 Fan Shi
Fan Shi Mei Zhao
Mei Zhao Shudan Zheng
Shudan Zheng Lihong Zheng2
Lihong Zheng2 Haiqiang Wang
Haiqiang Wang
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Genet. , 11 September 2023
Sec. Genetics of Common and Rare Diseases
Volume 14 - 2023 | https://doi.org/10.3389/fgene.2023.1213916
Metabolism-related fatty liver disease (MAFLD) is the most common form of chronic liver disease in the world. Its pathogenesis is influenced by both environmental and genetic factors. With the upgrading of gene screening methods and the development of human genome project, whole genome scanning has been widely used to screen genes related to MAFLD, and more and more genetic variation factors related to MAFLD susceptibility have been discovered. There are genetic variants that are highly correlated with the occurrence and development of MAFLD, and there are genetic variants that are protective of MAFLD. These genetic variants affect the development of MAFLD by influencing lipid metabolism and insulin resistance. Therefore, in-depth analysis of different mechanisms of genetic variation and targeting of specific genetic variation genes may provide a new idea for the early prediction and diagnosis of diseases and individualized precision therapy, which may be a promising strategy for the treatment of MAFLD.
Metabolic associated fatty liver disease (MAFLD) is a clinicopathological syndrome characterized by diffuse hepatocyte steatosis and lipid accumulation, formerly known as non-alcoholic fatty liver disease (NAFLD) (Kawaguchi et al., 2022). In early 2020, an international panel of 30 experts from 22 countries issued an international expert consensus statement on a new definition of MAFLD, proposing comprehensive and simple diagnostic criteria for MAFLD, and officially renamending NAFLD as MAFLD to highlight the causative link between fatty liver disease and metabolic dysfunction (Eslam et al., 2020). MAFLD can cause a range of diseases, including steatohepatitis, liver fibrosis, cirrhosis and eventually hepatocellular carcinoma, and has emerged as an important risk factor for liver failure and liver transplantation (Powell et al., 2021). MAFLD is a multi-systemic clinical disease that manifests itself not only in the liver but also has a wide range of extrahepatic manifestations, and findings have shown that patients with MAFLD are at increased risk for heart failure, obesity, type 2 diabetes mellitus, metabolic syndrome, chronic kidney disease, extrahepatic malignancies, cognitive impairment, polycystic ovary syndrome, osteoporosis, and hypothyroidism, among others (Kaya and Yilmaz, 2022; Pipitone et al., 2023; Wei et al., 2023). With changes in diet and lifestyle, the prevalence of MAFLD is increasing and it has gradually become the most common form of chronic liver disease worldwide, affecting more than one-third of the global population (Chan et al., 2022). Seriously endanger human health and cause huge economic burden to society (Eslam et al., 2020).
The pathogenesis of MAFLD is not fully understood. The widely accepted mechanism is the “two-strike” hypothesis, The first blow is liver lipid ectopic deposition and insulin resistance (IR), and the second blow is oxidative stress (Day and James, 1998). The liver is the central organ of lipid metabolism, secreting very low density lipoprotein (VLDL) on the one hand, and internalizing fatty acids and lipoproteins on the other (Heeren and Scheja, 2021), Liver is also the main target organ of insulin action, which plays a very important role in the control of blood glucose homeostasis. Hyperinsulinemia and IR are induced by increased levels of glucose and free fatty acids in the blood due to lack of exercise and a high sugar and fat diet. IR can reduce the intake of glucose by adipose tissue and skeletal muscle, induce hyperglycemia, promote the conversion of glucose into fatty acids (FAs) and triglycerides in hepatocytes, increase the new synthesis of liver fat, and lead to excess lipid deposition in the liver and fatty degeneration of the liver. However, the accumulation of lipid intermediates such as diglycerol ester (DAG) and ceramide in fatty liver leads to the inhibition of insulin signaling cascade, which induces the vicious cycle of IR and lipid deposition (Sakurai et al., 2021). Excessive accumulation of liver lipids can induce increased liver lipid toxicity and trigger cellular stress response, excessive elevation of oxidative stress level can lead to oxidative modification of specific DNA, protein and lipid metabolites, and thus cause cell damage. In addition, oxidative stress can also activate inflammatory bodies represented by NLRP3, release pro-inflammatory cytokines, induce hepatocyte necrosis, and promote the development of MAFLD (Clare et al., 2022).
The pathogenesis of MAFLD is complex, caused by an interaction between genetic, environmental, and metabolic disorders, The “two-strike” hypothesis is not a good explanation of the complexity of its pathogenesis, and has been gradually replaced by the “multiple strike” theory. The “multiple shocks” include genetic factors, insulin resistance, lipid metabolism disorders, hepatocyte steatosis, oxidative stress, inflammation, mitochondrial dysfunction, endoplasmic reticulum stress, epigenetic factors, intestinal flora disorders, etc. Multiple mechanisms work together to promote the occurrence and development of MAFLD (Bence and Birnbaum, 2021). With the continuous development of genetics, the genetic characteristics of MAFLD have been revealed to a large extent, and the role of genetic factors in the occurrence and development of MAFLD has been increasingly understood, which may involve intra-hepatic lipolysis, triglyceride output, hepatic mitochondrial oxidation or glucokinase activity (Sangro et al., 2023). Specific genetic risk variants have been shown to play a critical role in MAFLD and amplify the effect of MAFLD on disease outcome, increasing liver-related and overall mortality (Pingitore and Romeo, 2019).
MAFLD is a metabolic liver injury that is closely related to genetic susceptibility, and the clinical and phenotypic variation in patients may be caused by genetic variation factors (Dabravolski et al., 2021; Xia et al., 2021), and it is associated with a Single nucleotide polymorphism (SNP) in multiple related gene loci (Zhu et al., 2022). SNP refers to the DNA sequence polymorphism caused by the change of a single nucleotide at the genome level, which is mostly the conversion or reversal of a single base. With advances in high-throughput sequencing technology, genome-wide association Studies (GWAS) have provided insight into the heritage background of MAFLD (Sveinbjornsson et al., 2022). To identify the genetic variation associated with MAFLD, a study involving a large European population sample based on GWAS identified five genetic variation loci that may influence susceptibility to MAFLD, they were located near GCKR, TR1B1, TM6SF2, APOE and PNPLA3(Ghodsian et al., 2021). In addition, more and more genetic factors have been identified as genetic modification factors of MAFLD (Liao et al., 2022).
PNPLA3 protein, an enzyme with lipase activity for triglycerides and retinol esters and acyltransferase activity for phospholipids, is a key regulator of lipid droplets in hepatocytes and hepatic stellate cells, mapping of human PNPLA3 gene to chromosome 22, it has 9 exons encoding 481 amino acids, belongs to the Patatin-like phospholipase family, and is the most highly expressed in human hepatocytes and hepatic stellate cells (Pingitore and Romeo, 2019). PNPLA3 has attracted great attention in the field of liver since Romeo et al. discovered a non-synonymous variant (rs738409, I148M) in PNPLA3 via GWAS that is significantly associated with liver fat content (Romeo et al., 2008). The I148M variant of PNPLA3 is a SNP caused by the substitution of isoleucine (I) with methionine (M) in amino acid coding sequence 148(I148M) (Xiang et al., 2021). The incidence of PNPLA3I148M varies in different populations, and studies have shown a major association between the PNPLA3I148M variant and MAFLD in women, but not in men (Chen et al., 2015). The incidence of PNPLA3I148M is higher in Hispanic populations than in European-American and African American populations (Romeo et al., 2008). PNPLA3 I148M is a genetic variant highly associated with the occurrence and development of MAFLD (Basu Ray, 2019; Salari et al., 2021). In order to elucidate the mechanism by which genes and their variants affect disease development, human induced pluripotent stem cells (hiPSC) have been used to establish a PNPLA3-related NAFLD model. This in vitro system provides a platform for elucidation of the complete pathophysiology of the PNPLA3I148M variant in NAFLD and drug development. While minimizing reliance on live animal models, the results show that I148M variants lead to increased lipid accumulation and susceptibility to hepatotoxins (Tilson et al., 2021). Previous studies have demonstrated that PNPLA3(148M) variation is associated with a variety of liver diseases, including MAFLD, non-alcoholic steatohepatitis (NASH), fibrosis, cirrhosis, and hepatocellular carcinoma (HCC) (Dhar and Loomba, 2021), and increases the risk of liver-related death (Grimaudo et al., 2020; Stender and Loomba, 2020). The genetic locus has been identified as having the most powerful role in increasing liver disease (Shang and Mashek, 2020). Despite the importance of PNPLA3I148M, the underlying pathogenic mechanism behind this variation remains unclear and may be related to lipid metabolism disorder, inflammation, and pro-fibrosis (Figure 1).
