 Markus Ponleitner1†
Markus Ponleitner1† Daniela Maria Allmer2†
Daniela Maria Allmer2† Manfred Hecking2†Constantin Gatterer3†
Manfred Hecking2†Constantin Gatterer3† Senta Graf3†Mateja Smogavec4†Franco Laccone4†
Senta Graf3†Mateja Smogavec4†Franco Laccone4† Paulus Stefan Rommer1†
Paulus Stefan Rommer1† Gere Sunder-Plassmann2*†
Gere Sunder-Plassmann2*†- 1Department of Neurology, Comprehensive Center for Clinical Neurosciences and Mental Health, Medical University of Vienna, Vienna, Austria
- 2Division of Nephrology and Dialysis, Department of Medicine III, Medical University of Vienna, Vienna, Austria
- 3Division of Cardiology, Department of Medicine II, Medical University of Vienna, Vienna, Austria
- 4Institute for Human Genetics, Medical University of Vienna, Vienna, Austria
We describe the case of a 44-year-old male patient with a longstanding history of microhematuria and mildly impaired kidney function (CKD G2A1). The family history disclosed three females who also had microhematuria. Genetic testing by whole exome sequencing revealed two novel variants in COL4A4 (NM_000092.5: c.1181G>T, NP_000083.3: p.Gly394Val, heterozygous, likely pathogenic; Alport syndrome, OMIM# 141200, 203780) and GLA (NM_000169.3: c.460A>G, NP_000160.1: p.Ile154Val, hemizygous, variant of uncertain significance; Fabry disease, OMIM# 301500), respectively. Extensive phenotyping revealed no biochemical or clinical evidence for the presence of Fabry disease. Thus, the GLA c.460A>G, p.Ile154Val, is to be classified as a benign variant, whereas the COL4A4 c.1181G>T, p.Gly394Val confirms the diagnosis of autosomal dominant Alport syndrome in this patient.
1 Introduction
Autosomal dominant Alport syndrome, a collagen IV disease, and Fabry disease, a lysosomal storage disease, can lead to kidney failure and other organ manifestations (Kashtan et al., 2018; Li et al., 2022). Therefore, a timely diagnosis is mandatory to allow specific treatment for the delay of disease progression.
Fabry Disease constitutes a rare X-linked lysosomal storage disorder. Pathogenic GLA-variants cause significant reduction in α-galactosidase A-activity in males, resulting in accumulation of the pathogenic sphingolipid metabolites globotriaosylceramide (Gb3) and globotriaosylsphingosine (Lyso-Gb3) (Germain, 2010). Disease manifestation in affected patients includes damage to the heart, kidneys and central nervous system, among others (Arends et al., 2017). Phenotype-genotype correlations allow stratification into the most severe form of Fabry disease termed “classical”-form, as well as the “late onset”-form. Depending on X-inactivation, symptoms of female patients with pathogenic variants range from no signs of disease to severe affection comparable to classic males (Echevarria et al., 2016).
Pathogenic variants in the genes encoding the different α-chains (α3, α4, α5) of collagen type IV can cause Alport syndrome, which results in a dysfunctional glomerular basement membrane with organ manifestation including microhematuria, proteinuria and progressive loss of renal function, as well as sensorineural hearing loss and ocular pathologies in case of X-linked Alport syndrome (Kashtan et al., 2018).
To the best of our knowledge the variants in the GLA and COL4A4 genes detected in this patient, have not been described in the scientific literature or established databases so far. Thus, we give a detailed description of the clinical phenotype of this patient to classify his kidney disease correctly.
This description is of particular importance, because miss-classification of genetic variants would disconcert patients and may waste healthcare resources, especially in regard to expensive therapies for a rare disease.
2 Case description
We report on a 44-year-old male musician, born in Brazil, who had spent several years working and living in Austria at the time of diagnosis. He was referred to the Nephrology Outpatient Service at the Division of Nephrology and Dialysis, Department of Medicine III, of the Medical University of Vienna because of mildly impaired kidney function and microhematuria.
