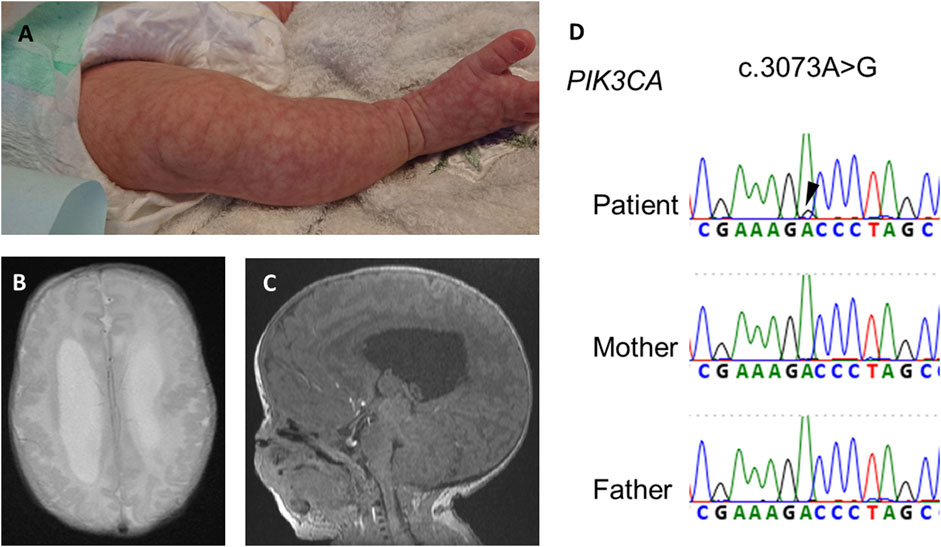
FIGURE 1. Physical and neuroimaging findings at the neonatal period. Cutis marmorata and sandal gap toe in the right leg (A), extensive bilateral polymicrogyria and ventricular dilatation [(B), axial t2-weighted image], and mild sagging of the central cerebellum [(C), sagittal T1-weighted image]. (D) Electropherogram of Sanger sequencing of the patient and his parents. Black arrowheads indicates a somatic mosaic PIK3CA variant.
At 2 months of age, she presented with hypoglycemia due to low serum cortisol. She was diagnosed with adrenal hormone deficiency and was treated with hydrocortisone. In addition, tracheostomy and ventriculoperitoneal shunt were performed for laryngomalacia and hydrocephalus, respectively, at 3 months of age.
At the age of 5 months, she suddenly developed tachycardia of 190–200 beats/min due to ectopic atrial tachycardia. Administrations of digoxin improved her cardiac rhythm, then returned to sinus rhythm. At 7 months of age, she exhibited labored respiration, tachycardia (200–220/min), hypoxia (SpO2 88%), and an accentuated second heart sound. Serum brain natriuretic peptide levels were 722 pg/ml, which suggested cardiac failure. The blood digoxin concentration was within the appropriate range (1.2 ng/ml). Chest X-ray showed cardiomegaly, pulmonary congestion, and consolidation (Figure 2B). Electrocardiogram revealed narrow QRS tachycardia with irregular RR intervals (Figure 2A-a). Echocardiography revealed D-shaped left ventricle (Figure 2C-a) and tricuspid regurgitation, and the estimated right ventricular pressure by the tricuspid regurgitation pressure gradient was 96 mmHg, while her systolic blood pressure was 84 mmHg. Pulmonary vein stenosis was not observed, nor was mitral valve or aortic stenosis. Echocardiography and contrast enhanced computed tomography (CT) revealed no portal vein abnormalities.