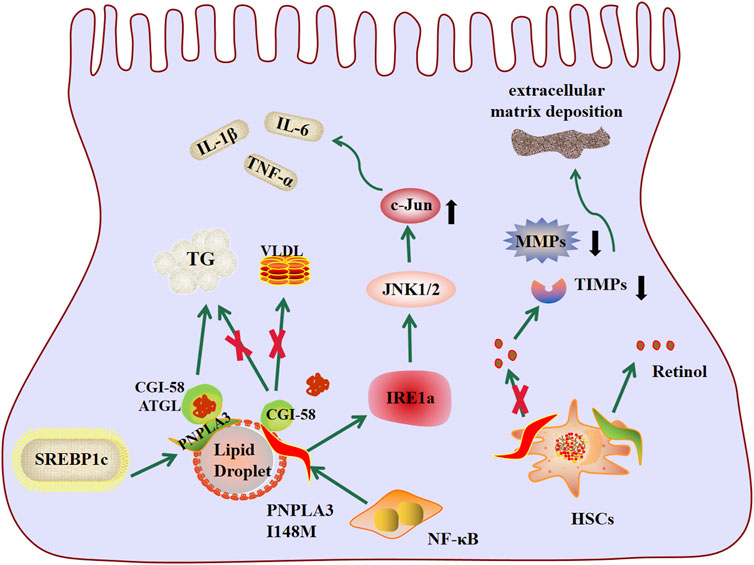
FIGURE 1. Schematic diagram of pathogenesis of genetic variation of PNPLA3 I148M.
In hepatocytes, PNPLA3 is regulated by carbohydrate response element-binding protein (ChREBP) and sterol regulatory element-binding protein 1c (SREBP1c), has hydrolase activity on triglycerides, and plays an important role in lipid drome remodeling and very low density lipoprotein (VLDL) secretion in hepatocytes (BasuRay et al., 2019). I148M replacement results in loss of PNPLA3 lipase activity, impaired lipid catabolism, lipid droplet remodeling and VLDL secretion, VLDL secretion is the pathway by which fat escapes from the liver, thus leading to the accumulation of triglycerides in hepatocytes (Qadri et al., 2020; Akkiz et al., 2021). In addition, I148M is associated with decreased ubiquitination, resulting in accumulation of PNPLA3 in the liver and decreased TG mobilization in lipid droplet (LD) (BasuRay et al., 2017). The efficacy of CGI-58 as a co-activator of adipose triglyceride lipase (ATGL) in promoting intracellular fat decomposition has been well known. CGI-58 was able to combine with ATGL to promote fat decomposition and lipid phagocytosis, leading to LD degradation (Yu et al., 2020). PNPLA3I148M may inhibit ATGL catalyzed lipolysis by binding with CGI-58, leading to inhibition of liphagy, and thus increasing liver LD accumulation. The ability of PNPLA3 I148M to promote steatosis was weakened in mice lacking liver CGI-58 (Wang et al., 2019), further consolidating the key role of CGI-58 in mediating the action of PNPLA3 I148M.
In addition to fat accumulation, inflammation is an important factor in the progression of MAFLD (Khanmohammadi and Kuchay, 2022). Overexpression of PNPLA3I148M leads to phosphorylation of STAT3 and activation of downstream inflammatory pathways, leading to the production of inflammatory substances such as IL-1β and IL-6, activates hepatic stellate cells, and further accelerates the process of NASH with fibrosis (Banini et al., 2021). NF-kB is the most important transcription factor that regulates inflammation and is involved in the progression of MAFLD. It was found that PNPLA3 I148M was activated by the transcription of NF-kB, and PNPLA3 I148M protein activated IRE1a signal of endoplasmic reticulum stress, then phosphorylated JNK1/2, upregulated the expression of c-Jun, and finally upregulated the expression of C-Jun-dependent inflammatory cytokines, such as TNF-α, and promote inflammation (Yuan et al., 2020). Enhanced IL-6/STAT3 signaling has been observed in PNPLA3I148M liver culture, which plays a central role in inflammation (Hirano, 2021), and can promote the progression of PNpla3I148M-induced MAFLD. IL-6 is a multipotent inflammatory cytokine involved in tissue homeostasis, regeneration and metabolism. IL-6 is significantly elevated in the liver of patients with MAFLD and correlates with severity, accelerating the progression of the disease, while blocking IL-6 signaling across the board reduces the progression of MAFLD (Park et al., 2023). Recruitment of these inflammatory cells eventually converges on hepatic stellate cells (HSCS), leading to activation.
Activation of hepatic stellate cells is a central factor in the progression of hepatic fibrosis. Upon liver injury, HSCs are activated, lose lipid-rich granules, and are transdifferentiated into a-smooth muscle actin (a-SMA)-positive myofibroblasts, which produce increased amount of ECM, proinflammatory, and profibrogenic cytokines, and cause liver fibrosis (Wang et al., 2022). In hepatic stellate cells, PNPLA3 is involved in retinol metabolism and has hydrolase activity on retinol ester, allowing the release of retinol from hepatic stellate cells. The hepatic stellate cells carrying PNPLA3I148M mutation lost the function of hydrolase activity, resulting in retinol intracellular retention. Impaired retinol production may lead to decreased secretion of matrix metalloproteinases and tissue inhibitors of metalloproteinases, resulting in extracellular matrix deposition and play an important role in hepatic stellate cell activation (Xiang et al., 2021). Human hepatic stellate cells with the PNPLA3(148M) variant showed higher expression of inflammatory cytokines and chemokines, and showed higher cell proliferation and migration (Bruschi et al., 2020). The genetic variation I148M has a significant effect on enhancing the pro-inflammatory response and pro-fibrosis characteristics of human hepatic stellate cells (Trepo et al., 2016; Bruschi et al., 2020). Thus, I148M variation in the PNPLA3 gene by regulating hepatic stellate cells is a risk factor for the development of severe hepatic fibrosis.
In conclusion, PNPLA3 is an important protein with a wide range of implications in metabolic liver diseases ranging from simple steatosis to cirrhosis and liver cancer, A better understanding of the biological function of PNPLA3 in lipid droplet metabolism will help advance the progress of disease treatment. This protein is an attractive target for the treatment of MAFLD, and targeting the PNPLA3(148M) variant is expected to be a promising direction for modern personalized medicine (Dong, 2019).
Transmembrane 6 superfamily member 2 (TM6SF2) is located on chromosome 19 and encodes proteins containing 375 or 377 amino acids, respectively. The protein is expected to have 7–10 transmembrane domains, but does not contain any known functional domains and is mainly expressed in the gut and liver, suggesting a metabolism-related function (Zhang et al., 2018). TM6SF2 is localized in the endoplasmic reticulum (ER) and ER-Golgi intermediate region and plays a key role in intracellular lipidization of very low density lipoproteins, thereby preventing fatty liver disease (Luo et al., 2022b).
The non-synonymous variant of TM6SF2 (E167K, rs58542926) is characterized by cytosine (C) mutation of nucleotide 499 to thymine (T), and lysine-altered glutamic acid encoding codon 167 (E167K) (Jiang et al., 2021). The misfolding of the E167K variant protein accelerates protein degradation, resulting in decreased TM6SF2 protein levels and gene function. The loss of function leads to a decrease in the number of lipoprotein particles responsible for lipidation and lipoprotein, resulting in very low density lipoprotein (VLDL) retention in the liver and increased TAG content in the liver. Therefore, it is considered to be an important risk factor for lipid metabolism-related diseases (Li et al., 2018). Studies have shown that liver specific TM6SF2 loss impairs VLDL secretion, promotes hepatic steatosis and fibrosis, and accelerates HCC development (Newberry et al., 2021). Loss of TM6SF2 also strongly affects the structure of endoplasmic reticulum and mitochondria, thereby increasing ER stress and oxidative stress. Endoplasmic reticulum stress is closely related to lipogenesis or lipolysis, interfering with lipid metabolism and ultimately leading to inflammation and liver cell damage (Flessa et al., 2022), playing an important role in the progression of MAFLD (Longo et al., 2022). Clinical and epidemiological studies have confirmed the role of the TM6SF2 variant in the development of MAFLD (Xue et al., 2022), and TM6SF2 has also been shown to play an important role in promoting liver fibrosis and hepatocellular carcinoma in mouse models (Luo et al., 2022a).