His history disclosed the presence of microhematuria for more than 20 years. According to the patient, his sister, mother and grand-mother (mother’s side)—all born in Brazil—also suffer(ed) from microhematuria, and kidney stones. The further familial history remained unrevealing, without juvenile cardiovascular events or kidney failure (Figure 1)
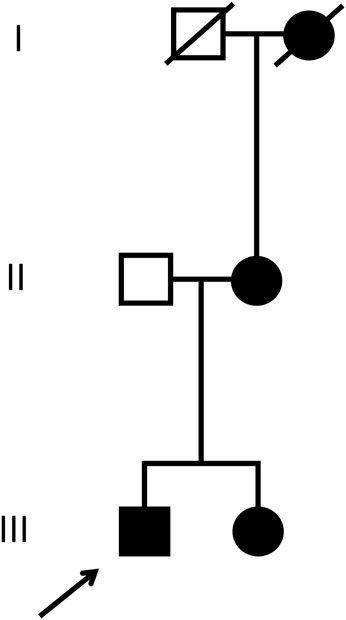
FIGURE 1. Pedigree. The patient discussed in the current case report (index patient) is marked with the arrow. Male individuals are shown as squares, females as circles. Affected (microhematuria) and healthy individuals are denoted with filled and empty symbols, respectively. Symbols of deceased individuals are crossed out.
During childhood, the patient had eye surgery due to strabismus. Four years before the current presentation, the patient suffered from cholecystitis and was treated with surgical cholecystectomy. Two years later, he received endoscopic retrograde cholangiopancreatography (ERCP) with stone-extraction due to gallstone-induced mild pancreatitis. No other previous illnesses were documented.
At the time we initiated the extensive workup, the patient reported no complaints and was feeling healthy. The physical examination was unremarkable, he was normotensive and has not been prescribed any medication.
3 Diagnostic assessment
Kidney and cardiac biomarkers over time are indicated in Table 1, confirming the presence of chronic kidney disease (CKD) stage G2A1 and persistent microhematuria, whereas serum concentrations of cardiac biomarkers were within the reference range.

TABLE 1. Renal and cardiac laboratory results.
Adhering to diagnostic standards in our clinic, whole exome sequencing was performed after proper patient counselling to further evaluate the suspected kidney disease in this patient and revealed two novel variants in COL4A4 (NM_000092.5: c.1181G>T, NP_000083.3: p.Gly394Val, heterozygous, varsome.com: likely pathogenic; Alport syndrome, OMIM# 141200, 203780; ClinVar ID SCV003918893) and GLA (NM_000169.3: c.460A>G, NP_000160.1: p.Ile154Val, hemizygous, varsome.com: variant of uncertain significance; Fabry disease, OMIM# 301500; ClinVar ID SCV003918894), respectively (Groopman et al., 2019).
These findings pointed to a diagnosis of Alport syndrome. The variant of uncertain significance in GLA, however, deserved further clinical assessments. Table 2 shows details of clinical and biochemical investigation, all of which did not support the diagnosis of Fabry disease.