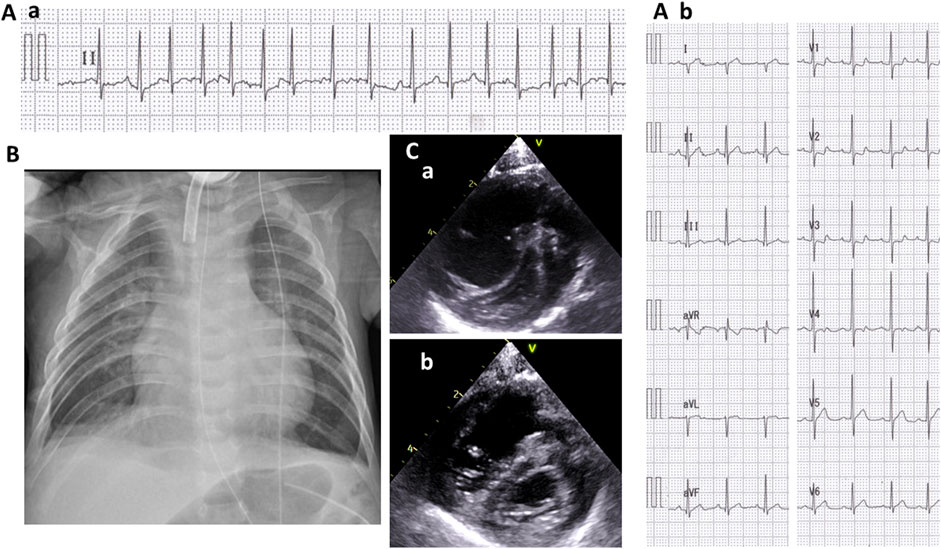
FIGURE 2. Cardiologic evaluation at 7 months old. A II-lead electrocardiogram (ECG) (A-a) when presenting with tachycardia and respiratory distress and a 12-lead ECG (A-b) after recovering sinus rhythm. A chest radiograph showing cardiomegaly, pulmonary congestion, and consolidation (B). A short-axis view of echocardiography when presenting tachycardia and respiratory distress (C-a) and after the initiation of treatment for pulmonary hypertension (C-b).
She was diagnosed with PAH and treated with rest and oxygen (FiO2 0.5). Her rhythm returned to the sinus rhythm (Figure 2A-b), and her heart rate reduced to 130–140/min. The shift of the ventricular septum improved (Figure 2C-b), and the estimated right ventricular pressure decreased to 50 mmHg.
Blood tests revealed normal routine biochemistry, hematology, negative antinuclear antibodies, normal total bile acid (8.7 μmol/l), and high KL-6 (567 U/ml). Echocardiography revealed a small atrial septal defect but no other cardiac malformations, such as pulmonary vein stenosis or left-sided heart diseases. Chest CT revealed atelectasis and inflammatory findings, but chronic lung disease and interstitial pneumonia were not suggested. Pulmonary venous-occlusive disease, chronic thromboembolic PAH, bronchopulmonary hypoplasia, and pulmonary artery hypoplasia were not observed. Taken together, these results suggest that her PH was due to PAH.
In addition to oxygen, sildenafil was administered, and her condition improved. We adjusted the ventilator FiO2 to 0.3, PIP/PEEP to 15/6 cmH2O, and estimated that the right ventricular pressure decreased to 55 mmHg on echocardiography. We increased sildenafil to 2.0 mg/kg/day eventually. She was weaned from mechanical ventilator at 14 months of age, while PAH persisted to require additional treatment with phosphodiesterase-5 inhibitor at 16 months of age.
After obtaining informed consent, we performed exome sequencing (xGen Exome Research Panel, Integrated DNA Technologies) for the proband using DNA extracted from blood and identified a somatic mosaic PIK3CA variant (NM_0062184.4: c.3073A>G, p.(Thr1025Ala)) in the patient. The variant allele fraction (VAF) of this variant was 9% (4/46, variant reads per total reads). The variant from exome sequencing was confirmed by Sanger sequencing (Figure 1D). The variant protein site of this case is located near His1047 within the kinase domain of PI3KCA. The kinase domain mutation His1047Arg induces a conformational change independently of RAS-binding to enhance PI3K/AKT/mTOR signaling (Zhao and Vogt, 2008).
We confirmed the VAF by deep sequencing, as described previously (Nakashima et al., 2014). Genomic DNA derived from blood leukocytes was PCR-amplified using the following primers: forward 5′-TCTAGCTTAAGCACAAAAGGATTT-3′ and reverse 5′-CAATCTTCAAAGTTTACCTTTTTGG-3′. A single-indexed sequencing library was prepared using the NEBNext Ultra II FS DNA Library Prep Kit for Illumina (NEW ENGLAND Bio Labs, Ipswich, MA) and sequenced on an Illumina MiSeq (Illumina, San Diego, CA) with 150-bp paired-end reads. Quality-filtered reads were mapped to the human reference genome sequence (GRCh 38) and aligned using BWA-MEM (Version 0.7.17), and the aligned read files in the BAM format were sorted and indexed using SAMtools. Data analysis, including allele counting, was performed by Integrative Genomics Viewer software. The deep sequencing revealed that the VAF of this variant was 9.17% (805/8771, variant reads per total reads).