The Glucose kinase regulatory protein gene (GCKR) is located on human chromosome 2p23.3, approximately 27 kb in length, consisting of 19 exons and 18 introns. The GCKR gene encodes a protein consisting of 625 amino acids called Glucose kinase regulatory protein (GKRP), which is a member of the glucose isomerase family. GKRP is a heat-resistant chemical protein. Glucose kinase (GCK), a phosphorylase related to glucose metabolism, phosphorylates glucose to form glucose 6-phosphate, thus regulating liver glucose metabolism and promoting liver lipid generation. By regulating the activity of GCK, GKRP can further affect the utilization of glucose by liver cells, regulate body metabolism and resynthesis of fat (Peter et al., 2011). In hypoglycemia, GKRP binds to GCK to inactivate it and thus prevent GCK from entering the nucleus, reducing glucose decomposition and raising blood sugar; When blood sugar rises, GKRP separates from GCK, and the GCK in the nucleus restarts and returns to the cytoplasm, promoting glycolysis and lowering plasma glucose levels. Thus, GCKR encodes GKRP, which is involved in the regulation of glucose homeostasis and blood glucose control, thereby regulating glucose flow into hepatocytes and thereby inducing new adipogenesis (DNL) (Meroni et al., 2021). A SNP in GCKR, rs1260326 c.1337C > T p. P446L, is a proline to leucine substitution encoding the amino acid position of GCKR protein 446 (P446L). GCKR P446L is a functionally deficient variant that increases fat production by inducing glycolysis (Carlsson et al., 2020). The variation of GCKR is associated with a variety of lipid metabolism disorders (Fernandes Silva et al., 2019), significantly affected the MAFLD (Yuan et al., 2022). A meta-analysis showed that both the GCKR rs780094 and rs1260326 polymorphisms were significantly associated with an increased risk of MAFLD (Li et al., 2021).
The apolipoprotein E (Apo E) gene is located in the 2 band of the 13 region on the long arm of human chromosome 19, containing 4 exons and 3 introns (19q13.2). Apo E is a polymorphic protein synthesized mainly by the liver that influences lipid metabolism by binding to chylomicron and LDL receptors to mediate the clearance of very low density lipoproteins in serum (Zhou et al., 2020). The genetic variation of Apo E is consistently associated with increased liver fat content (Nascimento et al., 2020). The variant APOE gene obstructs the clearance of circulating lipoproteins and may hinder the reuptake of lipids in the liver, thus failing to timely transport excess cholesterol in the blood, resulting in lipid accumulation. These findings may help to better elucidate the genetic susceptibility associated with the onset and progression of MAFLD (Jamialahmadi et al., 2021).
The nerve cell death inducing protein kinase 1 (TRIB1) gene is located on human chromosome 8 and encodes the tribbles protein (Zhang et al., 2019), Although proteins of the TRIB1 family are pseudokinases that lack typical phosphotransferase activity, this pseudokinase plays a key role in normal and disease biology through its function as signal transduction mediators and protein scaffolds (Zhang et al., 2021). TRIB1 is closely related to lipid metabolism and downregulates liver lipogenesis through multi-molecular interactions (Ishizuka et al., 2014). Promotes ubiquitination of the transcription factor CCAAT enhancer binding protein α, thereby promoting its degradation (Quiroz-Figueroa et al., 2021). Downregulated ChREBP inhibits the expression of lipogenic genes and reduces the secretion of very low density lipoprotein, thus reducing the accumulation of lipids in the liver (Iwamoto et al., 2015). The TRIB1 rs17321515 variant has been identified by GWAS as a risk site for MAFLD (Vujkovic et al., 2022). In addition, studies have confirmed that TRIB1 rs17321515 gene polymorphism increases the risk of MAFLD patients in Han Chinese (Liu et al., 2019a; Liu et al., 2019b).
The membrane bound O-acyltransferase 7 domain (MBOAT7), also known as lysophosphatidyl inositol acyltransferase 1 (LPIAT1), is a gene encoding acyltransferase activity and is located on human chromosome 19 (Sharma and Mandal, 2022). The acyltransferase activity encoding lysophosphatidyl inositol is involved in the conversion of fatty acids between phosphoesters and lysophosphoesters (Varadharajan et al., 2022). Experiments showed that the deficiency of MBOAT7 in mice remodeled PI and lysophosphatidylinositol (LPI) liver levels and promoted hyperinsulinemia and hepatic insulin resistance (Massey et al., 2023). MBOAT7 deficiency in mice and humans leads to fibrosis, suggesting that this may be an inflammation-independent hepatic fibrosis pathway mediated by lipid signaling (Thangapandi et al., 2021). rs641738C > T variation in the MBOAT7 gene caused damage to arachidonic acid phosphatidylinositol and promoted intracellular liver fat deposition, resulting in the conversion of saturated lysate phosphatidylinositol to triglycerides, thus promoting adipogenesis (Ismaiel and Dumitrascu, 2020). The study demonstrated that the rs641738C>T variant near MBOAT7 was a risk factor for the presence and severity of MAFLD in individuals of European ancestry (Teo et al., 2021).
The human HSD17B13 gene is located on chromosome 4q22.1, with a total length of about 17kb, 8 exons and 7 introns (Chen et al., 2020). HSD17B13 belongs to the HSD17B family. HSD17B13 is the most highly expressed lipid droplet associated protein enriched in liver with hepatocellular specificity. HSD17B1 is selectively expressed in hepatocytes and localized only to the surface of lipid droplets. HSD17B13 also acts as a retinoic dehydrogenase that converts retinol to retinoic acid (RA), and its elevated level is closely related to the development of MAFLD (Zhang et al., 2021). A genetic variant of HSD17B13 (rs72613567, T > TA) was first described in 2018 and was associated with a reduced risk of MAFLD (Abul-Husn et al., 2018), other variants of HSD17B13 (rs6834314, A > G and rs9992651, G > A) were later associated with lower inflammatory scores in patients with MAFLD, which may be related to protection against MAFLD (Motomura et al., 2021). A meta-analysis showed that a polymorphism of the HSD17B13rs72613567:TA allele variant was associated with a reduced risk of HCC and MAFLD in the entire study population (Tang et al., 2021). Other studies have found that there may be interaction between HSD17B13 rs72613567 gene variation and PNPLA3 rs738409, which directly affect the expression level of PNPLA3 mRNA in liver and reduce the activity of PNPLA3 p. I148 (Abul-Husn et al., 2018). In a multiracial cohort of Asian patients with NAFLD, variants of HSD17B13 rs72613567 and rs6834314 were negatively associated with MAFLD and NASH and were associated with a lower incidence of adverse liver outcomes (Ting et al., 2021). Therefore, HSD17B13 is considered as a potential therapeutic target for MAFLD. However, it should be noted that the HSD17B13rs72613567 variant appears to be a risk variant for liver fibrosis in the Chinese Han MAFLD population, which is inconsistent with previous conclusions, indicating that future studies need to be verified by different ethnic groups, and it is necessary to study different groups in genetic studies. To map the genome-wide association study signal in detail (Liu et al., 2021).
Klotho (KL) is a gene related to aging. It is located in 13q12. The total length of the 50 kb gene contains 4 introns and 5 exons, and has various biological effects such as antioxidant, anti-inflammatory and anti-apoptosis (Martin-Gonzalez et al., 2022). The human KL gene encodes alpha-Klotho protein, a multifunctional protein that regulates phosphate, calcium, and vitamin D metabolism, it is mainly expressed as a single transmembrane glycoprotein in kidney, parathyroid, brain and adipose tissue, and as a co-receptor of fibroblast growth factor 23(FGF23) (Kuro, 2019). A study using an animal model found that klotho knockout mice were extremely emaciated and significantly reduced fat accumulation in the liver, suggesting that it may play a direct role in the pathophysiology of fatty liver disease (Ohnishi et al., 2011). The KLrs495392 polymorphism was found to be protective against severe hepatic steatosis in patients with MAFLD and may reduce the risk of severe steatosis caused by the PNPLA3rs738409G variant (Liu et al., 2022). These data suggest that Klotho may be a therapeutic target for fatty liver disease and warrant further investigation (Chi et al., 2023).