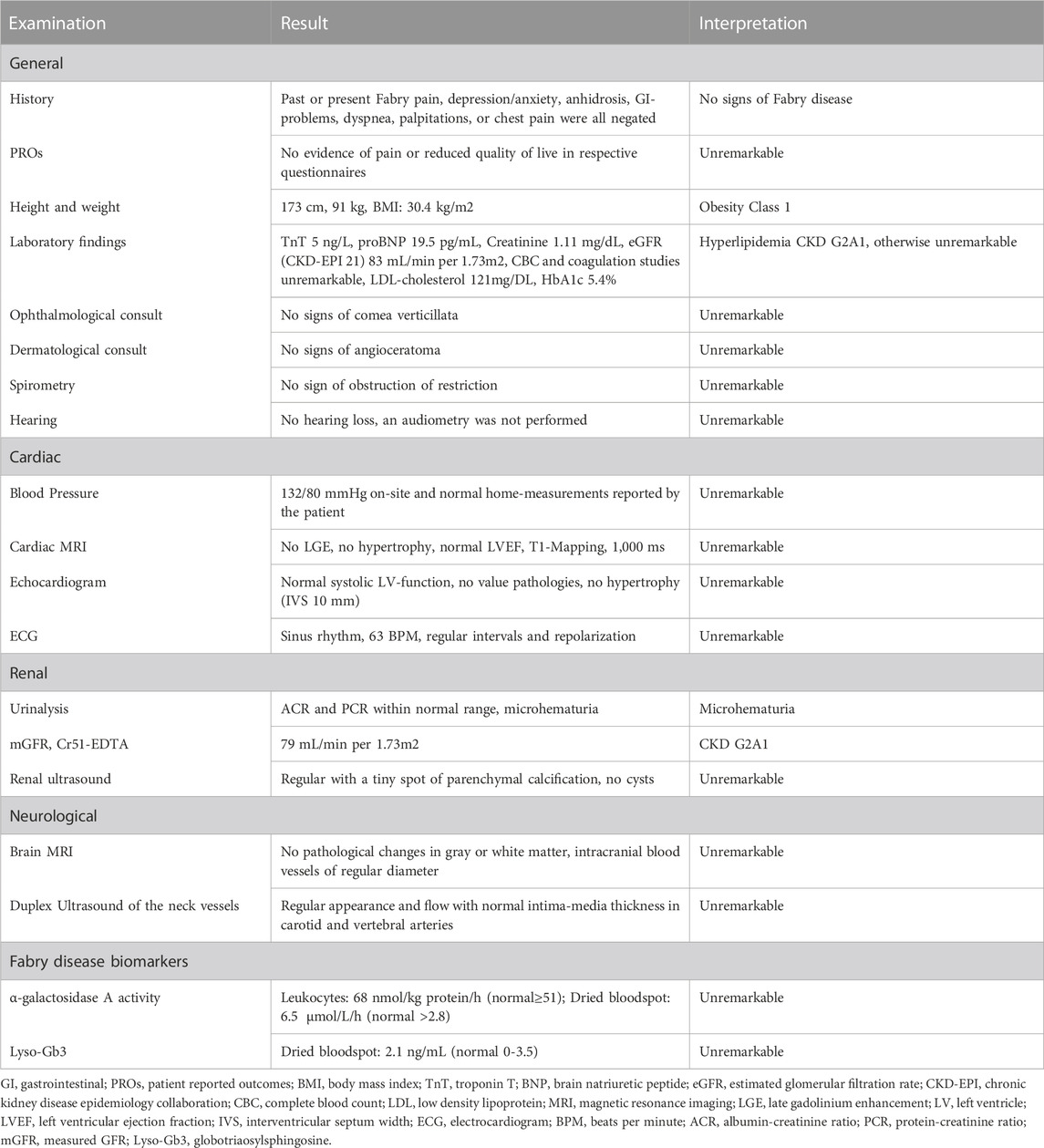
TABLE 2. Examination results.
4 Outcome
The patient received extensive counselling for his diagnosis of Alport syndrome. In addition, we provided the patient with advice regarding information for his relatives (all living abroad) and suggested they consult a nephrologist or geneticist for further consideration.
Furthermore, we scheduled doctor’s appointments annually to evaluate kidney function. To date, we did not initiate medical treatment of his disorder.
5 Discussion
Whole exome sequencing in this 44-year-old male patient who presented with CKD stage G2A1 and microhematuria suggested the potential presence of a kidney disease possibly caused by variants in two different genes, namely, autosomal dominant Alport syndrome and Fabry disease.