This somatic variant was not registered in the genome aggregation database (http://gnomad.broadinstitute.org/) and was predicted to be deleterious by multiple in silico pathogenicity prediction tools. The same variant has been identified in one case among 24 cases of MCAP with PIK3CA variants (Mirzaa G. M. et al., 2013), suggesting this somatic variant is causative for MCAP in this patient; however, there was no clinical information including the presence of PAH in the previous literature. Additionally, other case series of MCAP with PI3KCA variants identified no patient with PAH (Garde et al., 2021).
3 Discussion
The current case of MCAP presented with atrial tachycardia and severe PAH. PAH persisted even after weaning from respiratory support. Taken together with the 12-lead electrocardiogram findings, the preceding presence of atrial load findings due to elevated right ventricular pressure, absence of tachycardia during the neonatal period when tachycardia is more likely to occur, and resolving of tachycardia with improvement of transiently exacerbated PAH, we concluded the diagnosis was atrial tachycardia due to PAH. MCAP is an entity within the clinical spectrum of megalencephaly and overgrowth syndromes caused by constitutive hyperactivation of PI3K/AKT/mTOR signaling, which plays a crucial role in numerous cellular functions, including cell growth, proliferation, survival, migration, metabolism, angiogenesis, apoptosis, and brain development (Riviére et al., 2012).
PI3Ks are heterodimeric enzymes that convert phosphatidylinositol-4,5-bisphosphate (PIP2) to phosphatidylinositol-3,4,5-trisphosphate (PIP3), which interacts with AKT at the cellular membrane, where it is phosphorylated and activated. An important negative regulator of PI3K is tumor suppressor phosphatase and tensin homolog (PTEN), which dephosphorylates PIP3. AKT modulates a range of cellular metabolic and cell survival functions through indirect activation of mTOR, which controls cell proliferation, protein translation, and autophagy (Riviére et al., 2012; Weigelt and Downward, 2012).
PIK3CA variants associated with MCAP have been reported to cause PI3K hyperactivity and PI3K/AKT/mTOR signaling in cells derived from affected individuals (Loconte et al., 2015; Suzuki et al., 2017; Ranieri et al., 2018). Although in the current case, somatic mosaic expression of a pathogenic PIK3CA variant was identified in lymphocytes, various types of cells with increased PI3K/AKT/MTOR signaling may be distributed in various organs.
PAH is a progressive and fatal disease characterized by pulmonary vascular resistance and pulmonary arterial pressure (Morrell et al., 2009; Babicheva et al., 2021). PAH progression is related to pulmonary vasoconstriction, concentric pulmonary vascular remodeling, local thrombosis, and pulmonary artery wall thickening (Galiè et al., 2009; Babicheva et al., 2021). Wall thickening is driven by increased proliferation and inhibited apoptosis among pulmonary vascular endothelial cells (EC), pulmonary smooth muscle cells (PASMC), and fibroblasts (FB) (Maron et al., 2020; Ratwatte et al., 2020; Babicheva et al., 2021). Interestingly, the molecular basis of pulmonary artery wall thickening is closely related to PI3K/AKT/mTOR signaling (reviewed by Babicheva et al., 2021). PI3K functions downstream of BMPR2, a gene associated with hereditary PAH (Deng et al., 2000; Andruska and Spiekerkoetter, 2018). Although the genetic background for PAH has not yet been elucidated, BMPR2, KCNK3, ABCC8, TBX4, and SOX17 were known as familial PAH-related genes (Southgate et al., 2020). At least, no causative variant in these five genes was identified in the current case.
In cellular models of PAH, proliferation, and migration of pulmonary artery muscle cells were accelerated through AKT phosphorylation. The rescue could be achieved with PI3K inhibitors (ten Freyhaus et al., 2011). Several PI3K/AKT/MTOR signaling inhibitors have been tested in clinical trials for the treatment of PAH (reviewed by Babicheva et al., 2021). We also have a strong interest if PIK3CA inhibitor or mTOR inhibitor effectively works for her PAH; however, we could not use them since they were not approved by Pharmaceuticals and Medical Devices Agency in Japan.