In addition, there are many potential genetic modification factors. Klotho beta (KLB) is a protein-coding gene located on chromosome 4p14. The gene belongs to the glycoside hydrolase family 1 gene group. It is mainly expressed in fat and liver, KLB gene polymorphism is associated with obesity and liver inflammation, and may be involved in the pathogenesis of MAFLD (Ji et al., 2019). For example, the KLBrs17618244 gene variant was associated with liver damage in adult MAFLD patients, even more so in the presence of obesity stratification (Panera et al., 2021). An experiment based on MRI-PDFF and liver biopsy to study the clinical, laboratory, and genetic characteristics of MAFLD patients in A Chinese population found that patients with a variant allele of UQCC1 had low LFC, and UQCC1rs878639 (A>G) was identified as having a protective effect against MAFLD (Yang et al., 2022). Mitochondrial amidoxme reducing Component 1 (MARC1) variant rs2642438 increases phosphatidylcholine in human liver and reduces the severity of non-alcoholic fatty liver disease (Luukkonen et al., 2020).
In summary, it can be seen that genetic polymorphism is closely related to lipid metabolism, inflammation and fibrosis in MAFLD (Figure 2). These genetic variants are either risk factors or protective factors. Therefore, in-depth analysis of genetic variants can improve our understanding of the pathogenesis of the disease, and may provide new targets and more personalized treatment for future MAFLD treatment.
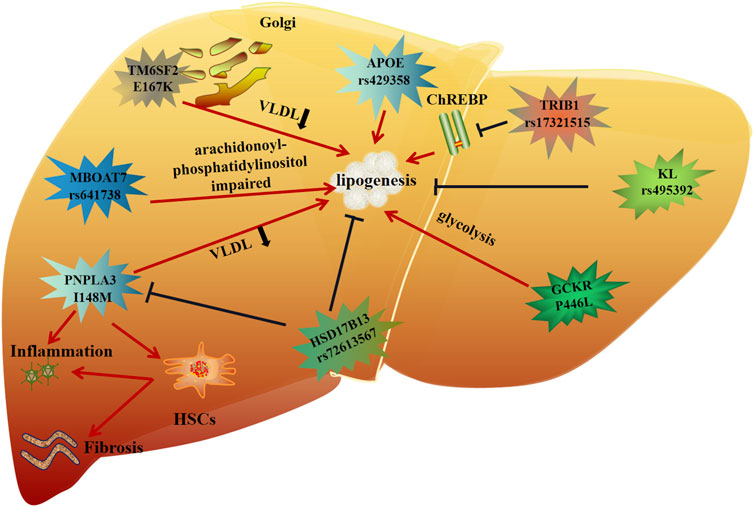
FIGURE 2. Molecular mechanism of genetic variation in MAFLD.
Currently, lifestyle interventions and weight loss remain the cornerstones of treatment for MAFLD (Stefano et al., 2023). Given the multifaceted pathophysiology of the disease, combination therapy such as lipid lowering, blood pressure lowering, glucose lowering, anti-obesity, antioxidant, anti-inflammatory and anti-fibrosis drugs may be considered a reasonable alternative (Makri et al., 2022). Some new signaling pathways and pharmacological targets are also being studied, such as cell therapy and intestinal flora regulation therapy (Raza et al., 2021), but no drug has been approved for the treatment of this disease alone. Genetic factors have been shown to be closely related to the onset and progression of MAFLD, suggesting that the exploration of genetic variants with diagnostic and therapeutic potential may have a promising clinical application prospect in the treatment of MAFLD (Romeo et al., 2020). Currently, the identification of people at risk for MAFLD patients is not accurate enough, and biomarkers to predict disease risk and treatment response are still lacking (Friedman et al., 2018). So on the one hand we can use these genetic factors to predict disease risk; on the other hand, therapeutic drugs can be developed by targeting genes that are particularly important.
Liver biopsy is the gold standard diagnostic method for MAFLD, but it is limited due to its high cost and the possibility of complications due to the invasive procedure (Tsai and Lee, 2018). Genetic methods, especially SNPs, have received increasing attention in recent years due to their non-invasive applications, which combine the effects of a single SNP into a single score for predicting the risk of MAFLD. An increasing number of studies have demonstrated the utility of genetic risk scores (GRS) (June 2021). The higher the GRS, the higher the risk of liver–related disease MAFLD (Wang et al., 2021). GRS tools can be used to screen large numbers of people and, for those at high risk, can be prevented through aggressive lifestyle changes, increased physical activity and dietary modifications. For older adults who already have some degree of liver disease, these relevant genomic features may also guide more rigorous monitoring of liver disease complications (cirrhosis, liver cancer). It has been shown that inclusion of PNPLA3, TM6SF2 and HSD17B13 as genetic risk factors in a risk stratification model may improve its prediction of MAFLD severity and advanced fibrosis (Paternostro et al., 2021). Especially for children and adolescents, this population has relatively few other risk factors, so early predictive diagnosis with genetic biomarkers and early lifestyle interventions for those at risk may benefit patients the most (Li et al., 2020). The GRS tool can provide recommendations for lifestyle changes in genetically predisposed patients, but the effects on liver-related mortality and liver cancer development need to be determined in long-term studies (June 2021). Polygenic Risk Scores (PRS) can be used to predict MAFLDs non-invasively. For the increasing incidence of MAFLD-associated HCC, it is also necessary to develop reliable PRS for in-depth understanding of the causal relationship between NAFLD and HCC and to improve HCC risk stratification (Bianco et al., 2021). In the future, robust prediction models should also integrate rare and common variations and other risk factors, requiring very large samples to be developed (Dessein, 2021).
Genetic variation plays an important role in the pathogenesis of MAFLD and its evolution into cirrhosis and HCC, so targeting the locus of genetic variation may be a promising therapeutic approach. Among them, antisense oligonucleotides (ASOs) are a novel therapeutic approach that targets homologous mRNA sequences, modulates gene expression or translation of related proteins, and ASOs can be easily injected under the skin and directly target mRNA molecules, resulting in faster and longer-lasting responses than directly inhibiting protein production (Scharner and Aznarez, 2021). Some studies have found that targeting PNPLA3I148M by ASOs at the RNA level may provide significant advantages for realizing long-term inhibition of the expression of risk variants in carriers (Cherubini et al., 2021). Liver targeting of triantennal n-acetyl galactosamine-coupled ASO mediated Pnpla3 silencing reduced liver lipogenesis and steatosis in mice carrying a human I148M mutant (Linden et al., 2019). More recently, ION839 (AZD2693), a PNpla3-targeting ASO, was used in patients with NASH and PNPLA3 I148M in a Phase I clinical trial (NCT04483947) (Xu et al., 2022). Although ASOs is a promising precision medicine drug, mild to moderate toxicity as well as some pro-inflammatory and platelet reduction manifestations can still be observed at long-term high doses, requiring further research (Goyenvalle et al., 2023). In addition to targeting PNPLA3 gene variants, liver-specific acetyl-CoA carboxylase (ACC) inhibitors (MK4074) block enhanced fatty acid synthesis, improve fatty acid beta-oxidation, and then reverse the NAFLD phenotype caused by TM6SF2 defects (Li et al., 2022). The protective HSD17B13 variant is also a potential therapeutic target, reproterol, a potential regulator of 17β-HSD13, was found to prevent MAFLD through phosphorylation of 17β-HSD13Ser33 mediated by PKA, so targeting the Ser33 phosphorylation site may be a potential treatment approach (Su et al., 2022).
Metabolically related fatty liver is a common chronic disease, but its pathogenesis remains unclear. Most current studies believe that it is the result of multiple factors such as genetic variation and environment. With the increasing prevalence of MAFLD worldwide, it is imperative to explore new and effective treatments. In the past decade, more and more genetic variants such as GCKR, TRIB1, TM6SF2, APOE, PNPLA3, HSD17B13, etc. have been discovered, which regulate the susceptibility and progression of MAFLD by affecting lipid metabolism, inflammatory response, insulin resistance, oxidative stress, liver fibrosis and other processes. Therefore, targeting specific genetic variants may provide new ideas for the early prediction and diagnosis of MAFLD and individualized precision therapy. For genes involved in the pathogenesis of MAFLD, drugs that silence or turn off the gene can be used to reduce the effect of the gene variant; for protective gene variants, the effect of protective mutations can be simulated to prevent MAFLD. However, it is important to note that further research is needed to expand on the molecular mechanisms associated with these new findings, for example, according to the current study, PNPLA3I148M is mainly involved in lipid accumulation in the liver, but the specific mechanisms need further research to clarify and further validate these results. All new therapies need to be extensively evaluated for safety and efficacy in clinical trials. To sum up, this is a very promising research area. Although still in its infancy, this field may open up a whole new avenue for the treatment of MAFLD, and there are many more potential therapeutic targets waiting to be explored and discovered in the future, giving us hope to conquer MAFLD in the future.