The likely pathogenic COL4A4-variant that was identified in the patient is located in the “triple helical domain” (consisting of repeats of the pattern Gly-X-Y) of the extra-cellular matrix protein collagen type IV-α4. The triple helical domain allows 3 type IV collagen monomers to wind together into a triple helix and form a protomer. The resulting “triple helix” mediates structural integrity and interaction with various proteins. The absence of the glycine side chain which normally lies within the helix, facing away from the surface, allows a flexible but tightly packed conformation of the triple helix. The X and Y positions in the motif are variable (often proline), are presented on the outside of the helix and can be modified. This variability in the Gly-X-Y repeat is possible without greatly affecting the stability of the triple helix. However, replacement of the glycine residue significantly affects the integrity of the triple helix and consequently the extracellular matrix, as it can no longer be packed as tightly, and protein-protein interactions and modification of the outer side chains are impaired (Fidler et al., 2018; Gibson et al., 2022).
The second genetic variant of concern in this patient, GLA c.460A>G, p.lle154Val, was categorized as variant of uncertain significance. The Ile154 is part of a helix of the N-terminal (β/α)8-barrel structure of α-galactosidase A which is located on the outside of the protein. The side chain of the affected isoleucine itself also faces more outwards and does not seem to play a role in the proteins (α-galactosidase A’s) hydrophobic core. Also, the Ile154 lies between two aspartic acid residues and itself is a hydrophobic amino acid, which is replaced by valine, another hydrophobic amino acid (Garman, 2007; Sugawara et al., 2008). Conservation analysis by MutationTaster revealed that valine also occurs at this position in C. elegans, whereas in D. melanogaster the position is occupied by lysine, a basic amino acid. According to SIFT, this position may vary in other species; in humans, chimpanzees, macaques and mice, however, the isoleucine is conserved.
Although hematuria has been described in case-reports of Fabry-patients (Solis et al., 2010; Minami et al., 2021), it is not considered a hallmark manifestation of kidney involvement due to Fabry disease, but of Alport syndrome. In line with the retained α-galactosidase A activity, no pathological findings specific for Fabry disease were obtained by extensive phenotyping of our patient (detailed in Table 2). It is therefore easily conceivable that the microhematuria in conjunction with the slightly impaired kidney function seen in our patient is not caused by Fabry disease, but instead associated with the novel COL4A4-variant, causing autosomal dominant Alport Syndrome (Kashtan et al., 2018). Furthermore, normal enzyme activity found in this patient permits interpretation of the GLA-variant as non-pathogenic (Ortiz et al., 2018).
The lack of a pathological examination of renal tissue may be considered as a limitation to our study. Indeed, a kidney biopsy would have put the results of genetic testing in better context with the renal pathology in this patient. We would have expected findings in line with Alport disease, i.e., alterations of the glomerular basement membrane and no evidence for pathological glycolipid storage, i.e., zebra bodies in podocytes and other kidney cells by electron microscopy, which occur in Fabry disease (Rumpelt, 1980; Askari et al., 2007). In light of the unimpaired α-galactosidase A-function in this hemizygous male, the kidney biopsy would not be required for establishing the diagnosis of Fabry disease. The kidney biopsy, however, was denied by the patient.
Furthermore, one could argue that the clinical work-up in a male patient with preserved α-galactosidase A activity (see Table 2) can be considered superfluous. However, given the controversial discussion on the pathogenicity of other GLA variants with variable enzyme activity, such as p.Arg118Cys or p.Asp313Tyr, our case should be considered informative for the medical community (Lenders et al., 2013; Ferreira et al., 2015; Oder et al., 2016; 2018; Koulousios et al., 2017; Talbot and Nicholls, 2019).
Taken together, the phenotyping of the presented case suggests classification of the novel GLA-variant (NM_000092.5: c.460A>G, NP_000083.3: p.Ile154Val) as benign and classification of the novel COL4A4-variant (NM_000169.3: c.1181G>T, NP_000160.1: p.Gly394Val) as likely pathogenic. In the context of the work-up presented in this case, the clinical phenotype of Alport syndrome associated with this novel COL4A4-variant seems to be mild.
Patient consent
The patient provided written informed consent for the publication of this case report.
Data availability statement
The datasets for this article are not publicly available due to concerns regarding participant/patient anonymity. Requests to access the datasets should be directed to the corresponding author.