On the other hand, the frequency of PAH in MCAP patients remains unclear. Among 75 MCAP patients, four children presented with cardiac arrhythmia and died suddenly. PAH was not mentioned in these cases (Lapunzina et al., 2004). It is not clear why PAH is uncommon in MCAP, but it may be associated with variability in somatic mosaic tissue distribution as a feature of MCAP. There may be other cases of PAH that are milder and not identified. We did not directly prove that PAH in her was caused by the PIK3CA variant because we could not obtain any biopsy sample of the proliferative tissue from her lung. Analysis of lung tissue at autopsy in multiple cases of MCAP with PI3KCA variants would be necessary to prove causality.
Some congenital disorders, including Down syndrome (DS), are at a greater risk of PAH than the general population, partly due to upper airway obstruction and congenital heart disease (Hawkins et al., 2011; Pandit and Fitzgerald, 2012). Indeed, the frequency of early progression of histopathological pulmonary arteriopathy did not differ between patients with and without DS (Masaki et al., 2018). In contrast, the current patient had already undergone tracheostomy at 2 months of age and had a stable airway without hypoxia or hypercapnia for more than 3 months. In addition, apparent lung damage (e.g., pneumonia, atelectasis, or emphysema) was not identified.
4 Conclusion
We have reported the case of an MCAP patient with a somatic PIK3CA variant, whose condition was complicated by severe PAH. Although PAH in MCAP is a rare complication, we should pay attention to cardiological findings, including the atrial load findings due to elevated right ventricular pressure which could be screened by chest radiography and electrocardiogram in patients with MCAP.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
Ethics statement
The studies involving human participants were reviewed and approved by Human Genome/Gene Analysis Research Ethics Committee of Showa University. Written informed consent was obtained from the patient's legal guardian/next of kin for the publication of this case report.
Author contributions
YY and TS: Study conceptualization, analysis of clinical manifestations, and manuscript preparation. MK, MN, KW, and HS: Analysis and interpretation of the genetic data. HK, KO, and RE: Analysis of clinical manifestations. TU, MK, and HH revised the manuscript and supervised the study. All authors contributed to the article and approved the submitted version.
Funding
This study was supported by the Japan Society for the Promotion of Science (grant nos. JP16H05160, JP16K09975, JP20K08236, and JP21K07694), MHLW Research programs on rare and intractable diseases (JPMH20FC1039 and JPMH20FC1046), and an HUSM Grant-in-Aid from Hamamatsu University School of Medicine.
Acknowledgments
We are grateful to the patient and her families for their cooperation during this study.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Andruska, A., and Spiekerkoetter, E. (2018). Consequences of BMPR2 deficiency in the pulmonary vasculature and beyond: Contributions to pulmonary arterial hypertension. Int. J. Mol. Sci. 19 (9), 2499. doi:10.3390/ijms19092499
Babicheva, A., Makino, A., and Yuan, J. X. (2021). mTOR signaling in pulmonary vascular disease: Pathogenic role and therapeutic target. Int. J. Mol. Sci. 22 (4), 2144. doi:10.3390/ijms22042144
Deng, Z., Morse, J. H., Slager, S. L., Cuervo, N., Moore, K. J., Venetos, G., et al. (2000). Familial primary pulmonary hypertension (gene PPH1) is caused by mutations in the bone morphogenetic protein receptor-II gene. Am. J. Hum. Genet. 67 (3), 737–744. doi:10.1086/303059