FS and HW concepted and designed the review. FS, SZ, and MZ wrote the manuscript. FS, HW, and LZ revised the manuscript. All authors contributed to the article and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abul-Husn, N. S., Cheng, X., Li, A. H., Xin, Y., Schurmann, C., Stevis, P., et al. (2018). A protein-truncating HSD17B13 variant and protection from chronic liver disease. N. Engl. J. Med. 378, 1096–1106. doi:10.1056/NEJMoa1712191
Akkiz, H., Taskin, E., Karaogullarindan, U., Delik, A., Kuran, S., and andKutlu, O. (2021). The influence of RS738409 I148M polymorphism of patatin-like phospholipase domain containing 3 gene on the susceptibility of non-alcoholic fatty liver disease. Med. Baltim. 100, e25893. doi:10.1097/MD.0000000000025893
Banini, B. A., Kumar, D. P., Cazanave, S., Seneshaw, M., Mirshahi, F., Santhekadur, P. K., et al. (2021). Identification of a metabolic, transcriptomic, and molecular signature of patatin-like phospholipase domain containing 3-mediated acceleration of steatohepatitis. Hepatol. Baltim. Md 73, 1290–1306. doi:10.1002/hep.31609
Basu Ray, S. (2019). PNPLA3-I148M: a problem of plenty in non-alcoholic fatty liver disease. Adipocyte 8, 201–208. doi:10.1080/21623945.2019.1607423
Basuray, S., Smagris, E., Cohen, J. C., and andHobbs, H. H. (2017). The PNPLA3 variant associated with fatty liver disease (I148M) accumulates on lipid droplets by evading ubiquitylation. Hepatology 66, 1111–1124. doi:10.1002/hep.29273
Basuray, S., Wang, Y., Smagris, E., Cohen, J. C., and andHobbs, H. H. (2019). Accumulation of PNPLA3 on lipid droplets is the basis of associated hepatic steatosis. Proc. Natl. Acad. Sci. U. S. A. 116, 9521–9526. doi:10.1073/pnas.1901974116
Bence, K. K., and Birnbaum, M. J. (2021). Metabolic drivers of non-alcoholic fatty liver disease. Mol. Metab. 50, 101143. doi:10.1016/j.molmet.2020.101143
Bianco, C., Jamialahmadi, O., Pelusi, S., Baselli, G., Dongiovanni, P., Zanoni, I., et al. (2021). Non-invasive stratification of hepatocellular carcinoma risk in non-alcoholic fatty liver using polygenic risk scores. J. Hepatol. 74, 775–782. doi:10.1016/j.jhep.2020.11.024
Bruschi, F. V., Tardelli, M., Einwallner, E., Claudel, T., and andTrauner, M. (2020). PNPLA3 I148M up-regulates hedgehog and yap signaling in human hepatic stellate cells. Int. J. Mol. Sci. 21, 8711. doi:10.3390/ijms21228711
Carlsson, B., Linden, D., Brolen, G., Liljeblad, M., Bjursell, M., Romeo, S., et al. (2020). Review article: the emerging role of genetics in precision medicine for patients with non-alcoholic steatohepatitis. Aliment. Pharmacol. Ther. 51, 1305–1320. doi:10.1111/apt.15738
Chan, K. E., Koh, T. J. L., Tang, A. S. P., Quek, J., Yong, J. N., Tay, P., et al. (2022). Global prevalence and clinical characteristics of metabolic-associated fatty liver disease: A meta-analysis and systematic review of 10 739 607 individuals. J. Clin. Endocrinol. Metab. 107, 2691–2700. doi:10.1210/clinem/dgac321
Chen, H., Zhang, Y., Guo, T., Yang, F., Mao, Y., Li, L., et al. (2020). Genetic variant rs72613567 ofHSD17B13gene reduces alcohol-related liver disease risk in Chinese Han population. Liver Int. 40, 2194–2202. doi:10.1111/liv.14616
Chen, L.-Z., Xin, Y.-N., Geng, N., Jiang, M., Zhang, D.-D., and andXuan, S.-Y. (2015). PNPLA3 I148M variant in nonalcoholic fatty liver disease: demographic and ethnic characteristics and the role of the variant in nonalcoholic fatty liver fibrosis. World J. Gastroenterology 21, 794–802. doi:10.3748/wjg.v21.i3.794
Cherubini, A., Casirati, E., Tomasi, M., and andValenti, L. (2021). PNPLA3 as a therapeutic target for fatty liver disease: the evidence to date. Expert Opin. Ther. Targets 25, 1033–1043. doi:10.1080/14728222.2021.2018418
Chi, Z., Teng, Y., Liu, Y., Gao, L., Yang, J., and andZhang, Z. (2023). Association between klotho and non-alcoholic fatty liver disease and liver fibrosis based on the NHANES 2007–2016. Ann. Hepatology 28, 101125. doi:10.1016/j.aohep.2023.101125
Clare, K., Dillon, J. F., and andBrennan, P. N. (2022). Reactive oxygen species and oxidative stress in the pathogenesis of MAFLD. J. Clin. Transl. Hepatol. 10, 939–946. doi:10.14218/JCTH.2022.00067
Dabravolski, S. A., Bezsonov, E. E., Baig, M. S., Popkova, T. V., Nedosugova, L. V., Starodubova, A. V., et al. (2021). Mitochondrial mutations and genetic factors determining NAFLD risk. Int. J. Mol. Sci. 22, 4459. doi:10.3390/ijms22094459
Day, C. P., and James, O. F. (1998). Steatohepatitis: a tale of two "hits. Gastroenterology 114, 842–845. doi:10.1016/s0016-5085(98)70599-2
Dessein, A. (2021). Clinical utility of polygenic risk scores for predicting NAFLD disorders. J. Hepatol. 74, 769–770. doi:10.1016/j.jhep.2021.02.005
Dhar, D., and Loomba, R. (2021). Emerging metabolic and transcriptomic signature of PNPLA3-associated NASH. Hepatology 73, 1248–1250. doi:10.1002/hep.31735
Dong, X. C. (2019). PNPLA3-A potential therapeutic target for personalized treatment of chronic liver disease. Front. Med. (Lausanne) 6, 304. doi:10.3389/fmed.2019.00304
Eslam, M., Newsome, P. N., Sarin, S. K., Anstee, Q. M., Targher, G., Romero-Gomez, M., et al. (2020). A new definition for metabolic dysfunction-associated fatty liver disease: an international expert consensus statement. J. Hepatology 73, 202–209. doi:10.1016/j.jhep.2020.03.039
Eslam, M., Sanyal, A. J., George, J., and andInternational Consensus, P. (2020). MAFLD: A consensus-driven proposed nomenclature for metabolic associated fatty liver disease. Gastroenterology 158, 1999–2014.e1. doi:10.1053/j.gastro.2019.11.312
Fernandes Silva, L., Vangipurapu, J., Kuulasmaa, T., and andLaakso, M. (2019). An intronic variant in the GCKR gene is associated with multiple lipids. Sci. Rep. 9, 10240. doi:10.1038/s41598-019-46750-3
Flessa, C.-M., Kyrou, I., Nasiri-Ansari, N., Kaltsas, G., Kassi, E., and andRandeva, H. S. (2022). Endoplasmic reticulum stress in nonalcoholic (metabolic associated) fatty liver disease (NAFLD/MAFLD). J. Cell Biochem. 123, 1585–1606. doi:10.1002/jcb.30247
Friedman, S. L., Neuschwander-Tetri, B. A., Rinella, M., and andSanyal, A. J. (2018). Mechanisms of NAFLD development and therapeutic strategies. Nat. Med. 24, 908–922. doi:10.1038/s41591-018-0104-9
Ghodsian, N., Abner, E., Emdin, C. A., Gobeil, E., Taba, N., Haas, M. E., et al. (2021). Electronic health record-based genome-wide meta-analysis provides insights on the genetic architecture of non-alcoholic fatty liver disease. Cell Rep. Med. 2, 100437. doi:10.1016/j.xcrm.2021.100437
Goyenvalle, A., Jimenez-Mallebrera, C., Van Roon, W., Sewing, S., Krieg, A. M., Arechavala-Gomeza, V., et al. (2023). Considerations in the preclinical assessment of the safety of antisense oligonucleotides. Nucleic Acid. Ther. 33, 1–16. doi:10.1089/nat.2022.0061