Ethics statement
Ethical review and approval was not required for the study on human participants in accordance with the local legislation and institutional requirements. The patients/participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article. Written informed consent was obtained from the participant/patient(s) for the publication of this case report.
Author contributions
MP collected the data and drafted and revised the manuscript. DA assisted in biochemical interpretation of detected variants mutations and drafted the respective parts of the manuscript. MH assisted in manuscript revision. CG helped in data acquisition and manuscript revision. SG helped in assessment of data and manuscript revision. MS and FL did the genetic analysis. PSR assisted in revision of the manuscript. GSP conceptualized the project and helped in revising the manuscript. All authors contributed to the article and approved the submitted version.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2023.1211858/full#supplementary-material
References
Arends, M., Wanner, C., Hughes, D., Mehta, A., Oder, D., Watkinson, O. T., et al. (2017). Characterization of classical and nonclassical Fabry disease: A multicenter study. J. Am. Soc. Nephrol. 28, 1631–1641. doi:10.1681/ASN.2016090964
Askari, H., Kaneski, C. R., Semino-Mora, C., Desai, P., Ang, A., Kleiner, D. E., et al. (2007). Cellular and tissue localization of globotriaosylceramide in Fabry disease. Virchows Arch. 451, 823–834. doi:10.1007/s00428-007-0468-6
Echevarria, L., Benistan, K., Toussaint, A., Dubourg, O., Hagege, A. A., Eladari, D., et al. (2016). X-chromosome inactivation in female patients with Fabry disease. Clin. Genet. 89, 44–54. doi:10.1111/cge.12613
Ferreira, S., Ortiz, A., Germain, D. P., Viana-Baptista, M., Caldeira-Gomes, A., Camprecios, M., et al. (2015). The alpha-galactosidase A p.Arg118Cys variant does not cause a Fabry disease phenotype: Data from individual patients and family studies. Mol. Genet. Metabolism 114, 248–258. doi:10.1016/j.ymgme.2014.11.004
Fidler, A. L., Boudko, S. P., Rokas, A., and Hudson, B. G. (2018). The triple helix of collagens - an ancient protein structure that enabled animal multicellularity and tissue evolution. J. Cell Sci. 131, jcs203950. doi:10.1242/jcs.203950
Garman, S. C. (2007). Structure-function relationships in alpha-galactosidase A. Acta Paediatr. 96, 6–16. doi:10.1111/j.1651-2227.2007.00198.x
Gibson, J. T., Huang, M., Shenelli Croos Dabrera, M., Shukla, K., Rothe, H., Hilbert, P., et al. (2022). Genotype-phenotype correlations for COL4A3-COL4A5 variants resulting in Gly substitutions in Alport syndrome. Sci. Rep. 12, 2722. doi:10.1038/s41598-022-06525-9
Groopman, E. E., Marasa, M., Cameron-Christie, S., Petrovski, S., Aggarwal, V. S., Milo-Rasouly, H., et al. (2019). Diagnostic utility of exome sequencing for kidney disease. N. Engl. J. Med. 380, 142–151. doi:10.1056/NEJMoa1806891
Kashtan, C. E., Ding, J., Garosi, G., Heidet, L., Massella, L., Nakanishi, K., et al. (2018). Alport syndrome: A unified classification of genetic disorders of collagen IV α345: A position paper of the Alport syndrome classification working group. Kidney Int. 93, 1045–1051. doi:10.1016/j.kint.2017.12.018
Koulousios, K., Stylianou, K., Pateinakis, P., Zamanakou, M., Loules, G., Manou, E., et al. (2017). Fabry disease due to D313Y and novel GLA mutations. BMJ Open 7, e017098. doi:10.1136/bmjopen-2017-017098