Galiè, N., Hoeper, M. M., Humbert, M., Torbicki, A., Vachiery, J. L., Barbera, J. A., et al. ESC Committee for Practice Guidelines (CPG) (2009). Guidelines for the diagnosis and treatment of pulmonary hypertension: The task force for the diagnosis and treatment of pulmonary hypertension of the European society of cardiology (ESC) and the European respiratory society (ERS), endorsed by the international society of heart and lung transplantation (ISHLT). Eur. heart J. 30 (20), 2493–2537. doi:10.1093/eurheartj/ehp297
Garde, A., Guibaud, L., Goldenberg, A., Petit, F., Dard, R., Roume, J., et al. (2021). Clinical and neuroimaging findings in 33 patients with MCAP syndrome: A survey to evaluate relevant endpoints for future clinical trials. Clin. Genet. 99 (5), 650–661. doi:10.1111/cge.13918
Harada, A., Miya, F., Utsunomiya, H., Kato, M., Yamanaka, T., Tsunoda, T., et al. (2015). Sudden death in a case of megalencephaly capillary malformation associated with a de novo mutation in AKT3. Child's Nerv. Syst. ChNS official J. Int. Soc. Pediatr. Neurosurg. 31 (3), 465–471. doi:10.1007/s00381-014-2589-y
Hawkins, A., Langton-Hewer, S., Henderson, J., and Tulloh, R. M. (2011). Management of pulmonary hypertension in Down syndrome. Eur. J. Pediatr. 170 (7), 915–921. doi:10.1007/s00431-010-1378-1
Isenović, E. R., Kedees, M. H., Tepavcević, S., Milosavljević, T., Korićanac, G., Trpković, A., et al. (2009). Role of PI3K/AKT, cPLA2 and ERK1/2 signaling pathways in insulin regulation of vascular smooth muscle cells proliferation. Cardiovasc. hematological Disord. drug targets 9 (3), 172–180. doi:10.2174/187152909789007034
Lapunzina, P., Gairí, A., Delicado, A., Mori, M. A., Torres, M. L., Goma, A., et al. (2004). Macrocephaly-cutis marmorata telangiectatica congenita: Report of six new patients and a review. Am. J. Med. Genet. Part A 130A (1), 45–51. doi:10.1002/ajmg.a.30235
Loconte, D. C., Grossi, V., Bozzao, C., Forte, G., Bagnulo, R., Stella, A., et al. (2015). Molecular and functional characterization of three different postzygotic mutations in PIK3CA-related overgrowth spectrum (PROS) patients: Effects on PI3K/AKT/mTOR signaling and sensitivity to PIK3 inhibitors. PloS one 10 (4), e0123092. doi:10.1371/journal.pone.0123092
Maron, B. A., Brittain, E. L., Hess, E., Waldo, S. W., Barón, A. E., Huang, S., et al. (2020). Pulmonary vascular resistance and clinical outcomes in patients with pulmonary hypertension: A retrospective cohort study. Lancet. Respir. Med. 8 (9), 873–884. doi:10.1016/S2213-2600(20)30317-9
Masaki, N., Saiki, Y., Endo, M., Maeda, K., Adachi, O., Akiyama, M., et al. (2018). Is trisomy 21 a risk factor for rapid progression of pulmonary arteriopathy? - revisiting histopathological characteristics using 282 lung biopsy specimens. Circulation J. official J. Jpn. Circulation Soc. 82 (6), 1682–1687. doi:10.1253/circj.CJ-17-0754
Mirzaa, G., Graham, J. M., and Keppler-Noreuil, K. (2013). “PIK3CA-Related overgrowth spectrum,” in GeneReviews® [internet]. M. P. Adam, D. B. Everman, and G. M. Mirzaa Editors (Seattle, WA: University of Washington, Seattle), 1993–2022. Available at: https://www.ncbi.nlm.nih.gov/books/NBK153722/.
Mirzaa, G. M., Rivière, J. B., and Dobyns, W. B. (2013). Megalencephaly syndromes and activating mutations in the PI3K-AKT pathway: MPPH and MCAP. Am. J. Med. Genet. Part C, Seminars Med. Genet. 163C (2), 122–130. doi:10.1002/ajmg.c.31361
Mirzaa, G., Timms, A. E., Conti, V., Boyle, E. A., Girisha, K. M., Martin, B., et al. (2016). PIK3CA-associated developmental disorders exhibit distinct classes of mutations with variable expression and tissue distribution. JCI insight 1 (9), e87623. doi:10.1172/jci.insight.87623
Morrell, N. W., Adnot, S., Archer, S. L., Dupuis, J., Lloyd Jones, P., MacLean, M. R., et al. (2009). Cellular and molecular basis of pulmonary arterial hypertension. J. Am. Coll. Cardiol. 54 (1), S20–S31. doi:10.1016/j.jacc.2009.04.018