Grimaudo, S., Pipitone, R. M., Pennisi, G., Celsa, C., Camma, C., Di Marco, V., et al. (2020). Association between PNPLA3 rs738409 C>G variant and liver-related outcomes in patients with nonalcoholic fatty liver disease. Clin. Gastroenterol. Hepatol. 18, 935–944. doi:10.1016/j.cgh.2019.08.011
Heeren, J., and Scheja, L. (2021). Metabolic-associated fatty liver disease and lipoprotein metabolism. Mol. Metab. 50, 101238. doi:10.1016/j.molmet.2021.101238
Hirano, T. (2021). IL-6 in inflammation, autoimmunity and cancer. Int. Immunol. 33, 127–148. doi:10.1093/intimm/dxaa078
Ishizuka, Y., Nakayama, K., Ogawa, A., Makishima, S., Boonvisut, S., Hirao, A., et al. (2014). TRIB1 downregulates hepatic lipogenesis and glycogenesis via multiple molecular interactions. J. Mol. Endocrinol. 52, 145–158. doi:10.1530/JME-13-0243
Ismaiel, A., and Dumitrascu, D. L. (2020). Genetic predisposition in metabolic-dysfunction-associated fatty liver disease and cardiovascular outcomes—systematic review. Eur. J. Clin. Investigation 50, e13331. doi:10.1111/eci.13331
Iwamoto, S., Boonvisut, S., Makishima, S., Ishizuka, Y., Watanabe, K., and andNakayama, K. (2015). The role of TRIB1 in lipid metabolism; from genetics to pathways. Biochem. Soc. Trans. 43, 1063–1068. doi:10.1042/BST20150094
Jamialahmadi, O., Mancina, R. M., Ciociola, E., Tavaglione, F., Luukkonen, P. K., Baselli, G., et al. (2021). Exome-wide association study on alanine aminotransferase identifies sequence variants in the GPAM and APOE associated with fatty liver disease. Gastroenterology 160, 1634–1646.e7. doi:10.1053/j.gastro.2020.12.023
Ji, F., Liu, Y., Hao, J.-G., Wang, L.-P., Dai, M.-J., Shen, G.-F., et al. (2019). KLB gene polymorphism is associated with obesity and non-alcoholic fatty liver disease in the Han Chinese. Aging (Albany NY) 11, 7847–7858. doi:10.18632/aging.102293
Jiang, X., Qian, H., and andDing, W. X. (2021). New glance at the role of TM6SF2 in lipid metabolism and liver cancer. Hepatology 74, 1141–1144. doi:10.1002/hep.31851
Jun, D. W. (2021). An analysis of polygenic risk scores for non-alcoholic fatty liver disease. Clin. Mol. Hepatol. 27, 446–447. doi:10.3350/cmh.2021.0133
Kawaguchi, T., Tsutsumi, T., Nakano, D., and andTorimura, T. (2022). MAFLD: renovation of clinical practice and disease awareness of fatty liver. Hepatol. Res. 52, 422–432. doi:10.1111/hepr.13706
Kaya, E., and Yilmaz, Y. (2022). Metabolic-associated fatty liver disease (MAFLD): A multi-systemic disease beyond the liver. J. Clin. Transl. Hepatol. 10, 329–338. doi:10.14218/JCTH.2021.00178
Khanmohammadi, S., and Kuchay, M. S. (2022). Toll-like receptors and metabolic (dysfunction)-associated fatty liver disease. Pharmacol. Res. 185, 106507. doi:10.1016/j.phrs.2022.106507
Kuro, O. M. (2019). The Klotho proteins in health and disease. Nat. Rev. Nephrol. 15, 27–44. doi:10.1038/s41581-018-0078-3
Li, J., Hua, W., Ji, C., Rui, J., Zhao, Y., Xie, C., et al. (2020). Effect of the patatin-like phospholipase domain containing 3 gene (PNPLA3) I148M polymorphism on the risk and severity of nonalcoholic fatty liver disease and metabolic syndromes: A meta-analysis of paediatric and adolescent individuals. Pediatr. Obes. 15, e12615. doi:10.1111/ijpo.12615
Li, J., Zhao, Y., Zhang, H., Hua, W., Jiao, W., Du, X., et al. (2021). Contribution of Rs780094 and Rs1260326 polymorphisms in GCKR gene to non-alcoholic fatty liver disease: A meta-analysis involving 26,552 participants. Endocr. Metab. Immune Disord. Drug Targets 21, 1696–1708. doi:10.2174/1871530320999201126202706
Li, T. T., Li, T. H., Peng, J., He, B., Liu, L. S., Wei, D. H., et al. (2018). TM6SF2: A novel target for plasma lipid regulation. Atherosclerosis 268, 170–176. doi:10.1016/j.atherosclerosis.2017.11.033
Liu, W. Y., Zhang, X., Li, G., Tang, L. J., Zhu, P. W., Rios, R. S., et al. (2022). Protective association of Klotho rs495392 gene polymorphism against hepatic steatosis in non-alcoholic fatty liver disease patients. Clin. Mol. Hepatol. 28, 183–195. doi:10.3350/cmh.2021.0301
Li, Z. Y., Wu, G., Qiu, C., Zhou, Z. J., Wang, Y. P., Song, G. H., et al. (2022). Mechanism and therapeutic strategy of hepatic TM6SF2-deficient non-alcoholic fatty liver diseases via in vivo and in vitro experiments. World J. Gastroenterol. 28, 2937–2954. doi:10.3748/wjg.v28.i25.2937
Liao, S., An, K., Liu, Z., He, H., An, Z., Su, Q., et al. (2022). Genetic variants associated with metabolic dysfunction-associated fatty liver disease in western China. J. Clin. Lab. Anal. 36, e24626. doi:10.1002/jcla.24626
Linden, D., Ahnmark, A., Pingitore, P., Ciociola, E., Ahlstedt, I., Andreasson, A. C., et al. (2019). Pnpla3 silencing with antisense oligonucleotides ameliorates nonalcoholic steatohepatitis and fibrosis in Pnpla3 I148M knock-in mice. Mol. Metab. 22, 49–61. doi:10.1016/j.molmet.2019.01.013
Liu, Q., Liu, S. S., Zhao, Z. Z., Zhao, B. T., Du, S. X., Jin, W. W., et al. (2019a). TRIB1 rs17321515 gene polymorphism increases the risk of coronary heart disease in general population and non-alcoholic fatty liver disease patients in Chinese Han population. Lipids Health Dis. 18, 165. doi:10.1186/s12944-019-1108-2
Liu, Q., Xue, F., Meng, J., Liu, S. S., Chen, L. Z., Gao, H., et al. (2019b). TRIB1 rs17321515 and rs2954029 gene polymorphisms increase the risk of non-alcoholic fatty liver disease in Chinese Han population. Lipids Health Dis. 18, 61. doi:10.1186/s12944-019-1001-z
Liu, W. Y., Eslam, M., Zheng, K. I., Ma, H. L., Rios, R. S., Lv, M. Z., et al. (2021). Associations of hydroxysteroid 17-beta dehydrogenase 13 variants with liver histology in Chinese patients with metabolic-associated fatty liver disease. J. Clin. Transl. Hepatol. 9, 194–202. doi:10.14218/JCTH.2020.00151
Longo, M., Meroni, M., Paolini, E., Erconi, V., Carli, F., Fortunato, F., et al. (2022). TM6SF2/PNPLA3/MBOAT7 loss-of-function genetic variants impact on NAFLD development and progression both in patients and in in vitro models. Cell Mol. Gastroenterol. Hepatol. 13, 759–788. doi:10.1016/j.jcmgh.2021.11.007
Luo, F., Oldoni, F., and andDas, A. (2022a). TM6SF2: A novel genetic player in nonalcoholic fatty liver and cardiovascular disease. Hepatol. Commun. 6, 448–460. doi:10.1002/hep4.1822
Luo, F., Smagris, E., Martin, S. A., Vale, G., Mcdonald, J. G., Fletcher, J. A., et al. (2022b). Hepatic TM6SF2 is required for lipidation of VLDL in a pre-golgi compartment in mice and rats. Cell Mol. Gastroenterol. Hepatol. 13, 879–899. doi:10.1016/j.jcmgh.2021.12.008
Luukkonen, P. K., Juuti, A., Sammalkorpi, H., Penttila, A. K., Oresic, M., Hyotylainen, T., et al. (2020). MARC1 variant rs2642438 increases hepatic phosphatidylcholines and decreases severity of non-alcoholic fatty liver disease in humans. J. Hepatol. 73, 725–726. doi:10.1016/j.jhep.2020.04.021