Lenders, M., Duning, T., Schelleckes, M., Schmitz, B., Stander, S., Rolfs, A., et al. (2013). Multifocal white matter lesions associated with the D313Y mutation of the α-galactosidase A gene. PLoS One 8, e55565. doi:10.1371/journal.pone.0055565
Li, X., Ren, X., Zhang, Y., Ding, L., Huo, M., and Li, Q. (2022). Fabry disease: Mechanism and therapeutics strategies. Front. Pharmacol. 13, 1025740. doi:10.3389/fphar.2022.1025740
Minami, M., Mizuma, E., Nakahara, M., Oda, Y., Yoshimine, H., Tokunaga, K., et al. (2021). A case of latent heterozygous Fabry disease in a female living kidney donor candidate. Cen. Case Rep. 10, 30–34. doi:10.1007/s13730-020-00510-9
Oder, D., Üçeyler, N., Liu, D., Hu, K., Petritsch, B., Sommer, C., et al. (2016). Organ manifestations and long-term outcome of Fabry disease in patients with the GLA haplotype D313Y. BMJ Open 6, e010422. doi:10.1136/bmjopen-2015-010422
Oder, D., Wanner, C., and Nordbeck, P. (2018). The D313Y genotype-Pathogenic mutation or polymorphism. Clin. Genet. 93, 1257. doi:10.1111/cge.13237
Ortiz, A., Germain, D. P., Desnick, R. J., Politei, J., Mauer, M., Burlina, A., et al. (2018). Fabry disease revisited: Management and treatment recommendations for adult patients. Mol. Genet. Metab. 123, 416–427. doi:10.1016/j.ymgme.2018.02.014
Rumpelt, H. J. (1980). Hereditary nephropathy (Alport syndrome): Correlation of clinical data with glomerular basement membrane alterations. Clin. Nephrol. 13, 203–207.
Solis, M. A., Pascual, B., Boscá, M., Ramos, V., Carda, C., Monteagudo, C., et al. (2010). New mutation in female patient with renal variant of Fabry disease and HIV. J. Nephrol. 23, 231–233.
Sugawara, K., Ohno, K., Saito, S., and Sakuraba, H. (2008). Structural characterization of mutant alpha-galactosidases causing Fabry disease. J. Hum. Genet. 53, 812–824. doi:10.1007/s10038-008-0316-9
Keywords: Alport syndrome, case report, chronic kidney disease, COL4A4, Fabry disease, GLA, microhematuria
Citation: Ponleitner M, Allmer DM, Hecking M, Gatterer C, Graf S, Smogavec M, Laccone F, Rommer PS and Sunder-Plassmann G (2023) Phenotyping of a novel COL4A4 and novel GLA variant in a patient presenting with microhematuria and mildly impaired kidney function: a case report. Front. Genet. 14:1211858. doi: 10.3389/fgene.2023.1211858
Received: 25 April 2023; Accepted: 18 May 2023;
Published: 01 June 2023.
Edited by:
Andrea Dardis, University Hospital of Udine, ItalyReviewed by:
Paula Rozenfeld, CONICET Instituto de Estudios Inmunológicos y Fisiopatalógicos (IIFP), ArgentinaClaudia Banescu, University of Medicine, Pharmacy, Sciences and Technology of TârguMureş, Romania
Copyright © 2023 Ponleitner, Allmer, Hecking, Gatterer, Graf, Smogavec, Laccone, Rommer and Sunder-Plassmann. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gere Sunder-Plassmann, Z2VyZS5zdW5kZXItcGxhc3NtYW5uQG1lZHVuaXdpZW4uYWMuYXQ=
†ORCID: Markus Ponleitner, orcid.org/0000-0002-3709-1845; Daniela Maria Allmer, orcid.org/0009-0000-8060-0118; Manfred Hecking, orcid.org/0000-0002-8047-2395; Constantin Gatterer, orcid.org/0000-0003-1029-085X; Senta Graf, orcid.org/0000-0002-4443-8667; Mateja Smogavec, orcid.org/0000-0002-1459-4802; Franco Laccone, orcid.org/0000-0001-9466-6441; Paulus Stefan Rommer, orcid.org/0000-0001-5209-6647; Gere Sunder-Plassm, orcid.org/0000-0002-9253-9921