Nakamura, K., Kato, M., Tohyama, J., Shiohama, T., Hayasaka, K., Nishiyama, K., et al. (2014). AKT3 and PIK3R2 mutations in two patients with megalencephaly-related syndromes: MCAP and MPPH. Clin. Genet. 85 (4), 396–398. doi:10.1111/cge.12188
Nakashima, M., Miyajima, M., Sugano, H., Iimura, Y., Kato, M., Tsurusaki, Y., et al. (2014). The somatic GNAQ mutation c.548G>A (p.R183Q) is consistently found in Sturge-Weber syndrome. J. Hum. Genet. 59 (12), 691–693. doi:10.1038/jhg.2014.95
Pandit, C., and Fitzgerald, D. A. (2012). Respiratory problems in children with Down syndrome. J. Paediatr. child health 48 (3), E147–E152. doi:10.1111/j.1440-1754.2011.02077.x
Ranieri, C., Di Tommaso, S., Loconte, D. C., Grossi, V., Sanese, P., Bagnulo, R., et al. (2018). In vitro efficacy of ARQ 092, an allosteric AKT inhibitor, on primary fibroblast cells derived from patients with PIK3CA-related overgrowth spectrum (PROS). Neurogenetics 19 (2), 77–91. doi:10.1007/s10048-018-0540-1
Ratwatte, S., Anderson, J., Strange, G., Corrigan, C., Collins, N., Celermajer, D. S., et al. (2020). Pulmonary arterial hypertension with below threshold pulmonary vascular resistance. Eur. Respir. J. 56 (1), 1901654. doi:10.1183/13993003.01654-2019
Rivière, J. B., Mirzaa, G. M., O'Roak, B. J., Beddaoui, M., Alcantara, D., Conway, R. L., et al. (2012). De novo germline and postzygotic mutations in AKT3, PIK3R2 and PIK3CA cause a spectrum of related megalencephaly syndromes. Nat. Genet. 44 (8), 934–940. doi:10.1038/ng.2331
Southgate, L., Machado, R. D., Gräf, S., and Morrell, N. W. (2020). Molecular genetic framework underlying pulmonary arterial hypertension. Nat. Rev. Cardiol. 17 (2), 85–95. doi:10.1038/s41569-019-0242-x
Suzuki, Y., Enokido, Y., Yamada, K., Inaba, M., Kuwata, K., Hanada, N., et al. (2017). The effect of rapamycin, NVP-BEZ235, aspirin, and metformin on PI3K/AKT/mTOR signaling pathway of PIK3CA-related overgrowth spectrum (PROS). Oncotarget 8 (28), 45470–45483. doi:10.18632/oncotarget.17566
ten Freyhaus, H., Dagnell, M., Leuchs, M., Vantler, M., Berghausen, E. M., Caglayan, E., et al. (2011). Hypoxia enhances platelet-derived growth factor signaling in the pulmonary vasculature by down-regulation of protein tyrosine phosphatases. Am. J. Respir. Crit. care Med. 183 (8), 1092–1102. doi:10.1164/rccm.200911-1663OC
Weigelt, B., and Downward, J. (2012). Genomic determinants of PI3K pathway inhibitor response in cancer. Front. Oncol. 2, 109. doi:10.3389/fonc.2012.00109
Yano, S., and Watanabe, Y. (2001). Association of arrhythmia and sudden death in macrocephaly-cutis marmorata telangiectatica congenita syndrome. Am. J. Med. Genet. 102 (2), 149–152. doi:10.1002/ajmg.1428
Keywords: megalencephaly-capillary malformation syndrome, overgrowth syndrome, pulmonary arterial hypertension, PIK3CA, PI3K/Akt/mTOR pathway
Citation: Yoh Y, Shiohama T, Uchida T, Ebata R, Kobayashi H, Okunushi K, Kato M, Watanabe K, Nakashima M, Saitsu H and Hamada H (2023) Case report: Progressive pulmonary artery hypertension in a case of megalencephaly-capillary malformation syndrome. Front. Genet. 14:1221745. doi: 10.3389/fgene.2023.1221745
Received: 12 May 2023; Accepted: 24 July 2023;
Published: 08 August 2023.
Edited by:
Jared C. Roach, Institute for Systems Biology (ISB), United StatesReviewed by:
Adrienne Hammill, Cincinnati Children’s Hospital Medical Center, United StatesAvinash Vijay Dharmadhikari, Children’s Hospital of Los Angeles, United States
Copyright © 2023 Yoh, Shiohama, Uchida, Ebata, Kobayashi, Okunushi, Kato, Watanabe, Nakashima, Saitsu and Hamada. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tadashi Shiohama, YXN1aGFfaGFyZUB5YWhvby5jby5qcA==