Makri, E. S., Makri, E., and andPolyzos, S. A. (2022). Combination therapies for nonalcoholic fatty liver disease. J. Pers. Med. 12, 1166. doi:10.3390/jpm12071166
Martin-Gonzalez, C., Espelosin-Ortega, E., Abreu-Gonzalez, P., Fernandez-Rodriguez, C., Vera-Delgado, V. E., Gonzalez-Navarrete, L., et al. (2022). Klotho levels and their relationship with inflammation and survival among alcoholic patients. Biomolecules 12, 1151. doi:10.3390/biom12081151
Massey, W. J., Varadharajan, V., Banerjee, R., Brown, A. L., Horak, A. J., Hohe, R. C., et al. (2023). MBOAT7-driven lysophosphatidylinositol acylation in adipocytes contributes to systemic glucose homeostasis. J. Lipid Res. 64, 100349. doi:10.1016/j.jlr.2023.100349
Meroni, M., Longo, M., Tria, G., and andDongiovanni, P. (2021). Genetics is of the essence to face NAFLD. Biomedicines 9, 1359. doi:10.3390/biomedicines9101359
Motomura, T., Amirneni, S., Diaz-Aragon, R., Faccioli, L., Malizio, M., Coard, M., et al. (2021). Is HSD17B13 genetic variant a protector for liver dysfunction? Future perspective as a potential therapeutic target. J. Personalized Med. 11, 619. doi:10.3390/jpm11070619
Nascimento, J. C. R., Matos, G. A., Pereira, L. C., Mourao, A., Sampaio, A. M., Oria, R. B., et al. (2020). Impact of apolipoprotein E genetic polymorphisms on liver disease: an essential review. Ann. Hepatol. 19, 24–30. doi:10.1016/j.aohep.2019.07.011
Newberry, E. P., Hall, Z., Xie, Y., Molitor, E. A., Bayguinov, P. O., Strout, G. W., et al. (2021). Liver-specific deletion of mouse Tm6sf2 promotes steatosis, fibrosis, and hepatocellular cancer. Hepatology 74, 1203–1219. doi:10.1002/hep.31771
Ohnishi, M., Kato, S., Akiyoshi, J., Atfi, A., and andRazzaque, M. S. (2011). Dietary and genetic evidence for enhancing glucose metabolism and reducing obesity by inhibiting klotho functions. FASEB J. 25, 2031–2039. doi:10.1096/fj.10-167056
Panera, N., Meroni, M., Longo, M., Crudele, A., Valenti, L., Bellacchio, E., et al. (2021). The KLB rs17618244 gene variant is associated with fibrosing MAFLD by promoting hepatic stellate cell activation. EBioMedicine 65, 103249. doi:10.1016/j.ebiom.2021.103249
Park, J., Zhao, Y., Zhang, F., Zhang, S., Kwong, A. C., Zhang, Y., et al. (2023). IL-6/STAT3 axis dictates the PNPLA3-mediated susceptibility to non-alcoholic fatty liver disease. J. Hepatol. 78, 45–56. doi:10.1016/j.jhep.2022.08.022
Paternostro, R., Staufer, K., Traussnigg, S., Stattermayer, A. F., Halilbasic, E., Keritam, O., et al. (2021). Combined effects of PNPLA3, TM6SF2 and HSD17B13 variants on severity of biopsy-proven non-alcoholic fatty liver disease. Hepatol. Int. 15, 922–933. doi:10.1007/s12072-021-10200-y
Peter, A., Stefan, N., Cegan, A., Walenta, M., Wagner, S., Königsrainer, A., et al. (2011). Hepatic glucokinase expression is associated with lipogenesis and fatty liver in humans. J. Clin. Endocrinol. Metabolism 96, E1126–E1130. doi:10.1210/jc.2010-2017
Pingitore, P., and Romeo, S. (2019). The role of PNPLA3 in health and disease. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 1864, 900–906. doi:10.1016/j.bbalip.2018.06.018
Pipitone, R. M., Ciccioli, C., Infantino, G., La Mantia, C., Parisi, S., Tulone, A., et al. (2023). Mafld: a multisystem disease. Ther. Adv. Endocrinol. Metab. 14, 20420188221145549. doi:10.1177/20420188221145549
Powell, E. E., Wong, V. W., and andRinella, M. (2021). Non-alcoholic fatty liver disease. Lancet 397, 2212–2224. doi:10.1016/S0140-6736(20)32511-3
Qadri, S., Lallukka-Bruck, S., Luukkonen, P. K., Zhou, Y., Gastaldelli, A., Orho-Melander, M., et al. (2020). The PNPLA3-I148M variant increases polyunsaturated triglycerides in human adipose tissue. Liver Int. 40, 2128–2138. doi:10.1111/liv.14507
Quiroz-Figueroa, K., Vitali, C., Conlon, D. M., Millar, J. S., Tobias, J. W., Bauer, R. C., et al. (2021). TRIB1 regulates LDL metabolism through CEBPα-mediated effects on the LDL receptor in hepatocytes. J. Clin. Invest. 131, e146775. doi:10.1172/JCI146775
Raza, S., Rajak, S., Upadhyay, A., Tewari, A., and andAnthony Sinha, R. (2021). Current treatment paradigms and emerging therapies for NAFLD/NASH. Front. Biosci. (Landmark Ed. 26, 206–237. doi:10.2741/4892
Romeo, S., Kozlitina, J., Xing, C., Pertsemlidis, A., Cox, D., Pennacchio, L. A., et al. (2008). Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat. Genet. 40, 1461–1465. doi:10.1038/ng.257
Romeo, S., Sanyal, A., and andValenti, L. (2020). Leveraging human genetics to identify potential new treatments for fatty liver disease. Cell Metab. 31, 35–45. doi:10.1016/j.cmet.2019.12.002
Sakurai, Y., Kubota, N., Yamauchi, T., and andKadowaki, T. (2021). Role of insulin resistance in MAFLD. Int. J. Mol. Sci. 22, 4156. doi:10.3390/ijms22084156
Salari, N., Darvishi, N., Mansouri, K., Ghasemi, H., Hosseinian-Far, M., Darvishi, F., et al. (2021). Association between PNPLA3 rs738409 polymorphism and nonalcoholic fatty liver disease: a systematic review and meta-analysis. BMC Endocr. Disord. 21, 125. doi:10.1186/s12902-021-00789-4
Sangro, P., De La Torre Aláez, M., Sangro, B., and andD’avola, D. (2023). Metabolic dysfunction–associated fatty liver disease (MAFLD): an update of the recent advances in pharmacological treatment. J. Physiology Biochem. 2023. doi:10.1007/s13105-023-00954-4
Scharner, J., and Aznarez, I. (2021). Clinical applications of single-stranded oligonucleotides: current landscape of approved and in-development therapeutics. Mol. Ther. 29, 540–554. doi:10.1016/j.ymthe.2020.12.022
Shang, L., and Mashek, D. G. (2020). The underpinnings of PNPLA3-mediated fatty liver emerge. Hepatol. Baltim. Md 71, 375–377. doi:10.1002/hep.30888
Sharma, D., and Mandal, P. (2022). NAFLD: genetics and its clinical implications. Clin. Res. Hepatol. Gastroenterol. 46, 102003. doi:10.1016/j.clinre.2022.102003
Stefano, J. T., Duarte, S. M. B., Ribeiro Leite Altikes, R. G., and andOliveira, C. P. (2023). Non-pharmacological management options for MAFLD: a practical guide. Ther. Adv. Endocrinol. Metab. 14, 20420188231160394. doi:10.1177/20420188231160394
Stender, S., and Loomba, R. (2020). PNPLA3 genotype and risk of liver and all-cause mortality. Hepatology 71, 777–779. doi:10.1002/hep.31113
Su, W., Wu, S., Yang, Y., Guo, Y., Zhang, H., Su, J., et al. (2022). Phosphorylation of 17β-hydroxysteroid dehydrogenase 13 at serine 33 attenuates nonalcoholic fatty liver disease in mice. Nat. Commun. 13, 6577. doi:10.1038/s41467-022-34299-1
Sveinbjornsson, G., Ulfarsson, M. O., Thorolfsdottir, R. B., Jonsson, B. A., Einarsson, E., Gunnlaugsson, G., et al. (2022). Multiomics study of nonalcoholic fatty liver disease. Nat. Genet. 54, 1652–1663. doi:10.1038/s41588-022-01199-5
Tang, S., Zhang, J., Mei, T. T., Zhang, W. Y., Zheng, S. J., and andYu, H. B. (2021). Association of HSD17B13 rs72613567: tA allelic variant with liver disease: review and meta-analysis. BMC Gastroenterol. 21, 490. doi:10.1186/s12876-021-02067-y
Teo, K., Abeysekera, K. W. M., Adams, L., Aigner, E., Anstee, Q. M., Banales, J. M., et al. (2021). rs641738C>T near MBOAT7 is associated with liver fat, ALT and fibrosis in NAFLD: A meta-analysis. J. Hepatology 74, 20–30. doi:10.1016/j.jhep.2020.08.027
Thangapandi, V. R., Knittelfelder, O., Brosch, M., Patsenker, E., Vvedenskaya, O., Buch, S., et al. (2021). Loss of hepatic Mboat7 leads to liver fibrosis. Gut 70, 940–950. doi:10.1136/gutjnl-2020-320853
Tilson, S. G., Morell, C. M., Lenaerts, A. S., Park, S. B., Hu, Z., Jenkins, B., et al. (2021). Modeling PNPLA3-associated NAFLD using human-induced pluripotent stem cells. Hepatology 74, 2998–3017. doi:10.1002/hep.32063
Ting, Y.-W., Kong, A. S.-Y., Zain, S. M., Chan, W.-K., Tan, H.-L., Mohamed, Z., et al. (2021). Loss-of-function HSD17B13 variants, non-alcoholic steatohepatitis and adverse liver outcomes: results from a multi-ethnic asian cohort. Clin. Mol. Hepatology 27, 486–498. doi:10.3350/cmh.2020.0162
Trepo, E., Romeo, S., Zucman-Rossi, J., and andNahon, P. (2016). PNPLA3 gene in liver diseases. J. Hepatol. 65, 399–412. doi:10.1016/j.jhep.2016.03.011
Tsai, E., and Lee, T. P. (2018). Diagnosis and evaluation of nonalcoholic fatty liver disease/nonalcoholic steatohepatitis, including noninvasive biomarkers and transient elastography. Clin. Liver Dis. 22, 73–92. doi:10.1016/j.cld.2017.08.004
Varadharajan, V., Massey, W. J., and andBrown, J. M. (2022). Membrane-bound O-acyltransferase 7 (MBOAT7)-driven phosphatidylinositol remodeling in advanced liver disease. J. Lipid Res. 63, 100234. doi:10.1016/j.jlr.2022.100234
Vujkovic, M., Ramdas, S., Lorenz, K. M., Guo, X., Darlay, R., Cordell, H. J., et al. (2022). A multiancestry genome-wide association study of unexplained chronic ALT elevation as a proxy for nonalcoholic fatty liver disease with histological and radiological validation. Nat. Genet. 54, 761–771. doi:10.1038/s41588-022-01078-z
Wang, J., Conti, D. V., Bogumil, D., Sheng, X., Noureddin, M., Wilkens, L. R., et al. (2021). Association of genetic risk score with NAFLD in an ethnically diverse cohort. Hepatol. Commun. 5, 1689–1703. doi:10.1002/hep4.1751
Wang, M., Li, L., Xu, Y., Du, J., and andLing, C. (2022). Roles of hepatic stellate cells in NAFLD: from the perspective of inflammation and fibrosis. Front. Pharmacol. 13, 958428. doi:10.3389/fphar.2022.958428
Wang, Y., Kory, N., Basuray, S., Cohen, J. C., and andHobbs, H. H. (2019). PNPLA3, CGI-58, and inhibition of hepatic triglyceride hydrolysis in mice. Hepatology 69, 2427–2441. doi:10.1002/hep.30583
Wei, Z., Huang, Z., Song, Z., Zhao, W., Zhao, D., Tan, Y., et al. (2023). Metabolic dysfunction-associated fatty liver disease and incident heart failure risk: the kailuan cohort study. Diabetology Metabolic Syndrome 15, 137. doi:10.1186/s13098-023-01102-0
Xia, M., Zeng, H., Wang, S., Tang, H., and andGao, X. (2021). Insights into contribution of genetic variants towards the susceptibility of MAFLD revealed by the NMR-based lipoprotein profiling. J. Hepatol. 74, 974–977. doi:10.1016/j.jhep.2020.10.019
Xiang, H., Wu, Z., Wang, J., and andWu, T. (2021). Research progress, challenges and perspectives on PNPLA3 and its variants in Liver Diseases. J. Cancer 12, 5929–5937. doi:10.7150/jca.57951
Xu, X., Poulsen, K. L., Wu, L., Liu, S., Miyata, T., Song, Q., et al. (2022). Targeted therapeutics and novel signaling pathways in non-alcohol-associated fatty liver/steatohepatitis (NAFL/NASH). Signal Transduct. Target Ther. 7, 287. doi:10.1038/s41392-022-01119-3
Xue, W. Y., Zhang, L., Liu, C. M., Gao, Y., Li, S. J., Huai, Z. Y., et al. (2022). Research progress on the relationship between TM6SF2 rs58542926 polymorphism and non-alcoholic fatty liver disease. Expert Rev. Gastroenterol. Hepatol. 16, 97–107. doi:10.1080/17474124.2022.2032661
Yang, A., Zhu, X., Zhang, L., Zhang, Y., Zhang, D., Jin, M., et al. (2022). Non-invasive evaluation of NAFLD and the contribution of genes: an MRI-PDFF-based cross-sectional study. Hepatol. Int. 16, 1035–1051. doi:10.1007/s12072-022-10355-2
Yu, L., Li, Y., Grise, A., and andWang, H. (2020). CGI-58: versatile regulator of intracellular lipid droplet homeostasis. Adv. Exp. Med. Biol. 1276, 197–222. doi:10.1007/978-981-15-6082-8_13
Yuan, F., Gu, Z., Bi, Y., Yuan, R., Niu, W., Ren, D., et al. (2022). The association between rs1260326 with the risk of NAFLD and the mediation effect of triglyceride on NAFLD in the elderly Chinese Han population. Aging (Albany NY) 14, 2736–2747. doi:10.18632/aging.203970
Yuan, S., Liu, H., Yuan, D., Xu, J., Chen, Y., Xu, X., et al. (2020). PNPLA3 I148M mediates the regulatory effect of NF-kB on inflammation in PA-treated HepG2 cells. J. Cell Mol. Med. 24, 1541–1552. doi:10.1111/jcmm.14839
Zhang, H. B., Su, W., Xu, H., Zhang, X. Y., and andGuan, Y. F. (2021). HSD17B13: A potential therapeutic target for NAFLD. Front. Mol. Biosci. 8, 824776. doi:10.3389/fmolb.2021.824776
Zhang, Q. H., Yin, R. X., Chen, W. X., Cao, X. L., and andWu, J. Z. (2019). TRIB1 and TRPS1 variants, G × G and G × E interactions on serum lipid levels, the risk of coronary heart disease and ischemic stroke. Sci. Rep. 9, 2376. doi:10.1038/s41598-019-38765-7
Zhang, X., Liu, S., Dong, Q., Xin, Y., and andXuan, S. (2018). The genetics of clinical liver diseases: insight into the TM6SF2 E167K variant. J. Clin. Transl. Hepatol. 6, 326–331. doi:10.14218/JCTH.2018.00022
Zhang, X., Zhang, B., Zhang, C., Sun, G., and andSun, X. (2021). Current progress in delineating the roles of pseudokinase TRIB1 in controlling human diseases. J. Cancer 12, 6012–6020. doi:10.7150/jca.51627
Zhou, T., Li, H., Zhong, H., Zhong, Z., and andLin, S. (2020). Association of apoE gene polymorphisms with lipid metabolism in renal diseases. Afr. Health Sci. 20, 1368–1381. doi:10.4314/ahs.v20i3.43
Keywords: MAFLD, genetic variation, PNPLA3, HSD17B13, therapeutic potential
Citation: Shi F, Zhao M, Zheng S, Zheng L and Wang H (2023) Advances in genetic variation in metabolism-related fatty liver disease. Front. Genet. 14:1213916. doi: 10.3389/fgene.2023.1213916
Received: 28 April 2023; Accepted: 30 August 2023;
Published: 11 September 2023.
Edited by:
Mujeeb Zafar Banday, Government Medical College (GMC), IndiaReviewed by:
Zaki A. Sherif, Howard University, United StatesCopyright © 2023 Shi, Zhao, Zheng, Zheng and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Haiqiang Wang, aGFpcWlhbmc5MTVAMTYzLmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.